

The predicted models of anti-colon cancer and anti-hepatoma activities of substituted 4-anilino coumarin derivatives using quantitative structure-activity relationship (QSAR)
⁎Corresponding authors. daratu.putri.fmipa@um.ac.id (Daratu Eviana Kusuma Putri), rvenkatmpharm@gmail.com (Venkatalakshmi Ranganathan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Objectives
The objective of this work was to predict anti-colon and anti-hepatoma cancer activity of newly designed substituted 4-anilino coumarin derivatives using quantitative structure–activity relationship (QSAR).
Methods
The optimization of the derivatives including molecular and electronic properties data to generate the QSAR were resulted from H-GGA DFT/BPV86 method calculation combined with Hartree-Fock 6-31G basis set. The QSAR models were delivered in multiple linear regression (MLR) and the drug design was accomplished by considering the descriptors constructing the best QSAR models for each biological activity.
Results
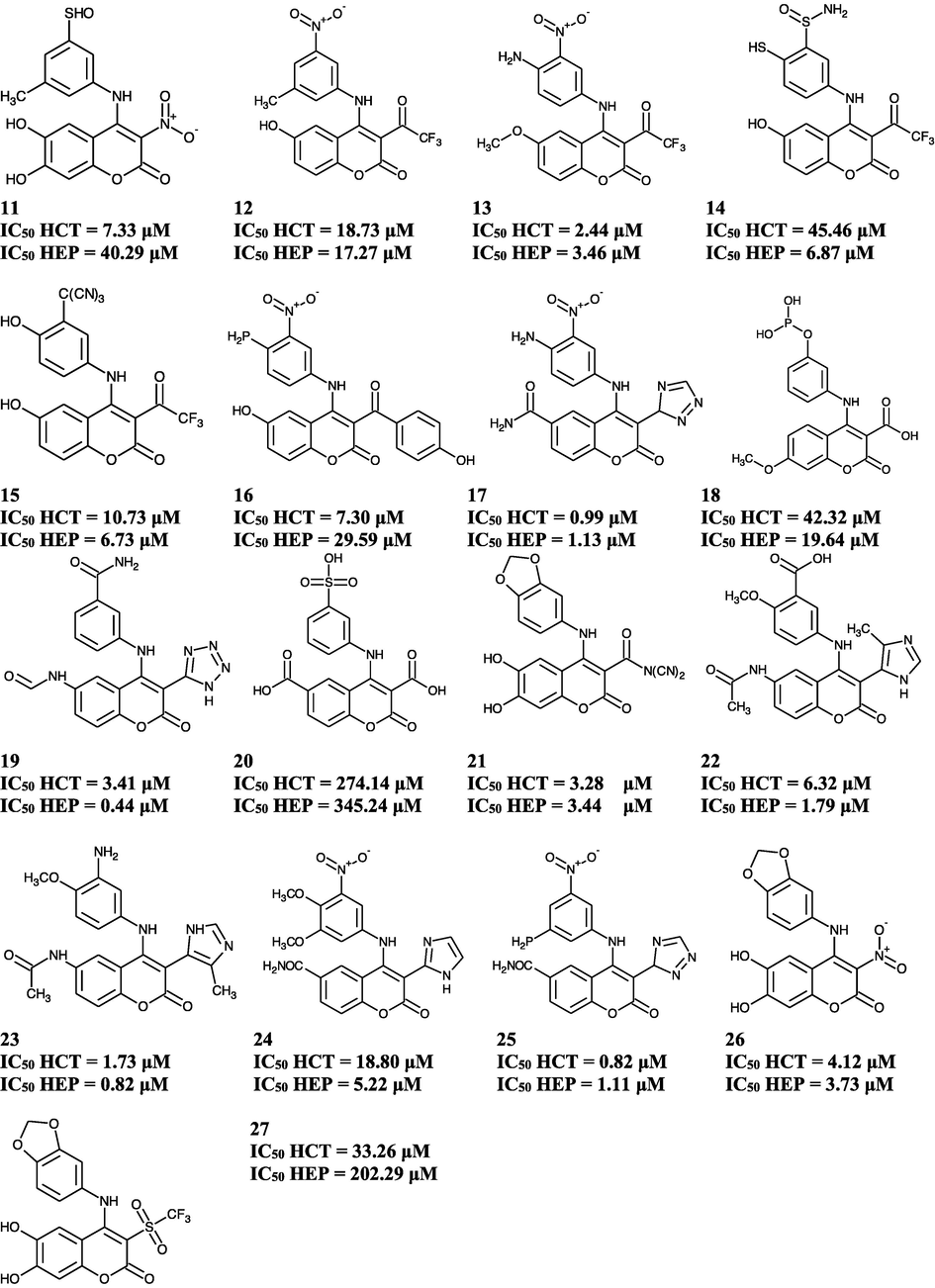
This research accomplished two best validated QSAR models for predicting anti-colon cancer and anti-hepatoma activities of substituted 4-anilino coumarin derivatives. There were 17 of newly designed substituted 4-anilino coumarin derivatives which their anticancer activity (Log IC50) were predicted using our both QSAR models. The QSAR predictions inform that the compound 25 and compound 19 have the best predicted anti-colon cancer and anti-hepatoma activities, respectively.
Conclusion
The results revealed that this approach can be used to assist in the action of anticancer drug discovery. Moreover, the retrosynthesis analysis of both compounds are also explained in a logic reverse of organic synthetic steps through Knoevenagel and Perkin reactions.
Keywords
Coumarin
Anti-cancer
QSAR
MLR

1 Introduction
Worldwide, cancer is one of seven leading death causes in more and less economically developed countries (Torre et al., 2015). Colon cancer and hepatocellular carcinoma are included as the most common human malignant diseases with the highest death cases (Ma et al., 2020). One of the most popular treatment of liver disease in early stage is liver transplantation but unfortunately it is limited to the lack of donors (Reddy et al., 2013). Thus, chemotherapy remains the main strategy for cancer treatment, but the phenomenon of drug resistance and the emergence of minimal side effect from new anti-cancer agents (Ma et al., 2020) are being the essential intention for cancer chemotherapy.
The propitious biological activity and simple synthetic modification have brought the scientists to design and develop the coumarin derivatives as pharmaceutical compounds (Cao et al., 2016). They exhibit variance biological activities such as anticoagulants (Horak et al., 2018), antidepressant (Sashidhara et al., 2011), antitumor (Cao et al., 2016), antioxidant (Erzincan et al., 2015) and so on. Cao et al. (2016) synthesized coumarin which exhibited potent anti-proliferative ability causing G2/M phase arrest. They claimed that this excellent activity came from the N-methyl linker group in coumarin hybrid. Luo et al. had synthesized coumarin scaffold and aniline moieties (substituted 4-anilino-coumarin derivatives) (Luo et al., 2017) which were in vitro cytotoxic activity evaluated against four human cancer cell lines (MCF-7, HepG2, HCTI16, and Panc-1). The effect of cell cycle arrest was assayed to MCF-7 (human breast cancer cell line) for the best compound which proving into concentration-dependently to ruin cell cycle at G2/M phase. Further, Luo et al. evaluated the best compound for the cytotoxicity test against HUVEC (human umbilical vein endothelial cells) which revealed normal cells were far less sensitive to the assayed compounds over cancer cells.
Quantitative Structure-Activity Relationship (QSAR) approach take the role to correlate the biological activity of compounds group with the computated molecular properties (Amin et al., 2021). Mladenovic et al. had used multiple linear regression (MLR) to perform the correlation between the antioxidant activity and various molecular descriptors (Mladenović et al., 2011). They employed DFT/B3LYP functional and 6-31G basis set to optimize the derivatives using Gaussian 03 software. Other research, Erzincan et al. (2015) created the QSAR equation and revealed that some physicochemical characters such as H-bond donor and lipophilic were important parameters in describing antioxidant ability.
QSAR modelling usually consists of four main steps: computing a series of descriptors, selecting some relevant descriptors, building the frequent nonlinear relationship between the descriptors and the biological activity, and validating the model.
2 Materials and methods
2.1 Hardware and software
The research used a PC with CPU @ 2.00 GHz RAM 4.00 GB Intel® Core™ i3-5005U. The softwares utilized were Hyperchem™ 8.0.10, Gaussian® 09 W, IBM® SPSS® Release 23, and ACD/ChemSketch 14.0. Overall research was being done at Austrian-Indonesian Centre for Computational Chemistry (AIC) Laboratory, Department Chemistry, Faculty of Mathematics and Science, Universitas Gadjah Mada.
2.2 Materials
A data set of 27 compounds (Luo et al., 2017) was used to generate the QSAR equations. The anticancer activity data were set as dependent variables (IC50) to HCT116 and HepG2 (μM) which were modified into logarithmic.
2.3 Method of calculation
Compound 33d as the most-desiring-activity compound was computed using PM6, DFT/BPV86, DFT/PBE, DFT/B3LYP (Van Bay et al., 2021), and Hartree-Fock (HF) method (Hmamouchi et al., 2016). The Basis set applied for HF and DFT calculation was 6-31G. 13C NMR chemical shifts were calculated by DFT/B3LYP method and 6-31G basis set. The best method with low correlation error between experimental and calculated chemical shift was selected.
2.4 Geometrical optimization and descriptors calculation
The 27 compounds of substituted 4-anilino coumarin derivatives were depicted employing GaussView 5.0 and optimized by Gaussian 09W application. From the calculations, ELUMO, EHOMO, delta EHOMO-ELUMO (ΔE), atomic charge (q), and dipole moment (μ) were obtained. The rest descriptors, volume (V), molecular mass (MW), refractivity (MR), partition coefficient (log P), polarisability (α), and surface area (SA) were calculated using Hyperchem™ 8.0.10.
2.5 Model development and validation
QSAR equations were created by utilizing multiple linear regression (MLR) using IBM® SPSS® Release 23. The regression development method was backward elimination with confident interval level 95%. The internal validation should meet the significance test: r2train > 0.6 (Golbraikh and Tropsha, 2000), SEE < 0.3 (Mishra et al., 2014), test Fcal/Ftab > 1, α(0.05) > sig(0.00) (Pramesti, 2017). The external validation used to eliminate the unqualified equations, r2test > 0.6, r2overall > 0.5, and RMSEtest < 0.3 are critical to be calculated (Golbraikh and Tropsha, 2000).
2.6 Drug design
The drug design was done by replacing the R1, R2, and R substituents from coumarin derivatives. Hydrophobic, electronic, and steric effects were the main properties to consider. To attain proper hydrophobic character, log P, R1, R2, and R of the newly designed molecules were replaced with certain moieties. To avoid unwanted value of Log P, Lipinski’s “rule of five” was implemented.
3 Results and discussion
3.1 Method of calculation
The compound used was 33d that was 4-(1,3-benzodioxol-5-ylamino)-6-methoxy-3-(trifluoroacetyl)–2H-chromen-2-one as it had the pre-eminent anti-cancer activity among the listed derivatives (Luo et al., 2017). In Fig. S1 (see the figure in Supplementary material), HF/6-31G method performs significant inaccuracy to predict the 13C NMR spectra than DFT and semiempirical method. High degree of error is proved by its high RMSE calculation which is 15.79 and low linearity between experimental and calculated chemical shifts was showed by r2 (0.9785) (Putri et al., 2019). This lack might due to this method’s limitation to carry the electron correlation in calculation process (Young, 2001). Electron correlation is important to improve the accuracy of computed energies and molecular geometries (Piris, 2017).
Semiempirical method (PM6 method) has better result than HF method expressed by higher r2 that is 0.9908. But, this method gives higher RMSE value (13.08) compared with several DFT methods indicates the higher error. The sp3 hybridization of nitrogen in the derivatives can be the main cause of false prediction from PM3 or AM1 calculation (Clark, 1993).
From Fig. S1, it can be seen that the three closest theoretical chemical shifts to the experiment are all from DFT methods (BPV86, PBE, and B3LYP). DFT functionals verily rely on organic compounds molecular structure (Van Bay et al., 2021). The determination coefficient of the three DFT functionals are 0.9834 (BPV86) (Putri et al., 2019), 0.9825 (PBE), and 0.9812 (B3LYP) indicating the pleasant coherence between theoretical and experimental chemical shifts. B3LYP RMSE calculation gives higher value comparing with PBE and BPV86. Despite, PBE method provides negligible difference in RMSE calculation (9.52) with BPV86 (9.27) (Putri et al., 2019) but its optimizing process is significantly more time consuming. Therefore, DFT/BPV86 using 6-31G basis set is the best method which offers the best prediction for the independent variables to be more accurate. In addition, BPV86 is included as hybrid GGA (generalized gradient approximation) functional (Sajjad et al., 2017) which integrate Becke and Perdew’s 1986 functional density with electron correlation replaced with Vosko.
3.2 Geometrical optimization and descriptors calculation
Generating QSAR equations always charge high attentions in the entire process. The Hartree-Fock complicity in Hybrid density functional (H-GGA) methods upgrades a conventional GGA used in this research (BPV86) into the next level sophistification for the derivatives. H-GGA let a percentage of Hartree-Fock exchange to be fitted semiempirically with experimental ionization potentials, total atomic energies, proton affinities and other data representating a group of small molecules (Becke, 1993).
The data of several distance modifications between two atoms are performed in Table S1 (see the table in Supplementary material). The distance between C7 and O without optimization is quite narrow (1.338 Å). Following the optimization, the distance becomes wider (1.412 Å) placing the atoms into more relax position and reduce the high level energy.
3.3 Model development and validation
The QSAR equations robustness is predicted by calculating the capability of the training set equation to predict log IC50 of the test set. Backward elimination has become the most promising method to progressively eliminate the descriptors and also gives better performance to correlation coefficient compared with forward selection and stepwise regression. In the end of the process, only a few descriptors are left in order to reduce redundant, the risk of overfitting from noisy and irrelevant descriptors.
3.4 Model development and validation of anti-colon cancer (anti-HCT)
The best five models resulted from various training and test set packs of anti-colon cancer activity are listed in Table S2 (see the table in Supplementary material). The lowest rtrain value is 0.872 and the lowest r2train is 0.760. It explains the goodness of statistical significance that is>87.2% and the r2train reveals the closeness of the data to the fitted regression. This study provided the lowest Fcal/Ftab values of all models is 3.588 explaining a good statistically significant. Every model is clustered as having good accuracy in predicting Log IC50 of anti-HCT.
The external validation was done. In Table 1, the data which satisfied the standard were merely model 4. Model 4 presented r2test 0.8307, with RMSE 0.177, and PRESS 0.157. The PRESS and RMSE value of this model was occupied as the smallest value considering that model 4 gave the least error. In addition, the accuracy of the model 4 was described by correlation coefficient (r = 0.882) that meant statistical significance>88% (Fig. S3, see the figure in Supplementary material). Therefore, model 4 was considered as a validated, robust, and predictive model. (Model 4, Table S2)
| Model | Test set | r2test | PRESS | RMSE |
|---|---|---|---|---|
| 1 | 23e, 14f, 23f, 23a, 14a | 0.5398 | 1.940 | 0.623 |
| 2 | 14e, 33b, 14f, 23a, 23i | 0.6033 | 0.217 | 0.208 |
| 3 | 14e, 33b, 14f, 23a, 23i | 0.8216 | 0.217 | 0.208 |
| 4 | 32c, 33b, 23f, 14i, 25c | 0.8307 | 0.157 | 0.177 |
| 5 | 32c, 14e, 14a, 14c, 14g | 0.8508 | 0.277 | 0.336 |
The descriptors that affect the molecule's activities are C15 and C17 of anilino group and Log P (Eq. 1, Fig. 3). This discovery is suitable with molecular docking of Cao et al. which shows that anilino forms hydrophobic interactions with amino acids residues Ala316 (Cao et al., 2016). The atomic charge shift significantly changes the interaction ability of this ring with its receptor. Another critical factor was the solubility. Unsatisfactory log P of a drug may cause it never reach the receptor. The calculated log IC50 values of model 4 of all compounds were compared with the experimental activities giving a slope of 0.7559 that shows that this model is able to favourably predict 75.59% of the data (Fig. 1).
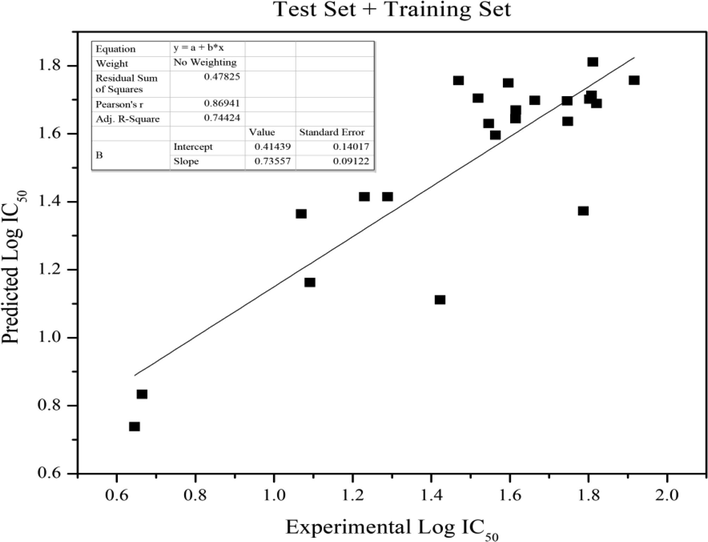
- The correlation of experimental and predicted Log IC50 of anti-HCT training and test set.
3.5 Model development and validation of Anti-Hepatoma activity (anti-HEP)
Table S3 (see the table in Supplementary material) provides the best models of various training and test sets for anti-hepatoma activity resulted by SPSS software. The lowest r values is 0.845 indicating a good statistical significance which is >84.5%. Every model in Table S3 shows a very good determination coefficient that the lowest is 0.747. The other case, the whole ratio values of Fcal/Ftab show numbers between 3.588 and 7.400 which meet the standard value. In addition, the highest SEE value among all candidate is 0.209 meaning the errors rate of all equation are quite small.
The external validation was done which model 1, 2, and 3, presented r2test > 0.6 (Table 2). Yet, the only accepted RMSE value was belonged to model 2 that was 0.298. This model also had the smallest PRESS values (0.445). The both values meant that its error was the mildest among the rest equations. Despite, the r2test (0.8223) occupied as the highest r2test among others. In addition, model 2 has statistical significance >86% (Fig. S5, see the figure in Supplementary material). Therefore, model 2 was considered as a robust, validated and predictive model.
| Model | Test set | r2 | PRESS | RMSE |
|---|---|---|---|---|
| 1 | 14b, 23b, 25a, 25d, 25e | 0.6394 | 0.639 | 0.358 |
| 2 | 14b, 23b, 25a, 25d, 25e | 0.8223 | 0.445 | 0.298 |
| 3 | 14a, 14e, 23b, 23e, 25c | 0.6905 | 1.376 | 0.525 |
| 4 | 14b, 14g, 23b, 23g, 25d | 0.4912 | 1.825 | 0.604 |
| 5 | 14e, 23e, 32c, 33b, 33e | 0.3052 | 3.781 | 0.870 |
The descriptors that affect the molecules activities are C2, C17, C18 and Log P. C17 and C18 build the anilino ring (Eq. 2, Fig. 3). According to Cao et al., this anilino ring is very important for derivative’s anticancer activity due to its hydrophobic interaction with amino acid Ala316. (Model 2, Table S3)
Log P descriptor could be further modified to provide better anticancer activity. The picture above was the comparison between calculated log IC50 from entire derivatives and observed values providing r2 = 0.6922. This determination coefficient indicates that the model was able to favourably predict 69.22% of the data (Fig. 2).
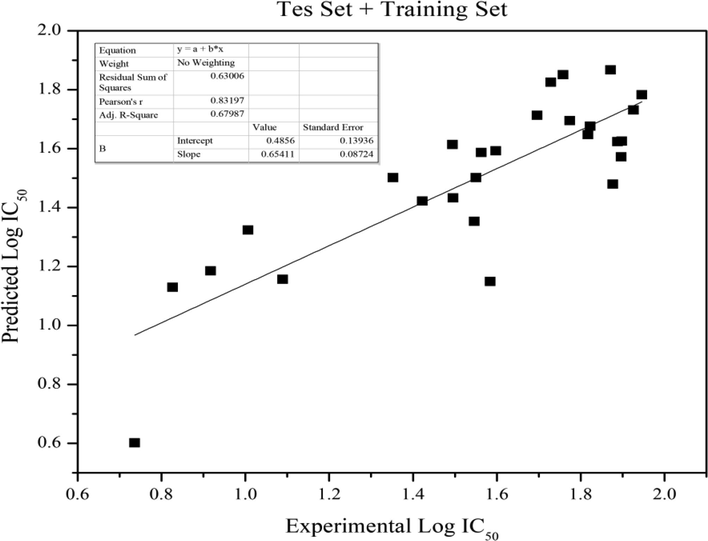
- The correlation of experimental and predicted Log IC50 of anti-hepatoma training and test set.
3.6 Drug design
Analyzing Eqs. 1 and 2, hydrophobic and electronic were the physicochemical properties that should be further discussed. The hydrophobic properties of a drug is vital to provide it’s easiness to cross cell membranes and in receptor interaction. Despite, a huge part of all living mass consists of water leading to every biochemical reactions are in aqueous phase. The new molecules should have certain moieties that alter the Log P becoming lower than the reference yet maximize its performance to cross lipid barriers. Therefore, Lipinski’s “rule of five” is also implemented to design new molecules.
A drug polarity is highly affected by the electronic. These characteristics give an effect on how easily a drug could pass cell membranes or how strongly it could bind to a receptor. According to Eqs. 1 and 2 (Fig. 3), the potential of the compounds is highly correlated with the partial atomic charge of the benzene rings. Hence, the moieties chosen to create new molecules with certain atomic charges. The overall result of new compound designs are presented in Fig. 4. The most promising structure for anti-coloncancer is structure 25 with a predicted IC50 = 820 nM and for anti-hepatoma is structure 19 with predicted IC50 = 440 nM.
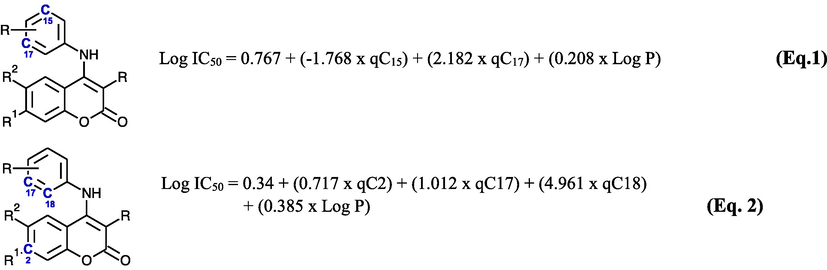
- Atomic charge points of each equation.
- New compound designs.
The retrosynthesis of newly designed compounds 25 and 19 is depicted in Figs. 5 and 7 respectively. Referring to them, aniline from the lactone (coumarin) ring should be disconnected firstly. The disconnection gives aniline derivative and 4-chlorocoumarin (Godhani et al., 2015; Holm and Straub, 2011). The substitution is applied to be easily performed via chloride than hydroxyl due to hydroxyl’s poor leaving group ability (Luo et al., 2017).
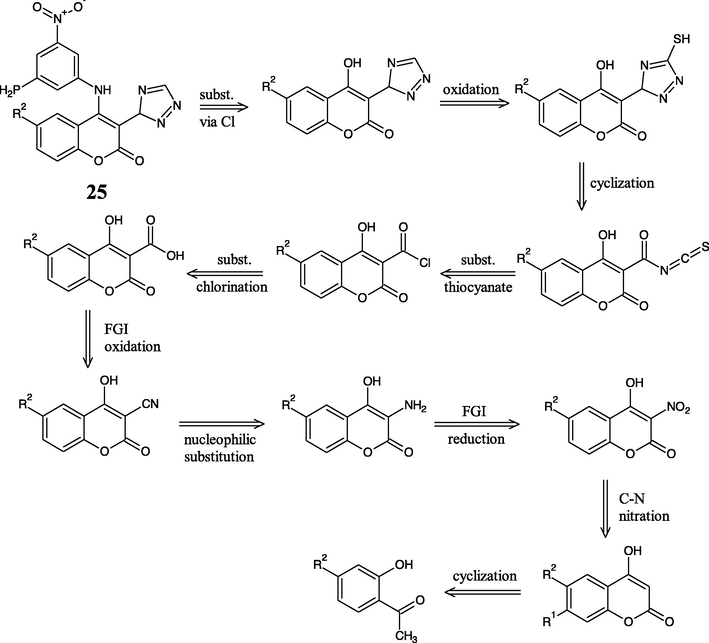
- Retrosynthesis of new designed compound 25.
The pentagonal ring of 1,2,4-triazole can be made by nucleophilic substitution of thiocyanate into chloro-carbonyl moiety followed with cyclization and oxidation (Godhani et al., 2015). The carboxylation into coumarin ring cannot be easily made, therefore it needs an FGI from –CN. The last disconnection builds O1-C2 and C3-C4 in the cyclic shape of the coumarin ring. The lactone ring cyclization requires an aromatic o-hydroxy carbonyl compound and a two-carbon fragment (C2 and C4 builder) in Knoevenagel and Perkin reactions. Despite, the reaction of aldehyde and triazide chlorosilane (TACS) was suggested via the formation of siloxy azide, and subsequent formation of gem-diazoalkane to give later cyclization to tetrazole derivatives (El-Ahl et al., 1995). These probable synthesis routes of the proposed compound has been completely written as Figs. 6 and 8.
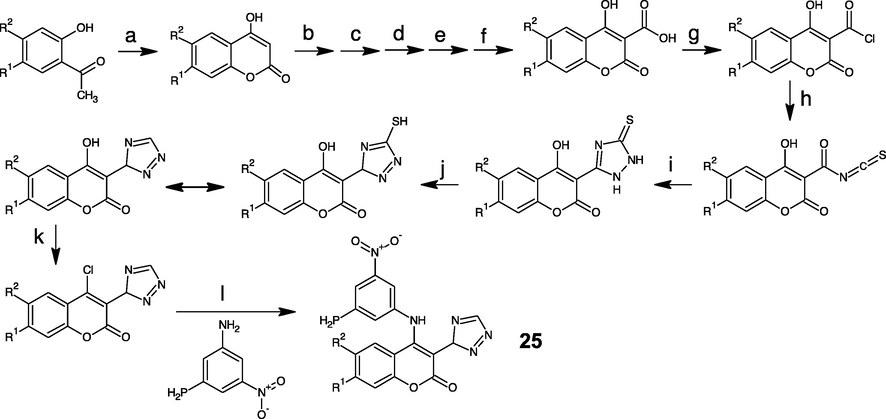
- Reagents and conditions of new anti-coloncancer compound (25) synthesis:(a) NaH, DEC, 120 °C, 3 h; (b) HNO3, H2SO4 (c) H2, Pd, C, Ac2O; (d) HCl, H2O; (e) HCl, NaNO2, Cu(I)CN; (f) NaOH; (g) SOCl2; (h) NH4SCN; (i) N2H4; (j) HNO3, H2O, 45 °C; (k) POCl3, 60 °C, 2 h; (l) K2CO3, DMF, 75 °C, 2 h. (Luo et al., 2017; Godhani et al., 2015; Holm and Straub, 2011; Warren, 1982; Potts, 1961).
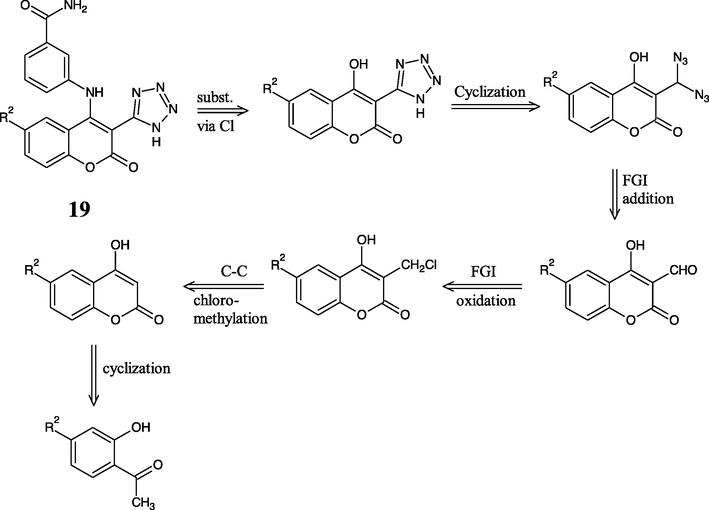
- Retrosynthesis of new designed compound 19.
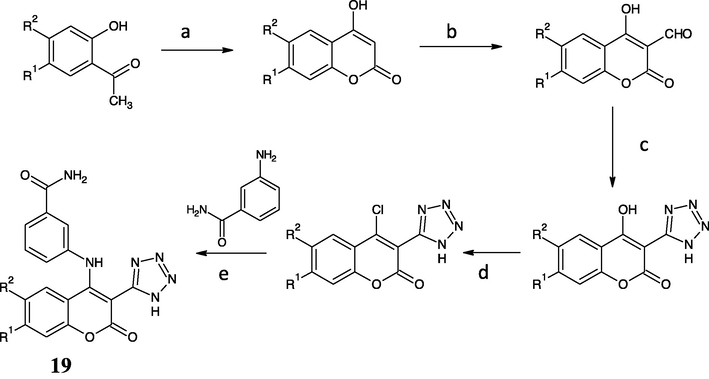
- Reagents and conditions of new anti-hepatoma compound (19) synthesis: (a) NaH, DEC, 120 °C, 3 h; (b) ZnCl2, CH2O, HCl; (c) SiCl4, NaN3, CH3CN, 25 °C, 12 h; (d) POCl3, 60 °C, 2 h; (e) K2CO3, DMF, 75 °C, 2 h (Luo et al., 2017; El-Ahl et al., 1995; Warren, 1982).
Cancer treatment like chemotherapy have major limitations. Chemotherapy strategy is powerful in killing cancer cells but also has curing problems caused by cytotoxic side effects on normal cells (Hendouei et al., 2019). This research has considered this risk by selecting the main molecule skeleton with low toxicity towards normal human cells (HUVEC). It is highly recommended to follow any cancer regimens regulation from the medication institution including any involvement of antipsychotic drug to relieve nausea and other side effect of the treatment. Furthermore, for those who are at high risk of chemotherapy it is recommended to consider colonoscopy for colon cancer screening and surveillance to detect the colonic lesions before receiving any treatments.
4 Conclusions
The anti-colon cancer and antihepatoma activity of substituted 4-anilino coumarin derivatives were successfully predicted using QSAR equation of multiple linear statistical analysis. The validated QSAR models designed the 17 new compounds of substituted 4-anilino coumarin derivatives and predicted their anti-colon cancer and anti-hepatoma activities. The best new compounds, 4-[(3-nitro-5-phosphanylphenyl)amino]-2-oxo-3-(3H-1,2,4-triazol-3-yl)–2H-chromene-6-carboxamide was predicted as anti-colon cancer with IC50 = 0.82 μM, and 3-{[6-(formylamino)-2-oxo-3-(1H-tetrazol-5-yl)–2H-chromen-4-yl]amino}benzamide as anti-hepatoma with IC50 = 0.44 μM.
Acknowledgments
We are grateful to Austrian-Indonesian Centre for Computational Chemistry (AIC) Laboratory, Department Chemistry, Faculty of Mathematics and Science, Universitas Gadjah Mada for providing their instrument facilities, PC, software computation for analysis data samples. The authors extend their appreciation to the Researchers supporting project number (RSP2022R470) King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Chemical-informatics approach to COVID-19 drug discovery: Monte Carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J. Biomol. Struct. Dyn.. 2021;39(13):4764-4773.
- [CrossRef] [Google Scholar]
- A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys.. 1993;98(2):1372-1377.
- [CrossRef] [Google Scholar]
- Design, Synthesis, and Evaluation of in Vitro and in Vivo Anticancer Activity of 4-Substituted Coumarins: A Novel Class of Potent Tubulin Polymerization Inhibitors. J. Med. Chem.. 2016;59(12):5721-5739.
- [CrossRef] [Google Scholar]
- Semiempirical Molecular Orbital Theory: Facts, Myths and Legends. In: Recent Experimental and Computational Advances in Molecular Spectroscopy. Dordrecht: Springer Netherlands; 1993. p. :369-380.
- [CrossRef] [Google Scholar]
- A facile and convenient synthesis of substituted tetrazole derivatives from ketones or α, β-unsaturated ketones. Tetrahedron Lett.. 1995;36(40):7337-7340.
- [CrossRef] [Google Scholar]
- QSAR models for antioxidant activity of new coumarin derivatives. SAR QSAR Environ. Res.. 2015;26(7-9):721-737.
- [CrossRef] [Google Scholar]
- Synthesis and biological screening of 1, 2, 4-triazole derivatives. Ind. J. Chem.. 2015;54:556-564.
- [Google Scholar]
- Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. Mol. Divers.. 2000;5:231-243.
- [CrossRef] [Google Scholar]
- Molecular mechanisms of anti-psychotic drugs for improvement of cancer treatment. Eur. J. Pharmacol.. 2019;856:172402.
- [CrossRef] [Google Scholar]
- Predictive modelling of the LD 50 activities of coumarin derivatives using neural statistical approaches: electronic descriptor-based DFT. Integr. Med. Res.. 2016;10:451-461.
- [CrossRef] [Google Scholar]
- Synthesis of N -substituted 1,2,4-triazoles. A review. Org. Prep. Proced. Int.. 2011;43(4):319-347.
- [CrossRef] [Google Scholar]
- Horak, K.E., Fisher, P.M., Hopkins, B., 2018. Pharmacokinetics of Anticoagulant Rodenticides in Target and Non-target Organisms 87–108. doi:10.1007/978-3-319-64377-9_4.
- Design, synthesis and biological evaluation of novel 3-substituted 4-anilino-coumarin derivatives as antitumor agents. Bioorg. Med. Chem. Lett.. 2017;27(4):867-874.
- [CrossRef] [Google Scholar]
- Anti-cancer potential of polysaccharide extracted from hawthorn (Crataegus.) on human colon cancer cell line HCT116 via cell cycle arrest and apoptosis. J. Funct. Foods. 2020;64:103677.
- [CrossRef] [Google Scholar]
- Exploring QSAR studies on 4-substituted quinazoline derivatives as antimalarial compounds for the development of predictive models. Med. Chem. Res.. 2014;23(3):1397-1405.
- [CrossRef] [Google Scholar]
- In Vitro Antioxidant Activity of Selected 4-Hydroxy-chromene-2-one Derivatives—SAR, QSAR and DFT Studies. Int. J. Mol. Sci.. 2011;12:2822-2841.
- [CrossRef] [Google Scholar]
- Global method for electron correlation. Phys. Rev. Lett.. 2017;119:063002
- [CrossRef] [Google Scholar]
- Pramesti, G., 2017. Statistika Penelitan dengan SPSS 24. PT. Gramedia, Jakarta.
- Study on anti-tumor activity of novel 3-substituted 4 anilino-coumarin derivatives using quantitative structure-activity relationship (QSAR) Mater. Sci. Forum. 2019;948:101-108.
- [CrossRef] [Google Scholar]
- Matching donor to recipient in liver transplantation: Relevance in clinical practice. World J. Hepatol.. 2013;5(11):603.
- [CrossRef] [Google Scholar]
- Benchmark study of structural and vibrational properties of scandium clusters. J. Mol. Struct.. 2017;1142:139-147.
- [CrossRef] [Google Scholar]
- Discovery and synthesis of novel 3-phenylcoumarin derivatives as antidepressant agents. Bioorg. Med. Chem. Lett.. 2011;21:1937-1941.
- [CrossRef] [Google Scholar]
- TD-DFT benchmark for UV-Vis spectra of coumarin derivatives. Vietnam J. Chem.. 2021;59:203-210.
- [CrossRef] [Google Scholar]
- Chemistry Computational Chemistry a Practical Guide for Applying Techniques to Real-World Problems. New York: New York. John Wiley & Sons; 2001.
- [CrossRef]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.101837.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




