

Translate this page into:
Trachelogenin from Urena lobata demonstrates promising potentials against SARS-CoV-2 Mpro (3CL Protease) of Endemic COVID
⁎Corresponding author. anilkumardurg1996@gmail.com (Anil Kumar)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
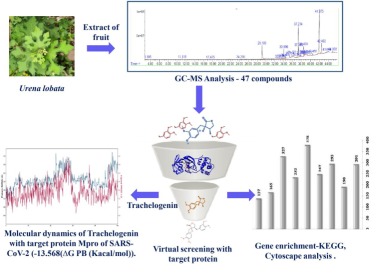
The global outbreak of acute respiratory syndrome, triggered by SARS-CoV-2, has presented a significant global challenge in the quest for compounds capable of either preventing the virus from entering host cells or impeding its replication within cells. The long persistence of the virus in the human body reported as “Endemic COVID” has compelled discoverers to find a competent compound to control the virus in endemic form. In the present study, the plant Urena lobata has been considered to search for competent molecules to inhibit virus replication, considering the anti-viral application of plants by traditional healers across the world. In our research, we identified 47 phyto-compounds present in the methanol and ethyl acetate extract obtained from the fruit of U. lobata. Among these compounds, 41 were reported in this plant for the first time. We found trachelogenin as the most potent compound against SARS-CoV-2Mpro, determined through evaluations of protein–ligand complex compatibility, drug-likeness, and assessments of molecular dynamics RMSD and RMSF. The trachelogenin shows binding ability with domain I and II of both subunits of Mpro protein with −13.568 (ΔG PB (Kacal/mol)).
Keywords
GC–MS
3CL protease
Urena lobata
RMSD
RMSF
Network pharmacology

1 Introduction
Although, World Health Organization has already declared the end of the SARS-CoV-2 pandemic but it is true that in several parts of the world, it is existing as endemic and causing a threat to human life. Nowadays reports are coming across the world that Endemic COVID is prevailing in human parts beyond their normal span of life in cells and causing various kinds of pathogenicity. SARS-CoV-2 belongs to the family of Beta corona viruses, as noted by Popoola et al.,(2022). To date, there have been reports of seven distinct strains of Human corona virus (HCoV). These include two strains from alphacorona viruses, namely 229E and NL63, and five strains from beta corona viruses, which encompasses OC43, HKU1, SARS, MERS, and COVID-19, as highlighted by Abdelli et al., (2021) and Weiss et al., (2005). SARS-CoV-2 possesses the largest RNA genome among viruses. It consists of approximately 30 kilobases of positive-sense single-stranded RNA, which is associated with nucleoprotein and features a 5′ cap and a 3′ poly-A tail, as described by Lundstrom (2020). The RNA of SARS-CoV-2 encodes two distinct groups of proteins. The first group comprises structural proteins, including the spike protein (S), matrix protein (M), and nucleoprotein (N). The second group consists of non-structural proteins (nsp), such as nsp5, also known as 3CL-protease (chymotrypsin-like protease) or Mpro (Main proteases)(Artika et al., 2020; Cascella et al., 2023; Ibrahim et al., 2021; Lundstrom, 2020; Shah et al., 2020). The protease mentioned above plays a crucial functional role in ORF1a and ORF1b, facilitating the generation of polypeptide ppla and pp1ab. These polypeptides are responsible for the synthesis of multiple non-structural proteins necessary for viral replication and maturation, including RNA-dependent RNA polymerase (RdRp)(Ibrahim et al., 2021). It has a conserved catalytic domain as reported by Hall & Ji, (2020) who has also reported that His41 and His163 are the key residues for this enzymatic activity. This makes SARS-CoV-2 Mpro a potential target for the drug development process. In this current study, the phytochemical compounds derived from Urana lobata, a plant of significant medicinal importance, were employed to investigate their potential in inhibiting the activity of the SARS-CoV-2 Mpro. Urena lobata, commonly known as Caesarweed, hibiscus bur, or Congo jute, member of Malvaceae family. It is shrub of ∼ 0.6–3 m of height with maximum ∼ 7 cm diameter of width, mostly grown in tropical and temperate zone of America, Africa, Bangladesh, China, India, Indonesia and Philippines with well-drained soil and direct sunlight region. The extract of plant root, and leaves are being used in various part of India, Africa and China as herbal medicine.U. lobata has a long history of utilization by diverse traditional healers to address a range of health conditions, including pneumonia, malaria, wounds, cough, toothache, venereal diseases, and rheumatism. It has been shown to possess potent anti-inflammatory effects (Su et al., 2018), additionally, the plant exhibits promising anti-diabetic properties, significant anti-pneumonia effects, and notable antioxidant properties, as evidenced by different researches (Ali et al., 2013; Lissy et al., 2006; Purnomo et al., 2015; Yang et al., 2017). U. lobata have shown effective curative properties against pneumonia and inflammation, and in COVID-19 also, pneumonia and inflammation are major pathogenic symptoms, so we hypothesize that this plant may have some active compounds that can be used for COVID-19 management. The objective of our study was to evaluate the inhibitory potential of U. lobata molecules against the SARS-CoV-2 Mpro. This research aimed to contribute to the management of the COVID-19 epidemic, particularly in addressing endemic COVID. To achieve this, we conducted docking studies targeting the active region of the SARS-CoV-2 Mpro with active compounds derived from U. lobata. The stability and dynamics of the protein–ligand complex were further confirmed through the analysis of Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF).
2 Materials and methods
2.1 Plant identification and extraction
In April 2021, the plant sample was collected from the Durg, Chhattisgarh, India (N 21° 11′ 49.51′', E 81° 17′ 50.837). The plant was authenticated by the Botanical Survey of India, Allahabad, (voucher specimen no. B.S.I/C.R.C. 2020–21/200) as U. lobata. The fruit samples were cleaned with double-distilled water, shade-dried for three weeks, and ground into powder. Using a Soxhlet extractor, polar and non-polar compounds were extracted with ethyl acetate and methanol at 60 °C for 5–10 h. The extracts were filtered and stored in sterile air-tight bottles for further use.
2.2 GC–MS analysis
The identification of organic substances and volatile compounds present in U. lobata were conducted using GC–MS analysis of the ethyl acetate and methanol extracts. Peak area, retention time, and molecular formula were used to confirm the presence of phyto chemical compounds in the test samples. The GC analysis utilized a SH-I-5Sil MS Capillary column measuring 30 m x 0.25 mm x 0.25um, with a splitless injection mode. Helium gas was used as the carrier gas at a flow rate of 1 mL/min, and the sample components were ionized at 70 eV. The GC oven temperature was initially set at 45 °C for 2 min, then ramped up to 140 °C at a rate of 5 °C/min, followed by a further increase to 280 °C, where it was held isothermally for 10 min. A sample injection volume of 2 μL was used, and the GC run time spanned from 9.10 min to 52.0 min. The identification of compounds was achieved by comparing their mass fragmentation patterns and retention times. To determine the structure of the identified compounds, their mass fragmentation patterns were matched with authentic compounds from the NIST 14.L library (2020) in the USA, in accordance with Mallard and Linstrom, (2001).
2.3 Drug-likeliness by Lipinski and Veber rules
The drug-likeliness of the compounds was evaluated based on the principles outlined by Lipinski's “Rule of Five.” These rules consider several factors, including the number of hydrogen bond donors not exceeding 5, the number of hydrogen bond acceptors not exceeding 10, the molecular weight not exceeding 500, and the logarithm of the partition coefficient (logP) not exceeding 5 and Veber (according to which compound should have less than or equal to 10 rotatable bond (RB) & less than or equal to 140 topological polar surface area (TSPA). Ligands that successfully pass through the filters possess a higher likelihood of demonstrating favorable oral bioavailability, hence were considered (Daina et al., 2017; Lipinski, 2004).
2.4 Pharmacological prediction
The compounds identified by GC–MS were searched in the PubChem database (pubchem.ncbi.nlm.nih.gov/) and retrieved in sdf format. These files were used to identify the target using the Swisstargetpredition tool (https://www.swisstargetprediction.ch/). The target predicated were then submitted to a string database (https://string-db.org/) to retrieve information related to interaction among the identified target. Multi-protein inputs were given into the database, Homo sapience as the target organism, and higher confidence scores were taken into search criteria after disconnected nodes were removed. This relationship between targets was downloaded in tsv format and imported into cytoscape for further analysis. The network was further analyzed and enrichment was performed to retrieve more information such as the involvement of identified target genes in different biological processes, cellular compartments, molecular functions, and pathways by using a string plug-in in the cytoscape.
2.5 Screening and dynamic simulation of phyto-compounds on SARS-CoV-2 Mpro
The structure of the compounds were obtained from the PubChem database and converted from sdf to mol2 format using OpenBabel(Boyle et al., 2011). Polar hydrogens were added, and the molecule was saved in pdbqt format using AutoDock Tool 1.5.6 (ADT) (Morris et al., 2009). The structure of SARS-CoV-2S Mpro (6LU7) was retrieved from the protein databank and prepared by removing water molecules and ligands, adding polar hydrogen atoms, charges, and converting it to pdbqt format using ADT. The molecular docking was conducted using Autodock vina following grid parameter as center_x = 15.21, center_y = 14.71, center_z = 15.88, size_x = 46.81, size_y = 39.19, size_z = 42.20. In our dynamic investigations, we employed the Schrodingers Desmond tool to assess the stability of the complex structure acquired from docking studies. Desmond conducted free energy calculations, factoring in the temperatures. To ensure neutrality, we used the OPLS (Optimized Potentials for Liquid Simulations) force field for both the protein and ligand, along with Sodium (Na+) and calcium ions (Cl-). A duration of 100 ns (ns) Molecular dynamics(MD) was run within an constant Number of Particles, Pressure, and Temperature (NPT) ensemble. We analyzed the data by evaluating the RMSD and RMSF of the protein and ligand.
3 Results
3.1 GCMS analysis
In GC–MS study of methanol extract, we identified 24 compounds (Table 1, Fig. 1),among which 10 compounds from methanol extract were 1,4-Benzenedicarboxylic acid, bis(2-ethylhexyl) ester (47.03 %); 9,12-Octadecadienoic acid (Z,Z)-, methyl ester (12 %); Dodecyl acrylate(10.92 %); Hexadecanoic acid, methyl ester (5.14 %); trans-13-Octadecenoic acid, methyl ester(4.77 %); 9,12-Octadecadienoic acid, ethyl ester(4.18 %); Hexanedioic acid, bis(2-ethylhexyl) ester(3.87 %); Propanoic acid, 3-mercapto-, dodecyl ester(2.13 %); Hexadecanoic acid, ethyl ester(1.13 %); Isopropyl linoleate(1.04 %); as a major compound and were comprised total 92.93 %. Note – Highlighted row in pink color showing the compounds present in both the extract. GCMS = gas chromatography mass spectrometry).


GC–MS graph of methanol extract of U. lobata.
In the ethyl-acetate extract, we detected 39 compounds (Table 1, Fig. 2,)among which 15 compounds were reported up to 82.56 %. These are 3-Chloropropionic acid, heptadecyl ester(22.81 %); Diisooctyl adipate(22.81 %); 9,12-Octadecadienoic acid (Z,Z)-, methyl ester(7.85 %); Propanoic acid, 3-mercapto-, dodecyl ester(5.22 %); 9,12-Octadecadienoic acid, ethyl ester(3.73 %); Hexadecanoic acid, methyl ester(3.40 %); 1-Dodecanol(3.02 %); Hexadecanoic acid, ethyl ester(2.85 %); 1,2,3-Propanetriol, 1-acetate(2.67 %); Hexanedioic acid, bis(2-ethylhexyl) ester(1.86 %); 9-Octadecenoic acid (Z)-, methyl ester(1.80 %); 1-Nonene, 4,6,8-trimethyl-(1.26 %); Hexanoic acid, 2-ethyl-, hexadecyl ester(1.21 %); Octane, 2-bromo-(1.07 %); Ethyl Oleate(1.00 %). After combining and removal of duplicates of all the compounds from both the extract, atotal of 47 compounds were found unique. Among these compounds, the majority were fatty acid esters in nature, and out of these 47, biologically active reported compounds were 19(Table S1 and Fig. 6).
GC–MS graph of ethyl-acetate extract of U. lobata.
Nine compounds highlighted in pink (Table 1) namely 1,4-Benzenedicarboxylic acid, bis(2-ethylhexyl) ester; 9,12-Octadecadienoic acid (Z,Z)-, methyl ester; Hexadecanoic acid, methyl ester; 9,12-Octadecadienoic acid, ethyl ester; Hexanedioic acid, bis(2-ethylhexyl) ester; Propanoic acid, 3-mercapto-, dodecyl ester; Hexadecanoic acid, ethyl ester; Methyl stearate; and Hexanoic acid, 2-ethyl-, hexadecyl ester were detected in both the extract. These compounds were found higher in methanol extract compared to ethyl-acetate extract except Propanoic acid, 3-mercapto-, dodecyl ester; Hexadecanoic acid, ethyl ester; and Hexanoic acid, 2-ethyl-, hexadecyl ester which were found higher in ethyl-acetate extract in terms of total peak percentage. All of these newly identified compounds and the compounds previously identified were considered for evaluation of drug-likeness properties using the Lipinski & Veber rule. Compounds were also checked for the presence of PAINS and Brenk alert and we found that 9 compounds fulfilled all the criteria of drug-likeness (Tables 2 and 3). (MW = Molecular weight; RB = Rotatable bond; HBA = Hydrogen bond donor; HBA = Hydrogen bond acceptor; TPSA = topological surface area; PAINS = Pan-assay interference compounds). (GI = Gastro-intestinal; BBB = blood brain barrier; PGP = P-glyco-protein; CYP = Cytochrome 450).
Name of the compounds
Molecular formula
MW
RB
HBA
HBD
TPSA
LOGP
Lipinski violations
Veber violations
PAINS alerts
Brenk alerts
Nonanoic acid, methyl ester
C10H20O2
172.26
8
2
0
26.3
2.58
0
0
0
0
1-Undecanol
C11H24O
172.31
9
1
1
20.23
3.13
0
0
0
0
1-Dodecanol
C12H26O
186.33
10
1
1
20.23
3.41
0
0
0
0
Phenol, 2,4-bis(1,1-dimethylethyl)-
C14H22O
206.32
2
1
1
20.23
3.87
0
0
0
0
Benzenepropanoic acid, 3,5-bis(1,1-dimethylethyl)-4-hydroxy-, methyl ester
C18H28O3
292.41
6
3
1
46.53
3.77
0
0
0
0
Kaempferol,
C15H10O6
286.24
1
6
4
111.13
−0.03
0
0
0
0
Trachelogenin
C21H24O7
388.41
7
7
2
94.45
1.32
0
0
0
0
Apigenin
C15H10O5
270.24
1
5
3
90.9
0.52
0
0
0
0
Chrysoeriol
C16H12O6
300.26
2
6
3
100.13
0.22
0
0
0
0
Name of the compounds
GI absorption
BBB permeant
PGP substrate
CYP
1A2 inhibitorCYP
2C19 inhibitorCYP
2C9 inhibitorCYP
2D6 inhibitorCYP
3A4 inhibitor
Nonanoic acid, methyl ester
High
Yes
No
No
No
No
No
No
1-Undecanol
High
Yes
No
Yes
No
No
No
No
1-Dodecanol
High
Yes
No
Yes
No
No
No
No
Phenol, 2,4-bis(1,1-dimethylethyl)-
High
Yes
No
No
No
No
Yes
No
Benzenepropanoic acid, 3,5-bis(1,1-dimethylethyl)-4-hydroxy-, methyl ester
High
Yes
No
No
No
No
Yes
No
Kaempferol,
High
No
No
Yes
No
No
Yes
Yes
Trachelogenin
High
No
Yes
No
No
No
Yes
Yes
Apigenin
High
No
No
Yes
No
No
Yes
Yes
Chrysoeriol
High
No
No
Yes
No
Yes
Yes
Yes
The ADME (Absorption, Distribution, Metabolism, and Excretion) properties of the nine compounds were additionally predicted using the swissADME tool. The gastrointestinal property of all compounds was high. Nonanoic acid, methyl ester; 1-Undecanol; 1-Dodecanol; Phenol, 2,4-bis(1,1-dimethylethyl)-; and Benzenepropanoic acid, 3,5-bis(1,1-dimethylethyl)-4-hydroxy-, methyl ester were found blood brain barrier permeable. Only Trachelogenin was found as a PGP substrate which is involved in the efflux of foreign compounds. The cytochrom 450 family, which is involved in the metabolism of various molecules of our body was found inhibited by various compounds such as CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4. 1-Undecanol; 1-Dodecanol; Kaempferol; Apigenin; and Chrysoeriol were identified as inhibitor of CYP1A2.None of the compounds was found involved in the inhibition of CYP2C19. Except for Nonanoic acid, methyl ester; 1-Undecanol; 1-Dodecanol the remaining all 6 compounds were predicted as an inhibitor of CYP2D6, and Only Chrysoeriol was identified as an inhibitor of CYP2C9Besides, Kaempferol, Trachelogenin, Apigenin, Chrysoeriol were found as inhibitor of CYP3A4.
3.2 Pharmacological prediction
In our study, 443 target proteins were identified using Swiss target prediction for all the compounds. Further protein–protein interaction analysis was performed using Cytoscape. Additionally, functional enrichment analysis of the proteins was conducted using the STRING app, which revealed a comprehensive range of biological processes (BP) totaling 2250, 145 cellular components (CC), 312 molecular functions (MF), and 192 KEGG pathways. The analysis of the top 10 significant BP (FDR < 0.05 and p < 0.01) (Fig. 3) revealed that the identified proteins predominantly participate in the regulation of biological qualities; response to − oxygen-containing compounds, chemicals, organic substances, and stimuli; cellular response to chemical stimuli, homeostatic processes, and various metabolic processes. Regarding the top 10 CC (FDR < 0.05 and p < 0.01), it was observed that these proteins are primarily localized in the plasma membrane, serve as intrinsic and integral components of the plasma membrane, are associated with the cell periphery, plasma membrane region, synapse, cell junction, membrane raft, and cytoplasm(Fig. 3). and perform MF (FDR < 0.05 and p < 0.01) like catalytic activities, signaling receptor activities, small molecule binding, ion binding, protein kinase activities, transmembrane signaling receptor activities, protein bindings, catalytic activities, acting on a protein, phosphotransferase activities, alcohol group as acceptor, as a major activity(Fig. 3).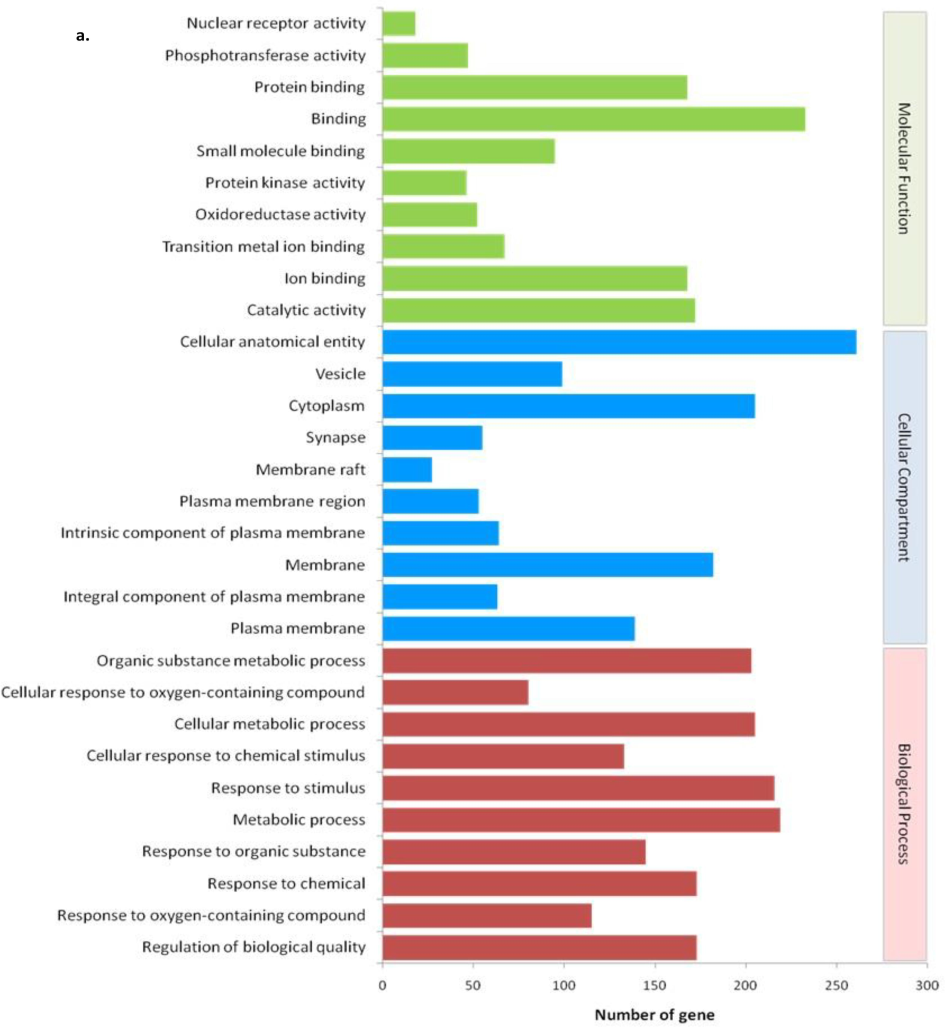
A. involvement of phyto-compounds in various biological process, cellular compartment, molecular function.b− Involvement of phyto-compounds in various pathways of KEGG for human (FDR* <= 0.05 & P# < 0.001) (KEGG = Kyoto Encyclopedia of Genes and Genomes; FDR* = False Discovery Rate; P# = probability).
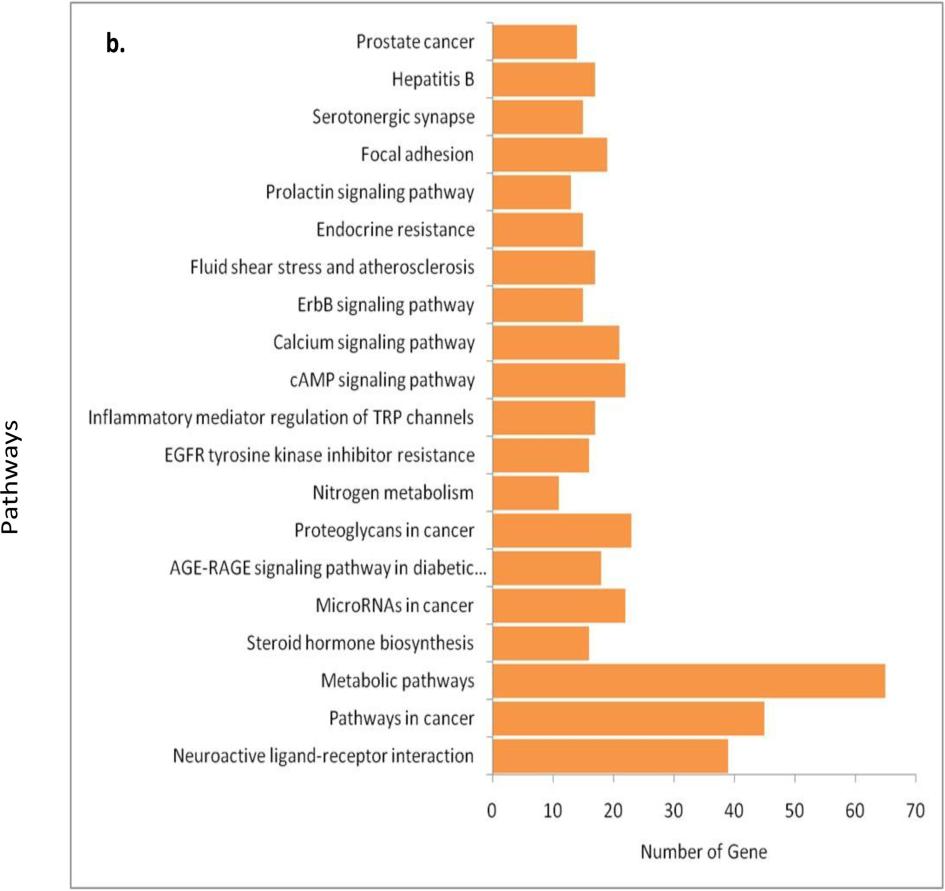
A. involvement of phyto-compounds in various biological process, cellular compartment, molecular function.b− Involvement of phyto-compounds in various pathways of KEGG for human (FDR* <= 0.05 & P# < 0.001) (KEGG = Kyoto Encyclopedia of Genes and Genomes; FDR* = False Discovery Rate; P# = probability).
In the 192 KEGG Pathways terms, top-20 were selected based on p-value which showed that these proteins were involved in neuroactive ligand-receptor interaction, cancer pathways, cAMP signaling pathway, calcium signaling, inflammatory mediator regulation of TRP channels, MAPK signaling, proteoglycans in cancer, metabolic pathways, serotonergic synapse, vascular smooth muscle contraction, microRNAs in cancer, sphingolipid EGFR tyrosine kinase inhibitor resistance, endocrine resistance, insulin resistance, signaling pathway, AGE-RAGE signaling pathway in diabetic complications, apoptosis, ras signaling pathway, focal adhesion, and Kaposi sarcoma-associated herpesvirus infection. On the basis of their degree value, the importance of nodes in the network showed SRC, HSP90AA1, STAT3, MAPK3, MAPK1, EGFR, AKT, JUN, ESR, IIK3CA as the potential key proteins target in the human body (Fig. 3).
3.3 Virtual screening of ligands
Virtual screening was performed to identify ligands with higher binding affinity to the protein, yielding promising results. Based on Hall and Ji (2020), amino acids His 41 and His 163 play key roles in protein function, and this site was used for screening compounds viz. Nonanoic acid, methyl ester; 1-Undecanol; 1-Dodecanol Benzenepropanoic acid, 3,5-bis(1,1-dimethylethyl)-4-hydroxy-, methyl ester;; Phenol, 2,4-bis(1,1-dimethylethyl)-;Kaempferol; Trachelogenin; Apigenin; and Chrysoeriol which are passed in drug-likeness filter of Lipinski and Veber (Table 2). The highest binding interaction was recorded for trachelogenin among all nine compounds (Table 4) by docking, based interaction score, and selected this compound for MD-simulation.
Molecule Name
Interacting Molecules
Affinity score
Phenol, 2,4-bis(1,1-dimethylethyl)-
GLY15, TRP31, ALA70, LYS97
−5.5
1-Undecanol
HIS41, MET49, MET165
−4.1
1-Dodecanol
HIS41, MET165,
−4.0
Nonanoic acid, methyl ester
TRP31, ALA70, GLY71, VAL73, PRO96, LYS97
−4.1
Benzenepropanoic acid, 3,5-bis(1,1-dimethylethyl)-4-hydroxy-, methyl ester
PHE140, HIS163, MET165, GLU66
−6.2
Trachelogenin
MET49, ASN142, GLY143, MET165, GLU166, LEU167, ARG188,
−6.9
Apigenin
PHE140, MET165, GLU166, GLN189, THR190,
−6.8
Chrysoeriol
LEU141, HIS163, MET165, GLU166, GLN189, THR190
−6.7
Kaempferol
MET49, LEU141, CYS145, HIS163, GLU166
−6.8
A MD-simulation was conducted to investigate the dynamic behavior of the system over time, providing insights into the flexibility and dynamics of the protein-drug interaction (Figs. 4-5). The RMSD analysis (Fig. 5), was performed on the protein backbone atoms, indicating the overall stability of the simulated system. Regarding the compound trachelogenin, it initially exhibited fluctuations but eventually reached a stable state. Furthermore, the RMSF analysis of the protein–ligand complex, specifically focusing on compound trachelogenin (Fig. 5), revealed some fluctuations at the terminal residues. This observation may be attributed to the inherent flexibility present in the head and tail region of the compound(Aris et al., 2021). The shaded green zone shown in the RMSF graph indicates the ligand binding residues. Fig. 5 shows the interacting residues of Mpro and the contact probability chart of residues with the ligand. ASN 142, GLY143, and GLU166 interacted with the ligand via hydrogen bonds throughout the simulation frame (green bar in Fig. 5). MET49, MET165, and LEU167 made contact with the ligand through hydrophobic interactions. Among these residues, MET165 had the most frequent hydrophobic contact with the ligand. Apart from these, there were other interacting residues such as THR26, LEU27, HIS41, PHE140, LEU141, SER144, CYS145, HIS163, LEU167, HIS172, VAL186, ASP187, GLN189, THR190, and GLN92, which contacted with ligand during the simulation but are less prominent.
Ligand interaction with the of SARs-CoV-2 Mpro residues (a) Ligand dock pose & interacting amino acid (b) Type of interaction with Mpro residues throughout the simulation.
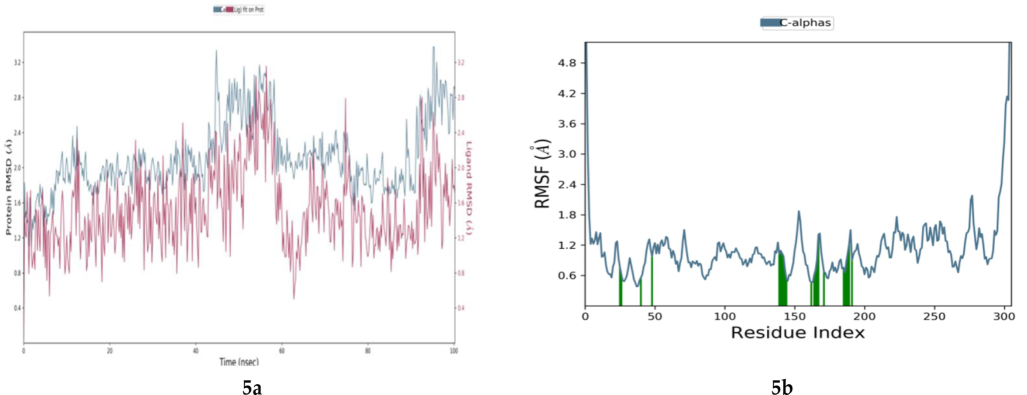
Molecular Dynamics of protein ligand complex. (a.) root mean square devation (RMSD) of protein (blue lines) and protein–ligand complex (red lines) (b.) root mean square fluctuation (RMSF) of SARs-CoV-2 Mpro. Green bar representing the interacting residue area in the protein ligand complex.
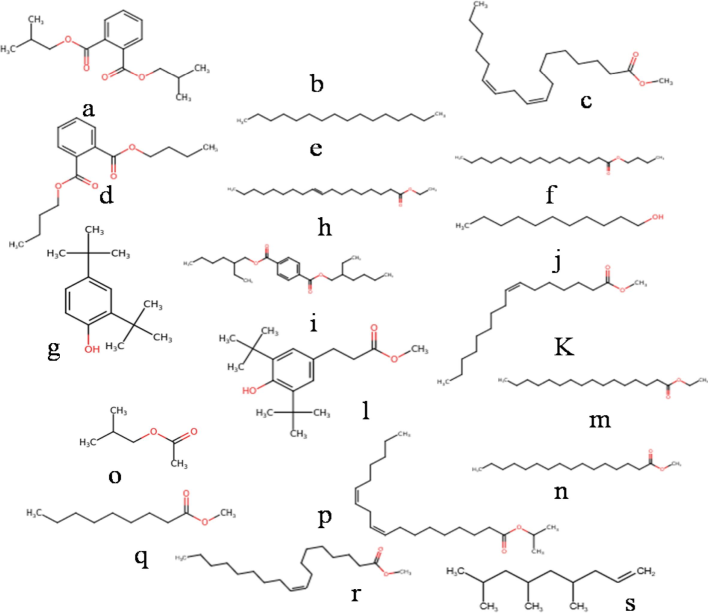
Structure of active phyto-compound reported for various ailments (a) 1,2-Benzenedicarboxylic acid, bis(2-methylpropyl) ester; (b) 1-Dodecanol; (c) 9,12-Octadecadienoic acid (Z,Z)-, methyl ester; (d) Dibutyl phthalate; (e) Hexadecane; (f) Hexadecanoic acid, butyl ester; (g) Phenol, 2,4-bis(1,1-dimethylethyl)-; (h) (E)-9-Octadecenoic acid ethyl ester; (i) 1,4-Benzenedicarboxylic acid, bis(2-ethylhexyl) ester; (j) 1-Undecanol; (k) 7-Hexadecenoic acid, methyl ester, (Z)-; (l) Benzenepropanoic acid, 3,5-bis(1,1-dimethylethyl)-4-hydroxy-, methyl ester; (m) Hexadecanoic acid, ethyl ester; (n) Hexadecanoic acid, methyl ester; (o) Isobutyl acetate; (p) Isopropyl linoleate; (q) Nonanoic acid, methyl ester; (r) 9-octadecenoic acid (Z)- methyl ester; (s) 1-Nonene, 4,6,8-trimethyl-.
4 Discussion
In our current research, the analysis of the methanol and ethyl-acetate extracts derived from the fruit of U.lobata revealed the presence of 47 phyto-compounds. These compounds exhibit diverse biological activities, including anti-quorum sensing, anti-biofilm, anti-bacterial, insecticidal, anti-androgenic, anti-cancer, lipase production stimulation, inhibition of glucose metabolism, anti-sickling, anti-inflammatory, allelochemical, anti-fungal, anti-neurodegenerative disorder, anti-diabetic, anti-oxidant was reported (Table S1 & Fig. 6). Ample evidence supports the potential of herbal products as a valuable source for the development of novel antiviral treatments (Ibrahim et al., 2021). Prior research has elucidated the viral mechanisms of SARS-CoV-2, encompassing crucial aspects such as viral entry, replication, assembly, release, and the interplay of viral proteins and enzymes (Weiss and Navas-Martin, 2005). Notably, key players in the virus's life cycle include SARS-CoV-2 Mpro, RdRp (RNA-dependent RNA polymerase), S protein (spike glycoprotein), and TMPRSS2 (transmembrane protease serine 2). Understanding these mechanisms is pivotal in exploring and harnessing the potential of herbal remedies in combating viral infections. In the network analysis SRC, HSP90AA1, STAT3, MAPK3, MAPK1, EGFR, AKT, JUN, ESR, and IIK3CA were identified as potential targets for the compounds we used in the present study. These proteins are mostly involved in cell growth, differentiation, metabolism, apoptosis, cytokines modulation(KEGG map05171 and map04151), etc. It is also reported that disturbances in the arrangement of tight junctions and elevation in the permeability of epithelial cells are common phenomena in acute and long-term inflammatory disorders, triggered by pollutants, microbial byproducts, oxidative agents, proteases, and cytokines(Basuroy et al., 2005; Petecchia et al., 2012; Turner, 2006). Petecchia et al., (2012) reported TNF alpha, IL-4 and IFN-gamma Cytokine-induced damage to the epithelium involves the breakdown of tight junctions and alteration in the permeability of the epithelial barrier, and the process is mediated by the EGFR-dependent MAPK/ERK1/2 signaling pathway. When bovine chondrocytes are stimulated with cytokines like IL-1 and TNFα, they produce ROS (reactive oxygen species). This leads to the expression of c-jun, which is regulated by its gene product through JNKs (c-Jun NH2-terminal kinases). H2O2 and NO, both types of ROS, also stimulate JNK activity. Flavonoid-containing enzymes are involved in the ROS-mediated signaling process, as suggested by the inhibitory effect of diphenyleneiodonium on JNK activation(Lo et al., 1996). In our study, we found potential antioxidant features in U. lobata and confirmed that the molecules of U. lobata have the property to interfere with the mechanism triggered by SARS-CoV-2 and to reduce its pathogenicity.
In the context of SARS-CoV-2 research and development, the spike protein and 3CLpro/Mpro are widely acknowledged as the primary pharmacological targets. These targets have garnered significant attention due to their crucial role in the pathogenesis and replication of the virus. Extensive research and development efforts are focused on these specific targets to identify potential therapeutic interventions against SARS-CoV-2(Liu et al., 2020). With various Medicinal properties in U. lobata we wonder whether the plant can also be used for anti-covid related constituents. We thus performed molecular docking with various molecules identified from U.lobata and SARS-CoV-2 3CLpro/Mpro to identify potential inhibition. The Mpro plays a crucial role in the maturation of various functionally active protein units. Due to the highly conserved nature of its active site, it is considered a promising target for potential interventions. In our study, we focused on identifying a potential anti-SARS-CoV-2 drug candidate that could form a stable complex with the targeted protein. The selection criteria for this candidate revolved around achieving the lowest possible binding score during binding interactions. The structural arrangement of SARS-CoV-2 Mpro encompasses three distinct domains: domain-I (residues 8–101), domain-II (residues 102–184), and domain-III (residues 201–303). Within the protein, the active site is situated at the interface of domain I and domain II. The active site can be further divided into several subunits, including hydrophobic pocket S1, which is formed by the side chains of Phe140, His163, and Glu166, along with the main chain atoms of Leu141, Met165, Glu166, and His172. Another subunit, S2, is composed of the side chains of His41, Met49, Pro52, Met165, and Gln189. Additionally, S3 sub-locations are formed by the side chains of Pro168, Gln189, and Gln192, along with the main chain atoms of Glu166, Gln189, and Thr190. The residues His41 and Cys145 combine to create the catalytic dyad, which plays a crucial role in the process of proteolysis. Recently Hall and Ji, (2020) identified amino acids His 41 and His 163 as important residues for protein function. Here molecular docking and binding interaction analysis revealed that compound trachelogenin shows the interaction in domain-I & domain-II subunits of the active region of the protein. Compound trachelogenin also interacted with LEU27, HIS41, and MET49, by hydrophobic interaction, in domain-I and hydrophobic interaction with CYS145, MET165, GLU166, LEU167, ARG188, and GLN189 residues detected in domain-II in the active pocket region(Hall and Ji, 2020).
Some similar studies have been reported previously but not with trachelogenin(Beck et al., 2020; Chen et al., 2005; Chowdhury et al., 2018; Hagar et al., 2020; Jo et al., 2020; Lung et al., 2020). In a study conducted by Chen et al.,(2005) a comprehensive examination of a library containing 720 natural molecules was carried out to assess their inhibitory effects on the SARS-CoV-1 3C-like Protease. The findings highlighted two particular compounds, namely tannic acid (IC50 = 3 µM) and 3-isotheaflavin-3-gallate (IC50 = 7 µM), demonstrating significant inhibitory properties against the target proteaseSeveral polyphenolic compounds found in black tea, namely theaflavin, theaflavin-30-monogallate, and theaflavin-3–30-digallate, have been documented for their antiviral properties against the hepatitis C virus, as reported by Choudhury et al.,(2015). In a study by Jo et al.(2020), among 64 flavonoid metabolites, herbacetin (−9.263), rhoifolin (−9.565), and pectolinarin (−8.054) exhibited significant inhibitory activity against the SARS-CoV Mpro protein. In a study by Hagar et al., (2020), hydroxychloroquine and remdesivir were reported to exhibit binding energies of −6.06 and −4.96 kcal/mol, respectively, against the Mpro of SARS-CoV-2. Back et al.,(2020) identified atazanavir as the most promising drug candidate against 3CLpro, with a binding affinity of −7.40 kcal/mol. This was followed by remdesivir (−6.40 kcal/mol), efavirenz (−5.40 kcal/mol), ritonavir (−6.80 kcal/mol), and dolutegravir (−7.20 kcal/mol).In our current journey of a potential lead, we found the potential binding interaction and energy value for compound Trachelogenin.
5 Conclusions
The primary objective of this study was to explore the natural compounds derived from U. lobata and assess their potential as inhibitors against the SARS-CoV-2 3CL protease. Our study revealed 47 compounds in the fruit of U. lobata, among them 19 were previously reported to have different biological activities and the rest were unexamined. All 47 compounds reported in the present study and 32 reported in the previous study were processed for network pharmacological study, of which 9 compounds passed the drug-likeness filter out of 79 ligands. A molecular docking study has identified Trachelogenin as the most active and suitable compound in the present study, and to assessthe stability of the protein–ligand complex, we examined the structural dynamics of the system using Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF) analyses. The highest inhibitory activity was found in Trachelogenin which suggests that Trachelogenin is the most appropriate lead compound to inhibit SARS CoV-2 and is considered for endemic COVID episode. It is also being reported from across the world and is supposed that the virus persists in the intestine of patients beyond their normal life cycle.
CRediT authorship contribution statement
Dinesh Kumar: Investigation. Somendra Kumar: Writing – original draft. Motiram Sahu: Writing – original draft. Chandramohan Govindasamy: Writing – review & editing, Conceptualization. Anil Kumar: Supervision, Conceptualization.
Funding
We acknowledge the University Grants Commission, India for providing financial research support (Id: MAY2018-373173); King Saud University for Researcher Supporting Project [ RSPD2025R712].
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from Western Algeria. J. Biomol. Struct. Dyn.. 2021;39:3263-3276.
- [CrossRef] [Google Scholar]
- Antioxidant and Cytotoxic Activities of Methanol Extract of Urena lobata (L) Leaves. The Pharma. Innovation. 2013;2
- [Google Scholar]
- Identification of potential riboflavin synthase inhibitors by virtual screening and molecular dynamics simulation studies. J. King Saud Univ. – Sci.. 2021;33:101270
- [CrossRef] [Google Scholar]
- MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem. J. 2005;393:69-77.
- [CrossRef] [Google Scholar]
- Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J.. 2020;18:784-790.
- [CrossRef] [Google Scholar]
- Cascella, M., Rajnik, M., Aleem, A., Dulebohn, S.C., Di Napoli, R., 2023. Features, Evaluation, and Treatment of Coronavirus (COVID-19), in: StatPearls. StatPearls Publishing, Treasure Island (FL).
- Inhibition of SARS-CoV 3C-like Protease Activity by Theaflavin-3,3’-digallate (TF3) Evid. Based Complement. Alternat. Med.. 2005;2:209-215.
- [CrossRef] [Google Scholar]
- Medicinal plants with potential anti-arthritic activity. J Intercult Ethnopharmacol. 2015;4:147-179.
- [CrossRef] [Google Scholar]
- Theaflavins, polyphenols of black tea, inhibit entry of hepatitis C virus in cell culture. PLoS One. 2018;13
- [CrossRef] [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [CrossRef] [Google Scholar]
- Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci.. 2020;21:3922.
- [CrossRef] [Google Scholar]
- A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis.. 2020;35:101646
- [CrossRef] [Google Scholar]
- Natural-like products as potential SARS-CoV-2 Mpro inhibitors: in-silico drug discovery. J. Biomol. Struct. Dyn.. 2021;39:5722-5734.
- [CrossRef] [Google Scholar]
- Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzyme Inhib. Med. Chem.. 2020;35:145-151.
- [CrossRef] [Google Scholar]
- The NIST Chemistry WebBook: A Chemical Data Resource on the Internet. J. Chem. Eng. Data. 2001;46:1059-1063.
- [CrossRef] [Google Scholar]
- Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol.. 2004;1:337-341.
- [CrossRef] [Google Scholar]
- Antioxidant potential of Sida retusa, Urena lobata AND Triumfetta rhomboidea. Anc. Sci. Life. 2006;25:10-15.
- [Google Scholar]
- Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci.. 2020;6:315-331.
- [CrossRef] [Google Scholar]
- Reactive Oxygen Species Mediate Cytokine Activation of c-Jun NH2-terminal Kinases *. J. Biol. Chem.. 1996;271:15703-15707.
- [CrossRef] [Google Scholar]
- The potential chemical structure of anti-SARS-CoV-2 RNA-dependent RNA polymerase. J. Med. Virol.. 2020;92:693-697.
- [CrossRef] [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30:2785-2791.
- [CrossRef] [Google Scholar]
- Cytokines induce tight junction disassembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab. Invest.. 2012;92:1140-1148.
- [CrossRef] [Google Scholar]
- West African medicinal plants and their constituent compounds as treatments for viral infections, including SARS-CoV-2/COVID-19. DARU J Pharm Sci. 2022;30:191-210.
- [CrossRef] [Google Scholar]
- Anti-diabetic potential of Urena lobata leaf extract through inhibition of dipeptidyl peptidase IV activity. Asian Pac. J. Trop. Biomed.. 2015;5:645-649.
- [CrossRef] [Google Scholar]
- In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci.. 2020;252:117652
- [CrossRef] [Google Scholar]
- Megastigmane glycosides from Urena lobata. Fitoterapia. 2018;127:123-128.
- [CrossRef] [Google Scholar]
- Molecular Basis of Epithelial Barrier Regulation: From Basic Mechanisms to Clinical Application. Am. J. Pathol.. 2006;169:1901-1909.
- [CrossRef] [Google Scholar]
- Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev. 2005;69:635-664.
- [CrossRef] [Google Scholar]
- The antibacterial effect of Urena lobata L. Fromv guangxi on mice with staphylococcus aureus pneumonia. Afr. J. Tradit. Complement. Altern. Med.. 2017;14:73-88.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2024.103572.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




