

Translate this page into:
The structural, electro-optical, charge transport and nonlinear optical properties of oxazole (4Z)-4-Benzylidene-2-(4-methylphenyl)-1,3-oxazol-5(4H)-one derivative
⁎Corresponding author at: Department of Chemistry, Faculty of Science, King Khalid University, Abha 61413, P.O. Box 9004, Saudi Arabia. Fax: +966 72418426. irfaahmad@gmail.com (Ahmad Irfan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The oxazole compounds are being used for multifunctional purposes ranging from organic light emitting diodes, organic thin film transistors, and photovoltaic to the nonlinear optical materials. In this study, several structural, electro-optical, charge transport and nonlinear optical properties of (4Z)-4-Benzylidene-2-(4-methylphenyl)-1,3-oxazol-5(4H)-one (BMPO) have been investigated. Density functional theory (DFT) and time dependent DFT are very accurate and reasonable approaches to optimize the ground and excited state geometries, respectively. Thus, in the present study DFT and TDDFT methods with the B3LYP/6-31G∗∗ levels of theory have been applied to shed some light on the structure-property relationship, frontier molecular orbitals (FMOs), optical properties. A clear intra-molecular charge transfer (ICT) from the highest occupied molecular orbitals (HOMOs) to the lowest unoccupied molecular orbitals (LUMOs) has been observed. The ionization potentials (IP), electron affinities (EA), total and partial densities of states have been discussed intensively. The electron reorganization energy of oxazole compound (BMPO) is smaller than the hole reorganization energy revealing that it might be good electron transport contender in OLED. The electron reorganization energy of BMPO is calculated to be 0.223 eV that is smaller than the perfluoropentacene (value is 0.250 eV), which is famous n-type semiconductor material. The first pathway of BMPO has almost comparable hole and electron transfer integral values whereas the calculated electron reorganization energy (0.223 eV) is considerably lower than the hole reorganization energy (0.381 eV) which leads to superior electron intrinsic mobility of the studied oxazole derivative as compared to the hole one. It is expected that BMPO might be excellent electron transport material.
Keywords
Semiconductors
Ab initio calculations
Electronic properties
Charge transport properties
Optical properties

1 Introduction
Organic semiconductor materials (OSMs) are enormously studied at both theoretical and experimental levels, since first reported in 1986 (Tsumura et al., 1986) due to light weight, low fabrication cost and large area bendy displays (Azam et al., 2014; Reshak et al., 2014). These advantages of OSMs give them edge over the silicon-based traditional inorganic semiconductors and attracted huge interest of academic researchers as well as their industrial partners because of their potential applications in photonics and electronic devices, such as organic field effect transistors (OFETs) (Irfan et al., 2016), organic light emitting diodes (OLEDs) (Ho et al., 2000; Tang and VanSlyke, 1987), organic photo-voltaic (OPVs) (Chaudhry et al., 2016; Ghebouli et al., 2016; Jin et al., 2016) and organic light-emitting transistors (OLETs) (Chaudhry et al., 2014; Irfan et al., 2015b).
Oxazole is the parent complex of a huge class of heterocyclic aromatic OSMs, which is azole with one oxygen and nitrogen separated by one carbon atom (Merkul and Muller, 2006). The oxazole derivatives gained significant attention as biologically active competitors, nonlinear optical materials and semiconducting devices (Wang et al., 2015). Previously, electron-deficient units were integrated to architect the n-type material (Lee et al., 2009). Earlier studies pointed out that the oxazole compounds delivered proficient electron transport as well as hole blocking properties (Zeng et al., 2015). The oxazole derivatives have been reported as good candidates for n-type OSMs due to their small reorganization energies and low energy levels of lowest unoccupied molecular orbitals (LUMOs) (Huong et al., 2014).
Hitherto, oxazole compound, i.e., (4Z)-4-Benzylidene-2-(4-methylphenyl)-1,3-oxazol-5(4H)-one (BMPO) has been synthesized and its crystal structure was reported (Asiri et al., 2015) as shown in Fig. 1. In the present study our aim is to shed light on the structural, electronic (highest occupied molecular orbitals (HOMOs), LUMOs, total/partial density of states (TDOS/PDOS)), molecular electrostatic potential (MEP), absorption (λabs) and fluorescence (λfl) spectra and charge transport properties (ionization potentials (IPs), electron affinities (EAs), hole/electron reorganization energies (λh/λe), hole/electron transfer integrals (Vh/Ve)), hole/electron intrinsic mobility and nonlinear optical properties of BMPO. No computational investigations of BMPO for above mentioned properties have been found in the literature so far. The paper is structured as follows: Section 2 presents an outline of the Density Functional Theory (DFT) and Time Dependent Density Functional Theory (TDDFT); Section 3 gives the frontier molecular orbitals, electronic, optical and nonlinear optical and charge transport properties; in Section 4 the major conclusions of the present investigation are presented.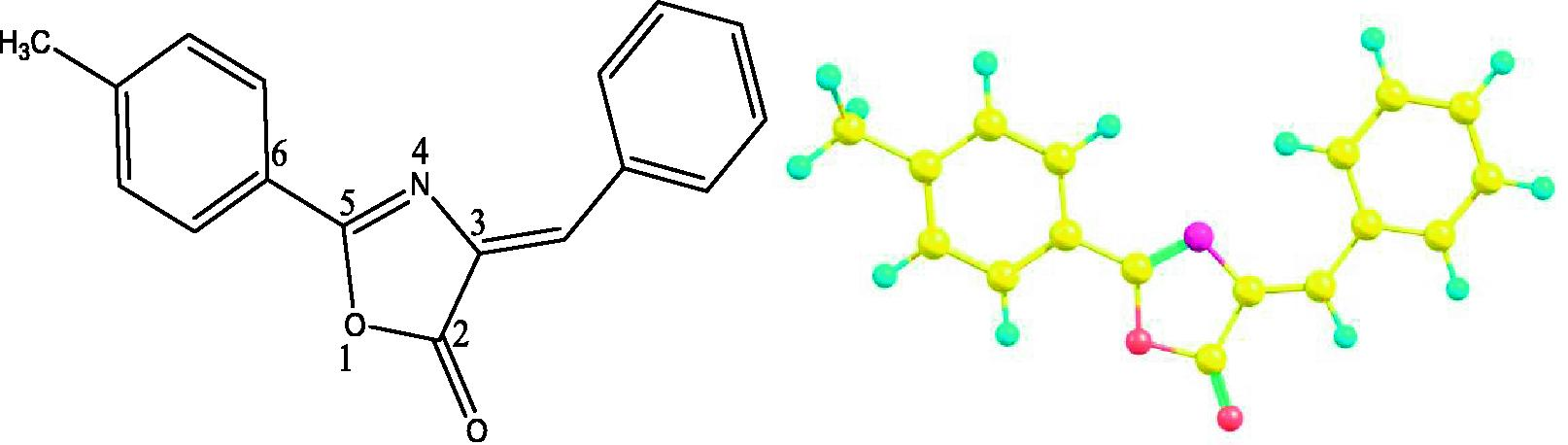
The oxazole derivative (BMPO) with numbering scheme (left) and optimized geometry (right).
2 Computational details
The B3LYP (Becke, 1993; Lee et al., 1988) delivers the best depiction among the standard DFT (Irfan et al., 2015a) functionals which is a good choice for small molecules (Cvejn et al., 2016; Irfan and Al-Sehemi, 2015; Irfan et al., 2015b; Wong and Cordaro, 2008). The B3LYP was used to predict and calculate the properties of interests which were in good agreement with experimental evidence. The charge transfer, structural and electro-optical properties as well as electron injection behavior have been studied by B3LYP and TD-B3LYP functionals which was found to be good and reliable approach (Zhang et al., 2008). The B3LYP hybrid functional along with the 6-31G∗∗ basis set have been used by Preat et al. to optimize the geometries at ground state and they concluded that this levels of theory (Preat et al., 2010, 2009) is suitable for small organic molecule.
Recently, Huong et al. stated that the geometries optimized at B3LYP/6-31G∗∗ levels of theory are good to reproduce the experimental data by shedding light on the electronic and charge transport properties data (Huong et al., 2013). The maximum absorption wavelengths of numerous organic dyes have been computed with an average deviation close to 0.20 eV with B3LYP functional (Guillaumont and Nakamura, 2000). The DFT has been applied by B3LYP/6-31G∗∗ levels of theory to optimize the S0 geometries, whereas the excited state (S1) geometries were optimized at TD-DFT (Matthews et al., 1996) using the TD/B3LYP with 6-31G∗∗ basis set. The same TD-DFT level was used to evaluate the spectra of absorption and emission as this has been verified to be a proficient method (Scalmani et al., 2006). Computational details about the transfer integrals, reorganization energies and intrinsic mobility calculations can be found in references and supporting information. All these quantum chemical calculations have been performed by using Gaussian09 package (Frisch et al., 2009).
The Density of States (DOS) is computed by GGA (generalized gradient approximation) at pw91 functional (Perdew et al., 1992) while the DNP basis set (Nadykto et al., 2008) has been used via DMol3 code (Delley, 2000) employed through the Accelrys Materials Studio package (Materials Studio Modeling, 2004).
3 Results and discussion
3.1 Geometries
The ground and excited state geometrical parameters of oxazole compound (BMPO) have been presented in Table S1. The optimized bond lengths in Å and bond angles in degrees have been compared with the geometrical parameters of the experimental crystal structure. The computed data, i.e., bond lengths and bond angles at B3LYP/6-31G∗∗ level of theory are in good agreement with the experimental crystal structure. In BMPO, maximum shortening from ground to excited state has been observed for C3—N4 bond length, i.e., 0.041 Å. The maximum lengthening from ground to excited state has been observed for C3—N4 bond length, i.e., 0.051 Å.
3.2 Electronic structure and photophysical properties
The distribution pattern of the HOMO−1, HOMO and LUMO at the ground and first excited states of the oxazole compound BMPO has been shown in Fig. 2. In BMPO, benzene is taking part in the formation of HOMO−1, benzene and oxazole of HOMO while LUMO is distributed on the central core between the benzene and methylphenol units revealing comprehensible intra-molecular charge transfer from HOMO−1 and HOMO to the LUMO. The calculated ground and excited states HOMO energies (EHOMO), LUMO energies (ELUMO) and HOMO–LUMO energy gaps (Eg) of BMPO at B3LYP/6-31G∗∗ and TD-B3LYP/6-31G∗∗ levels of theory have been presented in Table 1, respectively. In this study, we have compared the electron injection energy of BMPO. The electron injection energy can be evaluated as (=−ELUMO − (−work function of metal)). In the present case, we have considered the work function of aluminum which is −4.08 eV. The electron injection energy is around 1.66 eV (=−2.42 − (−4.08)) from the BMPO to aluminum electrode, respectively, while in another our study, we found electron injection barrier of isoxazole derivatives, i.e., 2-[(E)-(3,4-Dimethylisoxazol-5-yl)iminomethyl]phenol and 1-[(E)-(3,4-Dimethylisoxazol-5-yl)iminomethyl]-2-naphthol 1.98 and 1.92 eV, respectively. Thus it is expected that oxazole derivative (BMPO) might be a better electron charge transport contender than the above mentioned isoxazole derivatives. The ELUMO of BMPO is low-lying which revealed that this derivative would be thermodynamically more stable and charge transport can’t be quenched by losing the electron.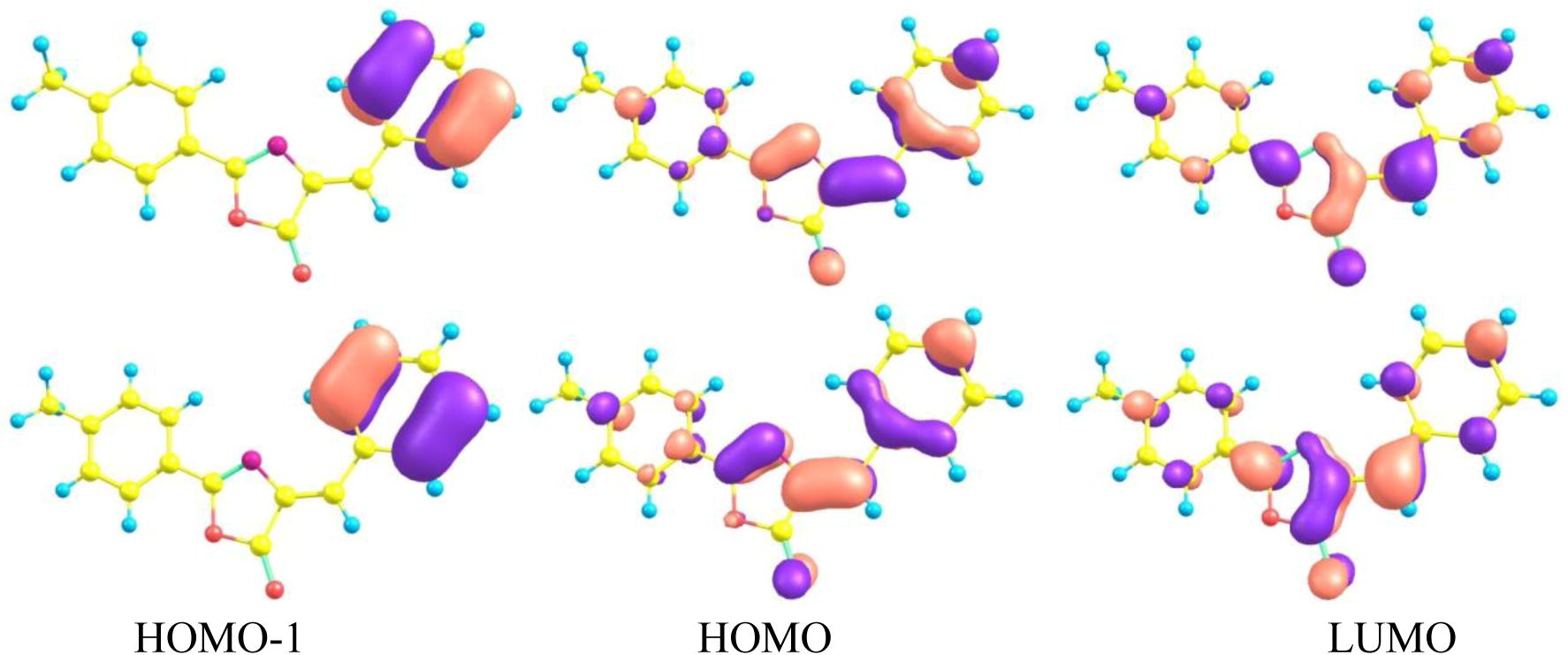
Distribution patterns of the HOMOs−1, HOMOs and LUMOs of BMPO at the ground states (bottom) and first excited state (top).
Ground states
Excited states
EHOMO
ELUMO
Eg
EHOMO
ELUMO
Eg
−5.86
−2.42
3.44
−5.52
−2.63
2.89
f
λa
Transition
f
λe
Transition
0.9328
371
H → L
0.9322
423
L → H
0.0216
311
H-1 → L
0.0237
326
L → H-1
The calculated absorption and emission wavelengths (λa and λe, respectively), oscillator strengths (f) and major transitions involved in the λa and λe at TD-B3LYP/6-31G∗∗ levels of theory have been presented in Table 1. The maximum λa and λe of BMPO have been observed 371 and 423 nm with the oscillator strength 0.9328 and 0.9322, respectively. The major transition in the absorption and emission has been noticed from H → L and L → H. The second major transition in the absorption and emission has been observed H-1 → L and L → H-1 with the oscillator strength 0.0216 and 0.0237 with the wavelength 311 and 326 nm, respectively. The Stokes shift in BMPO from λa to λe has been observed at 52 nm in the maximum wavelengths, respectively. It is estimated that BMPO might be good light violet emitter material.
3.3 Charge transport parameters
3.3.1 Ionization potential, electron affinity and reorganization energy
To understand the charge transfer nature of the organic semiconductor material IP and EA play significant role, i.e., smaller IP and greater EA values would lead to the proficient hole and electron injection ability by reducing the barrier, respectively. In Table 2, the adiabatic and vertical ionization potentials (IPa, IPv), adiabatic and vertical electron affinities (EAa, EAv) have been presented. The IPa/IPv of BMPO decreases 0.23/0.19 eV as compared to the 2-[(E)-(3,4-Dimethylisoxazol-5-yl)iminomethyl]phenol. Additionally, the EAa/EAv of 3 increases 0.41/0.49, and 0.24/0.30 than the 2-[(E)-(3,4-Dimethylisoxazol-5-yl)iminomethyl]phenol and 1-[(E)-(3,4-Dimethylisoxazol-5-yl)iminomethyl]-2-naphthol, respectively. These results indicate that electron injection barrier of BMPO would be smaller than the isoxazole derivatives, revealing that prior compound might be proficient electron transport material than the later ones.
System
IPa
EAa
IPv
EAv
λ (h)
λ (e)
BMPO
7.13
1.09
7.31
0.98
0.381
0.223
The hole reorganization energies (λ (h)) and electron reorganization energies (λ (e)) of oxazole (BMPO) have been tabulated in Table 2. In the present study, we have compared the (λ (h)) and (λ (e)) of studied compounds with each other as well as with some well-known hole and electron transport referenced compounds. The λ (e) of oxazole derivative BMPO is smaller than the λ (h) revealing that this compound might behave as better electron transporter. The computed λ (e) of BMPO is 0.223 eV which is smaller than the well-known n-type semiconductor material perfluoropentacene (λ (e) = 0.250 eV). Moreover, Huong et al. calculated the λ (e) of thiazole and oxazole based compounds from 0.21 to 0.37 eV (Huong et al., 2014). Again it can be seen that the λ (e) of 3 is smaller/comparable to the thiazole and oxazole derivatives. The mer-Alq3 is another renowned and frequently used electron transport material which have λ (e) 0.276 eV (Irfan et al., 2009). These results are illuminating that oxazole derivative BMPO would be an efficient electron transport material.
3.3.2 Transfer integrals and intrinsic mobility
Here, five distinct neighboring hopping pathways have been defined to study the charge transport performance of BMPO in detail. Table 3 displays the hole and electron transfer integrals determined by the scheme expressed in Eq. (6) (see supporting information). The highest values of hole and electron transfer integrals are 91.3 and 87.2 meV for first pathway of BMPO, respectively. The second, third, fourth and fifth pathways have the transfer integrals for hole/electron as 7.9, 7.1, −6.3 and −1.1 meV/−3.3, 12.6, −7.1 and −4.4 meV, respectively. The highest values of hole and electron transfer integrals for first pathway might be due to the close and face to face (stack) packing of the dimer with the mass center equals 1.84 Å. The other four pathways have the transverse kind of packing, which is the main reason behind their lower value of transfer integrals for hole as well as electron. All the dimers with their packing have been given in Fig. 3 for further clear understanding of the hopping pathways. The first pathway of BMPO has almost similar values of hole and electron transfer integrals; while the calculated λ (e) is 0.223 eV much lower than the λ (h) evaluated equals 0.381 eV, hence predicted that the BMPO would be good electron transport material.
Pathways
Transfer integrals (meV)
Mass centers (Å)
Intrinsic mobility (cm2/V·s)
Vh
Ve
Hole
Electron
i
91.3
87.2
1.84
7.3 × 10−2
0.40
ii
7.9
−3.3
4.00
1.9 × 10−5
3.8 × 10−6
iii
7.1
12.6
6.00
2.9 × 10−5
1.9 × 10−3
iv
−6.3
−7.1
6.40
2.0 × 10−5
2.1 × 10−4
v
−1.1
−4.4
6.50
1.9 × 10−8
3.2 × 10−5
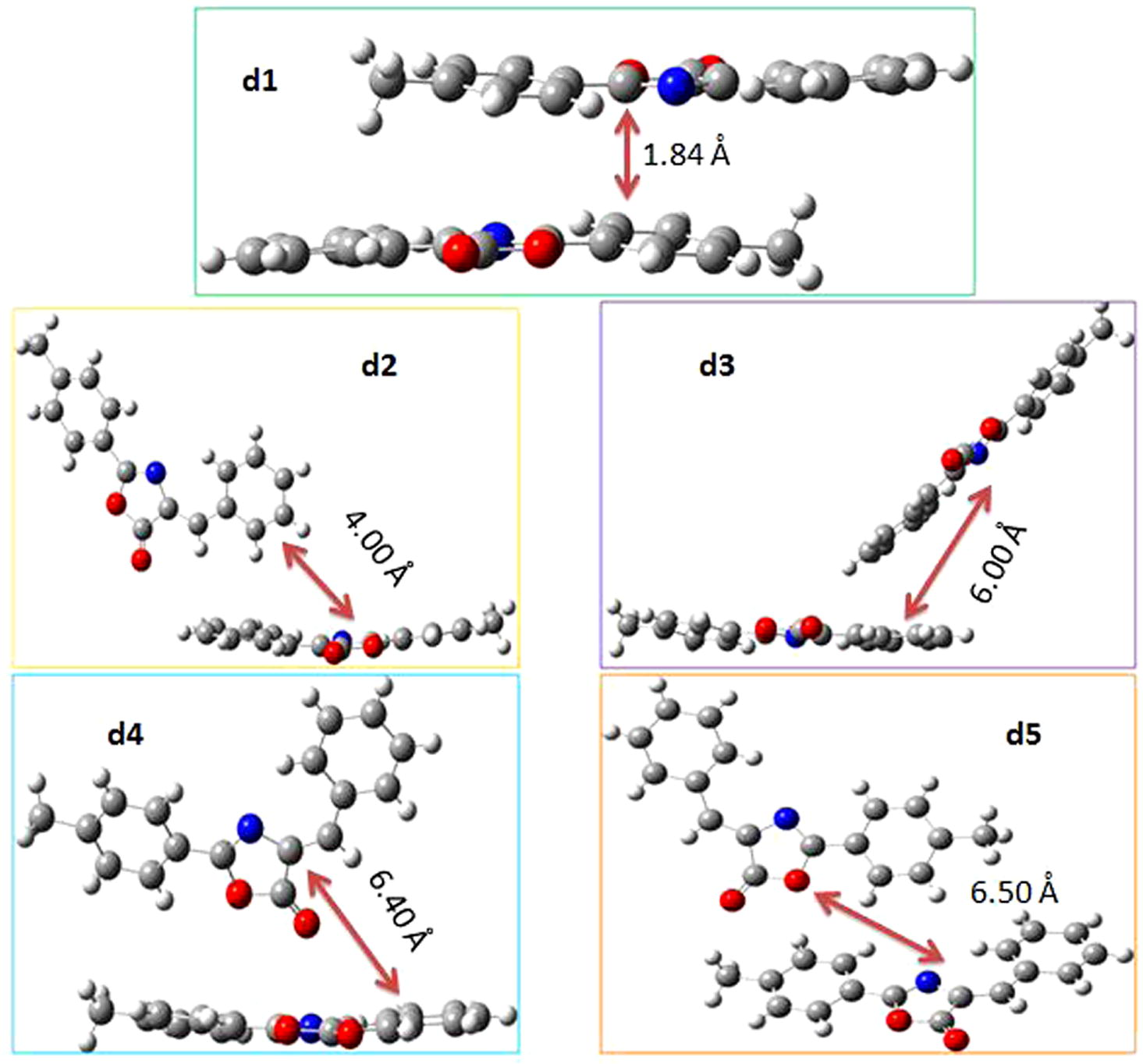
The dimers studied in current investigation to calculate the transfer integrals and intrinsic mobilities.
The hole and electron intrinsic mobilities of BMPO were estimated as stated in Eq. (8) (see supporting information) of computational details for five pathways and presented in Table 3. The first pathway of BMPO shows the highest value for electron intrinsic mobility as (0.40 cm2 V−1 s−1), which is because of the stack packing of the first dimer. The other four pathways were found with very low hole as well as electron intrinsic mobilities due to the side to side packing of the dimers (see Fig. 3) and not discussed in the text. The high value of electron intrinsic mobility as compared to the hole one illustrates that the BMPO would be better as electron transport material. We expect that the BMPO is a good electron transport material, hence supporting the predictions about BMPO in terms of ELUMO, IPv, EAv and hole/electron reorganization energies.
3.4 Nonlinear optical property
Recently, the field of nonlinear optical (NLO) materials designing has got a significant momentum due to the application in several fields including frequency doubling, fast data processing and SHG spectroscopy etc. There are many efforts devoted to get a NLO materials with required amplitude of NLO response (Levine and Bethea, 1975). In this regard, the organic class of materials is considered to be very promising because of their low cost, ease of fabrication as well as larger NLO responses (Badan et al., 1993; Zaitseva and Carman, 2001). In the present investigation, the BMPO molecule possesses electronic transitions involving intramolecular charge transfer (ICT) as illustrated by the pattern of its frontier molecular orbitals in ground and excited states, which indicate they have possible potential for efficient NLO phores. To check the possibility of BMPO for their potential applications as NLO materials, we have calculated their molecular electronic static first hyperpolarizability (βtot). The first hyperpolarizability and its components for BMPO were calculated using finite field (FF) approach. The B3LYP/6-31G∗∗ levels of theory combined with finite field (FF) approach have been used to calculate the first hyperpolarizability (βtot) of titled compound (Computational details/methodology has been given in supporting information).
It is important to mention here that the dipole moment of Urea molecule has been also calculated to be ∼ 4.24D and compared with its experimental results, which is found to be in good quantitative agreement with its experimental value of 4.56 D. For BMPO molecule, its total dipole moment is 3.475 D with its highest component of μy having a value of −3.228 D, where the negative sign implies that it is almost entirely directed along negative y-axis as shown in Fig. S1. As the electronic dipole moment is usually directed from negative to positive point charges, the Milliken population analysis indicates a significantly negatively charged central core along positive y-axis and positively charged outer wings with an average total positive charge along negative y-axis, resulting in a total dipole moment along negative y-axis. A similarly but somewhat different analysis can also be performed for polarizability values i.e. average polarizability (α0), anisotropy of polarizability (Δα) and hyperpolarizability (βtot) of BMPO molecule, which have non-zero amplitudes of 34.48 × 10−24, 161.29 × 10−24 and 12.07 × 10−30 esu, respectively as given in Table 4. The first hyperpolarizability (βtot) of BMPO, has non-zero amplitudes of 12.07 × 10−30 esu. The non-zero value of βtot shows that the titled molecules possess microscopic first static hyperpolarizability. The first hyperpolarizability value of BMPO is about 33 times larger than that of urea (a prototype NLO molecule) as calculated in present investigation at the same B3LYP/6-31G∗ levels of theory, which shows that BMPO can also be considered as a potential candidate for NLO applications.
Components
a. u.
esu (×10−24)
Components
a. u.
esu (×10−30)
αxx
410
60.75
βxxx
1007
8.70
αxy
−18.0
−2.67
βxxy
1076
9.30
αyy
220
32.60
βxyy
−229
−1.98
αxz
0.00
0.00
βyyy
127
1.10
αyz
0.00
0.00
βxxz
−18
−0.16
αzz
68.0
10.08
βxyz
−1
−0.01
α0
233
34.48
βyyz
−7
−0.06
Δα
1088
161.29
βxzz
−27
−0.23
μx
−0.498
−1.266Da
βyzz
−25
−0.22
μy
−1.270
−3.228D
βzzz
14
0.12
μz
0.081
0.206D
βtot
1397
12.07
μtot
1.367
3.475D
βtot
1215c
1112d
10.50c
9.610d
μtot (urea)
1.66
4.24D (4.56D)b
βtot (urea)
43
0.37
We have also calculated the hyperpolarizability of BMPO with LC-BLYP (u = 0.47) and CAM-B3LYP methods. A highly correlated MP2 method has been also used to assess the first hyperpolarizability. All the first hyperpolarizability values with different methods are compared with each other graphically as given in Fig. S2. It can be seen that there is no significant difference among these methods. The range corrected methods have reasonably reproduced the B3LYP first hyperpolarizability values with a difference of about 182 a. u. and 285 a. u. for CAM-B3LYP and LC-BLYP methods, respectively. Nevertheless, B3LYP method shows very close results to MP2 method with a difference of first hyperpolarizability value ∼ 23 a. u. However, we have also provided the CAM-B3LYP and LC-BLYP first hyperpolarizability values along with B3LPY in Table 4. We have provided the excel spread sheet along with step by step details in supporting information (Fig. S4) to calculated first hyperpolarizability amplitudes.
3.5 Densities of states
The partial and total DOS have been evaluated by implementing CASTEP module available in Materials Studio software program within the framework of DFT at GGA/PW91/DNP levels of theory to understand the electronic structure of BMPO comprehensively. The PDOS and TDOS with contributions from s, p-orbital of C, O and N atoms for BMPO are shown in Fig. S3. The peaks in the lower valence band at energy regime −18 to −10 eV are defined mostly by the s-orbitals of C atoms; the peaks between −22 and −20 eV are contribution from s-orbitals of O atom, whereas the peak between −19 and −18 eV from the s-orbitals of N atom. The major contributions from p-orbitals of C, O and N atoms are dominating from −10 to −0 eV in higher valence bands. In lower conduction bands, the peaks from 1 to 4 eV have major contribution from p-orbitals of C, O and N (see Fig. S3). The DOS profiles of BMPO display the upper valance and lower conduction bands are mainly of p-character, reflecting the major contribution of p-orbitals into the electronic properties of BMPO. Our analysis of the electronic properties shows that these materials with good electronic properties would be potential candidates for organic electro-optical device applications including OLEDs, OPVDs and OFETs.
3.6 Molecular electrostatic potential
The reactivity of the molecule can be explored by a very important feature termed as molecular electrostatic potential (MEP) surfaces. The areas with major positive potential are specified by blue color and these regions are preferred sites for nucleophilic attack, whereas the maximum negative potential sections have been presented in red color, which are favored site for electrophilic attack as shown in Fig. 4. The different colors are indicating the distinct intensities of MEP surfaces and increases in the order red < orange < yellow < green < blue. In MEP profile the blue and red colors demonstrate the strongest attraction and repulsion, respectively. The lone pair of electronegative atoms is mainly associated to the negative potential regions. The negative potential regions are on the O atom for BMPO molecule, while the intermediate electrostatic potentials are on benzene ring and shown in yellow color. The O atom in the BMPO molecule is representing the red color and will perform as electrophile regions. Similarly, the blue color indicates the nucleophile regions and illustrates the regions with electron insufficiency, which also represents the nucleonic energy of the LUMO orbitals. These outcomes reveal that the maximum repulsion will be shown by O atom.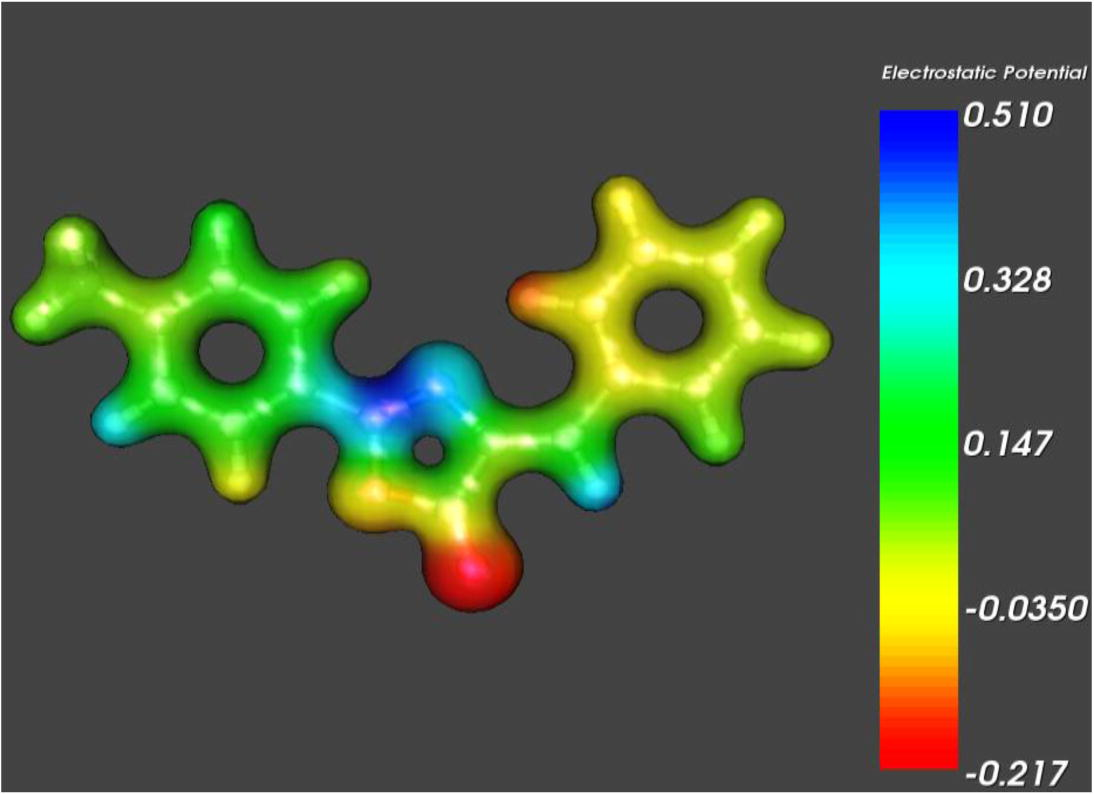
Molecular electrostatic potential of BMPO molecule.
4 Conclusions
The experimental crystallographic geometrical parameters for BMPO molecule have been reproduced by B3LYP/6-31G∗∗ levels of theory. A clear intramolecular charge transfer from HOMO−1 to LUMO as well as HOMO to LUMO has been observed. The smaller electron injection energy (1.66 eV) from BMPO to aluminum electrode than the isoxazole derivatives 2-[(E)-(3,4-Dimethylisoxazol-5-yl)iminomethyl]phenol (1.98 eV) and 1-[(E)-(3,4-Dimethylisoxazol-5-yl)iminomethyl]-2-naphthol (1.92 eV) anticipating that oxazole derivative (BMPO) might be better electron charge transport contender than the above mentioned isoxazole derivatives. The DOS profiles of BMPO display the upper valance and lower conduction bands are mainly of p-character, reflecting the major contribution of p-orbitals into the electronic properties of BMPO. The MEP outcomes reveal that the maximum repulsion will be shown by O atom. The TDDFT calculations showed 52 nm Stokes shift from λa to λe. It is also expected that BMPO might be good violet light emitter material. The computed λ (e) is 0.223 eV, which is smaller/comparable than/to the well-known n-type semiconductor materials perfluoropentacene (λ (e) = 0.250 eV), thiazole and oxazole based compounds (λ (e) = 0.21–0.37 eV) and mer-Alq3 (λ (e) = 0.276 eV). Hole and electron transfer integral values of BMPO are comparable while the electron reorganization energy is smaller than the hole one which leads to the superior electron intrinsic mobility. The stack packing of the first dimer (pathway) of BMPO is leading to the highest electron intrinsic mobility value (0.40 cm2 V−1 s−1) while side to side packing of the dimers (pathways 2–4) diminishing the hole as well as electron intrinsic mobility. The amplitudes of average polarizability (α0), anisotropy of polarizability (Δα) and hyperpolarizability (βtot) of BMPO molecule are found to be reasonably larger mounting to 34.48 × 10−24, 161.29 × 10−24 and 12.07 × 10−30 esu, respectively.
Acknowledgements
Authors would like to acknowledge the support of the King Khalid University for this research through a grant RCAMS/KKU/001-16 under the (Research Center for Advanced Materials Science) at King Khalid University, Kingdom of Saudi Arabia. Dr. A. Irfan acknowledges the Professor Zhang Jingping to provide technical support about M.S. calculations.
References
- Electronic structure, electronic charge density and optical properties of 3-methyl-1,4-dioxo-1,4-dohydronaphthalen-2-yl-sulfanyl (C13H10O4S) Int. J. Electrochem. Sci.. 2014;9:445-459.
- [Google Scholar]
- NLO properties of organic molecules and polymeric materials.American Chemical Society Symposium Series. Washington, DC: American Chemical Society; 1993.
- Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys.. 1993;98:5648-5652.
- [Google Scholar]
- Influence of push-pull configuration on the electro-optical and charge transport properties of novel naphtho-difuran derivatives: a DFT study. RSC Adv.. 2014;4:48876-48887.
- [Google Scholar]
- Optoelectronic properties of naphtho[2,1-b:6,5-b′]difuran derivatives for photovoltaic application: a computational study. J. Mol. Model.. 2016;22:1-13.
- [Google Scholar]
- Tripodal molecules with triphenylamine core, diazine peripheral groups and extended π-conjugated linkers. Dyes Pigm.. 2016;124:101-109.
- [Google Scholar]
- From molecules to solids with the DMol3 approach. J. Chem. Phys.. 2000;113:7756-7764.
- [Google Scholar]
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Jr., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R. L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, Ö., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J., 2009. Gaussian 09, Revision A. 01, Gaussian Inc.: Wallingford, CT.
- An ab initio study of the structural, elastic, electronic, optical properties and phonons of the double perovskite oxides Sr2AlXO6 (X= Ta, Nb, V) Mater. Sci. Semicond. Process.. 2016;42:405-412.
- [Google Scholar]
- Vibrational spectra, molecular structure, NBO, UV, NMR, first order hyperpolarizability, analysis of 4-Methoxy-4′-Nitrobiphenyl by density functional theory. Spectrochim. Acta Mol. Biomol. Spectrosc.. 2014;122:130-141.
- [Google Scholar]
- Calculation of the absorption wavelength of dyes using time-dependent density-functional theory (TD-DFT) Dyes Pigm.. 2000;46:85-92.
- [Google Scholar]
- Molecular-scale interface engineering for polymer light-emitting diodes. Nature. 2000;404:481-484.
- [Google Scholar]
- Π-conjugated molecules containing naphtho[2,3-b]thiophene and their derivatives: theoretical design for organic semiconductors. J. Phys. Chem. C. 2013;117:10175-10184.
- [Google Scholar]
- Theoretical design of n-tType organic semiconducting materials containing thiazole and oxazole frameworks. J. Phys. Chem. A. 2014;118:3335-3343.
- [Google Scholar]
- DFT investigations of the ground and excited state geometries of the benzothiazine and benzisothiazol based anticancer drugs. J. Saudi. Chem. Soc.. 2015;19:318-321.
- [Google Scholar]
- Push–pull effect on the charge transfer, and tuning of emitting color for disubstituted derivatives of mer-Alq3. Chem. Phys.. 2009;364:39-45.
- [Google Scholar]
- Electro-optical and charge injection investigations of the donor-π-acceptor triphenylamine, oligocene–thiophene–pyrimidine and cyanoacetic acid based multifunctional dyes. J. King Saud Univ.-Sci.. 2015;27:361-368.
- [Google Scholar]
- In-depth quantum chemical investigation of electro-optical and charge-transport properties of trans-3-(3,4-dimethoxyphenyl)-2-(4-nitrophenyl)prop-2-enenitrile. C.R. Chim.. 2015;18:1289-1296.
- [Google Scholar]
- The structural, electro-optical, charge transport and nonlinear optical properties of 2-[(3,5-dimethyl-1-phenyl-1H-pyrazol-4-yl)methylidene]indan-1,3-dione. Optik – Int. J. Light Elect. Optics. 2016;127:10148-10157.
- [Google Scholar]
- First-principle study of electrical and optical properties of (Al, Sn) co-doped ZnO. Optik. 2016;127:1988-1992.
- [Google Scholar]
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785-789.
- [Google Scholar]
- Synthesis, structural characterization, and unusual field-effect behavior of organic transistor semiconductor oligomers: inferiority of oxadiazole compared with other electron-withdrawing subunits. J. Am. Chem. Soc.. 2009;131:1692-1705.
- [Google Scholar]
- Second and third order hyperpolarizabilities of organic molecules. J. Chem. Phys.. 1975;63:2666-2682.
- [Google Scholar]
- Materials Studio Modeling, R.A.I., San Diego, 2004. Materials Studio Modeling, Release 3.0.1. Accelrys Inc., San Diego.
- Calculation of the photocurrent-potential characteristic for regenerative, sensitized semiconductor electrodes. Sol. Energy Mater. Sol. Cells. 1996;44:119-155.
- [Google Scholar]
- A new consecutive three-component oxazole synthesis by an amidation-coupling-cycloisomerization (ACCI) sequence. Chem. Commun. 2006:4817-4819.
- [Google Scholar]
- Computational quantum chemistry: a new approach to atmospheric nucleation. Adv. Quantum Chem.. 2008;55:449-478.
- [Google Scholar]
- (4Z)-4-Benzylidene-2-phenyl-1,3-oxazol-5(4H)-one. Corrigendum. Acta Crystallogr. Sect. E. 2015;71:e4.
- [Google Scholar]
- Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B. 1992;46:6671-6687.
- [Google Scholar]
- Enhanced efficiency of organic dye-sensitized solar cells: triphenylamine derivatives. J. Phys. Chem. C. 2009;113:16821-16833.
- [Google Scholar]
- Design of new triphenylamine-sensitized solar cells: a theoretical approach. Environ. Sci. Technol.. 2010;44:5666-5671.
- [Google Scholar]
- Thermoelectric properties, effective mass, chemical bonding, and optical properties of 1,3,6-trimetylo-alloxazine: C13H12N4O2. Int. J. Electrochem. Sci.. 2014;9:460-477.
- [Google Scholar]
- Geometries and properties of excited states in the gas phase and in solution: theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys.. 2006;124:094107-094115.
- [Google Scholar]
- Macromolecular electronic device: field-effect transistor with a polythiophene thin film. Appl. Phys. Lett.. 1986;49:1210-1212.
- [Google Scholar]
- Synthesis and bioactivity of novel pyrazole oxime derivatives containing oxazole ring. Chin. Chem. Lett.. 2015;26:672-674.
- [Google Scholar]
- Coumarin dyes for dye-sensitized solar cells: a long-range-corrected density functional study. J. Chem. Phys.. 2008;129:214703.
- [Google Scholar]
- Rapid growth of KDP-type crystals. Prog. Cryst. Growth Charact. Mater.. 2001;43:1-118.
- [Google Scholar]
- [3+2] cycloaddition/oxidative aromatization sequence via photoredox catalysis: one-pot synthesis of oxazoles from 2H-azirines and aldehydes. Org. Lett.. 2015;17:4070-4073.
- [Google Scholar]
- Theoretical studies on the geometrical and electronic structures of N-methyle-3,4-fulleropyrrolidine. J. Mol. Struct. (TheoChem). 2008;862:98-104.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.jksus.2016.10.004.
Appendix A
Supplementary material




