

Translate this page into:
Synthesis of 2-amino-5-methylpyridinium tetrachloridocadmate(II) (C6H9N2)2[CdCl4]: Structure, DFT-calculated descriptors and molecular docking study
⁎Corresponding author. issaoui_noureddine@yahoo.fr (Noureddine ISSAOUI)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
In this research paper, (C6H9N2)2[CdCl4], was effectively synthesized using the slow solvent evaporation procedure. Single crystal X-ray diffraction (scXRD) analysis revealed that the compound crystallizes in the triclinic system, specifically in the space group P . Powder XRD (PXRD) of the bulk material showed some minor impurities. The atomic arrangement of the title structure comprises discrete tetrahedral groups [CdCl4]2− linked to the organic entities through weak N(C)—H…Cl hydrogen bonds. Solid-state contacts were further studied through Hirshfeld surface analyses, complemented by 2D fingerprint plots. Computational results, obtained using the B3LYP tool with 6-311++G(d,p) + LANL2DZ mixed basis set, demonstrated consistent geometrical, vibrational, and electronic features to the experimental data. Non-covalent interactions were explored in depth using Atoms-In-Molecule (AIM) and Reduced Density Gradient (RDG) analyses. Thermogravimetry (TG) and Differential Scanning Calorimetry (DSC) analyses showed melting at 378 K and decomposition at around 540 K. Furthermore, the inhibition activity of the examined compound was explored in-silico through molecular docking studies targeting the inducible Nitric Oxide Synthase (iNOS) enzymes.
Keywords
Hirshfeld surface
DFT calculations
IR spectroscopy
AIM-RDG
HOMO-LUMO
Molecular docking

1 Introduction
A swiftly growing interest has recently emerged in investigating non-covalent interactions through both empirical and theoretical approaches. This surge in interest stems from their indispensable role in shaping the foundations of supramolecular chemistry. Among these interactions, hydrogen bonding stands out as a particularly influential force in crafting sophisticated assemblies, owing to its strength and precise directionality. Furthermore, hydrogen bonding profoundly impacts various domains, including molecular recognition, crystal engineering, and biological systems (Selvakumari et al., 2021; Rayes et al., 2021; Selvakumari et al., 2022). In this context, Organo-metal halide assembly of class I, in which weak non-covalent interactions serve as the linkage between organic and inorganic constituents, represents vibrant areas of chemical research and material science. In particular, the crystal chemistry of halogenocadmates(II) is of significant importance, as these compounds can assume a diverse range of coordinations, geometric arrangements, and the ability to crystallize within a diverse array of anionic structures (Karaa et al., 2023).
Additionally, pyridine derivatives, which play important roles in heterocyclic chemistry (Bis and Zaworotko, 2005; Fatima et al., 2022) and are commonly engaged in hydrogen-bond interactions, are recognized as significant structural moieties in medicinal chemistry. These compounds incorporate several alluring biological properties, notably anticancer and enzyme inhibition, making them a prime candidate for organic counterparts. In fact, a recent molecular docking review on neutral 2-amino-5-methyl pyridine (Kavitha and Alivelu, 2023) and on bis(2-amino-5-methylpyridinium) tetrachlorocuprate(II) (Gatfaoui et al., 2021) has assessed that the molecule meets drug-likeness criteria founded on the principles established by Lipinski and Veber. This suggests that the 2-amino-5-methyl pyridine molecule possesses the requisite physicochemical and pharmacokinetic properties, positioning it as a promising candidate for oral drug development.
In this contribution, we convey a comprehensive evaluation encompassing the growth of the material mentioned above and a comparative analysis between X-ray crystallographic data and structural data obtained through computational methods. The DFT/B3LYP-(6–311++G(d,p) + LANL2DZ) model chemistry was employed for the determination of geometrical parameters, vibrational properties, energy of the frontier molecular orbitals (FMOs), and the molecular electrostatic potential (MEP). Solvents exert a substantial influence on the stability and reaction rates of chemical processes (Medimagh et al., 2023; Godwini et al., 2023). This study delves into the intricate analysis of the title material's computed geometrical parameters, MEP reactive sites, and FMOs beyond the confines of the gas phase, encompassing diverse solvent environments, including water, methanol, dimethyl sulfoxide (DMSO), and acetone. The absorption spectrum obtained using the TD-DFT/BLYP calculation was compared to that obtained experimentally. Furthermore, although docking studies have rapidly permeated the field of biological research due to their computational efficiency (Chaudhary et al., 2023; Kose et al., 2018), only a limited number of publications have reported theoretical calculations regarding compounds with organic chlorocadmates (Jomaa et al., 2020; Jomma et al., 2021). In light of this, and considering the recent investigation of (C6H9N2)2[CuCl4] (Gatfaoui et al., 2021) compound with the same cation and the Cuprate metal targeting the iNOS enzyme, a docking study has been undertaken to estimate the inhibitory activity of (C6H9N2)2[CdCl4] and the impact of metal substitution on its binding capabilities in the iNOS enzyme's active site.
2 Experimental details
2.1 Synthesis
A solution with a metal/amine molar ratio of 1:2 was prepared by gradually adding CdCl2·H2O (0.201 g, 1 mmol) dissolved in dilute HCl (5 mL, 3 M) to a solution of 2-amino-5-methylpyridine (0.216 g, 2 mmol, purity 99 %) in ethanol while maintaining continuous stirring. This mixture was stirred for several minutes to obtain a homogeneous mixture.
2.2 X-ray data collection and physical measurements
The sample's morphology was initially observed using a JEOL JSM-6490LV scanning electron microscope spectrometer, operating at an acceleration voltage of 20 kV. Then, a high-quality and adequately sized single crystal of (C6H9N2)2[CdCl4] was selected for structural analysis. XRD data were acquired at 298 K using a D8 VENTURE Bruker AXS diffractometer equipped with a Multi-layer monochromator and CuKα radiation (λ = 1.54178 Å). Experimental conditions and Crystal data used for the collection of intensity data are presented in Table 1. Structural graphics were generated using the DIAMOND program (Bergerhoff et al., 1996). To ensure the accuracy and reliability of the remaining physical measurements, polycrystalline samples for PXRD were prepared by finely grinding the air-dried crystals. Subsequently, PXRD data were collected using a PANalyticalX'Pert Pro powder Diffractometer, employing Cu-Ka radiation (1.542 Å) within a 2θ range of 5–70°. The FT-IR spectrum was recorded with a Perkin-Elmer FT-IR 1000 spectrometer.
N° CCDC
2159048
Chemical formula
(C6H9N2)2[CdCl4]
Formula weight (g/mol)
472.50
Crystal system, space group
Triclinic, P
Temperature (K)
298
a (Å)
7.7127 (3)
b (Å)
8.6907 (3)
c (Å)
α (°)
β (°)
γ (°)15.0834 (5)
94.472 (2)
91.016 (2)
111.725 (1)
V (Å3)
935.16 (6)
Z
2
F(0 0 0)
468
µ (Cu Kα)
14.60 mm−1
Index ranges
–9 ≤ h ≤ 9, –10 ≤ k ≤ 10,
–17 ≤ l ≤ 17
Reflections collected
6929
Independent reflections
3237
Reflections with I > 2σ(I)
3023
Rint
0.040
(sin θ/λ)max (Å−1)
0.597
Absorption correction: multi-scan
Tmin = 0.443, Tmax = 0.753
Refined parameters
193
R[F2 > 2σ (F2)]
0.046
wR(F2)
0.135
Goodness of fit
1.12
Δρmax, Δρmin (e Å−3)
1.18, –1.08
Solide-state UV–Vis spectrum was recorded at room temperature using a Perkin Elmer Lambda 35 spectrophotometer. Synchronous TG and DSC analyses were conducted on a 7.4 mg sample in a platinum crucible under an air atmosphere. These analyses were performed using an SDT Q600 V20.9 Build 20′’ thermoanalyzer, with a heating rate of 10 K.min−1.
3 Computational details
The CIF format of (C6H9N2)2[CdCl4] served as the input file for molecular geometry optimization. This optimization was conducted in the gas phase and also within diverse solvent environments, such as water, methanol, DMSO, and acetone using Gaussian 16 (Frisch et al., 2016) with the GaussView 6 interface (Dennington et al., 2016). Density Functional Theory (DFT) calculations were carried out via the hybrid B3LYP functional (Lee et al., 1988) and the conventional 6–311++ G (d,p) basis set for all atoms, except cadmium, for which the LANL2DZ basis set and effective core potentials were utilized. Vibrational assignments were conducted with the VEDA4 program (Jamróz, 2004). 3D Hirshfeld surfaces and 2D fingerprints were generated using Crystal Explorer software (Wolff et al., 2013).
Topological features at the BCPs were assessed using the Multiwfn program (Lu and Chen, 2012), and the reduced density gradient of (C6H9N2)2[CdCl4] was graphed using both the Multiwfn and VMD programs (Humphrey et al., 1996). The optimized molecular structure was used to perform HOMO-LUMO distributions and MEP analysis using the Integral Equation Formalism Polarizable Continuum Model (IEFPCM). To explore the excited states, time-dependent DFT (TD-DFT) calculations were conducted on the optimized structure using the same level of theory. For molecular docking simulation, the iGEMDOCK program, which relies on the GEMDOCK scoring function (Yang et al., 2004), was employed. Graphical representations of ligands in the docked form were generated using the PyMol interface (Schrödinger, 2015) and Discovery Studio 4.1. The enzyme structure was obtained from the Research Collaboratory for Structural Bioinformatics (RCSB), Protein Data Bank (PDB).
4 Results and discussion
4.1 Powder X-ray diffraction and SEM
To ascertain the pureness of the synthesized (C6H9N2)2[CdCl4] powder, the experimental PXRD pattern was compared to that derived from the single crystal diffraction data (Fig.S1). Notably, the main peak positions shown in the PXRD pattern perfectly matches the simulated pattern, demonstrating that the synthesized bulk and the crystal data used for diffraction are homogenous with some minor impurities. SEM images (Fig.S2), were captured at various magnifications (500×, 1000×, 2000×, and 5000×). The SEM micrographs reveal that the surface morphology of the grown crystals appears as an assembly of homogeneous blocks with flat surfaces uniformly distributed in layers. This observation indicates the crystals’ high quality and suitability for X-ray diffraction studies.
4.2 Structure elucidation and molecular modeling
Single crystal XRD analysis discloses that the asymmetric unit of (C6H9N2)2[CdCl4] comprises one [CdCl4]2− anion and two crystallographically distinct 2-amino-5-methylpyridinium cations (Fig. 1). In both cations, the pyridine nitrogen atom is protonated in precedence to the primary amine. The molecular geometry optimization was performed in the gas phase as well as for a variety of solvents using the IEFPCM solvation model. The resulting molecular structure exhibits C1 point group symmetry, and the ground state energies are calculated as –795.22 a.u for the gas phase, –795.24 a.u for acetone, –795.23 a.u for DMSO, –795.22 a.u for water, and –795.23 a.u for methanol. The dipole moment was found to be 3.29 D for the gas phase, 1.92 D for acetone, 1.90 D for DMSO, 1.89 D for water, and 1.91 D for methanol. It is noteworthy that, while the solvent significantly influences the dipole moment of the investigated molecule, it remains indifferent to the total energy.![ORTEP (ellipsoids drawn at the 50 % probability level) representation of the asymmetric unit of (C6H9N2)2[CdCl4] with an atom-labeling scheme. Hydrogen atoms are depicted as spheres with arbitrary radii. Red dashed lines denote hydrogen bonds.](/content/185/2024/36/5/img/10.1016_j.jksus.2024.103178-fig2.png)
ORTEP (ellipsoids drawn at the 50 % probability level) representation of the asymmetric unit of (C6H9N2)2[CdCl4] with an atom-labeling scheme. Hydrogen atoms are depicted as spheres with arbitrary radii. Red dashed lines denote hydrogen bonds.
The crystal packing of (C6H9N2)2[CdCl4], observed along the bc plane (Fig. 2a), reveals parallel layers of cations and discrete anions stacked in positions z = 1/4 and 3/4. Each [CdCl4]2− anion forms hydrogen bonds with surrounding inclined rows of cations along the b-axis and with planar cations at z = 0, and ½ along the c-axis.![The arrangement of (C6H9N2)2[CdCl4] in the ( b → , c → ) plane is shown in view (a), while view (b) show a partial view of the crystal structure, highlighting the patterns of hydrogen bonding and π…π stacking interactions between adjacent aromatic rings.](/content/185/2024/36/5/img/10.1016_j.jksus.2024.103178-fig3.png)
The arrangement of (C6H9N2)2[CdCl4] in the (
plane is shown in view (a), while view (b) show a partial view of the crystal structure, highlighting the patterns of hydrogen bonding and π…π stacking interactions between adjacent aromatic rings.
The anions stacks, aligned parallel to the b-axis with a Cd—Cd distance of 6.740 Å, demonstrate no notable interactions between the [CdCl4]2– anions. Interatomic bond lengths and angles (Table S1) within the inorganic group perfectly align with analogous halogenocadmates (John et al., 2022; Hegde et al., 2021; Vassilyeva et al., 2021). Experimental Cd—Cl bond lengths range from 2.4236 (15) to 2.4934 (15) Å, while calculated lengths fall between 2.5442 and 2.5670 Å. Cl—Cd—Cl bond angles vary from 103.79 (6)° to 117.20 (7)°, and calculated angles range from 102.41 to 113.48°. The τ4 parameter, a metric for four-coordinate compounds (Yang et al., 2007), was employed to assess the geometry of (C6H9N2)2[CdCl4]. The resulting τ4 value of 0.90 indicates that the tetrahedral CdCl4 geometry exhibits slight distortion.
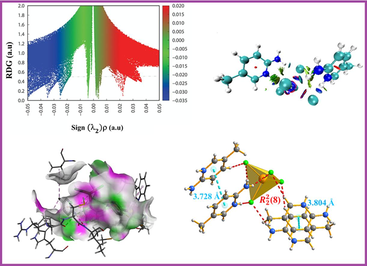
The (C—C, C—N) bond lengths of the 2-Amino-5-methylpyridinium cation are within the range observed in various 2-Amino-5-methylpyridinium metal–halide compounds (Jin et al., 2005; Carnevale et al., 2010; Ali et al., 2007; Tahenti et al., 2022). The C—C—C bond angles in this cation range from 115.8 (6)° to 122.9 (7)°. The optimized C—C—C bond angles vary from 116° to 122.1°. The protonation at atoms N1 and N3 leads to minor elevation in the N1—C1—C6 [123.4 (5)] and N3—C10—C12 [122.4 (7)] bond angles compared to the neutral 2-amino-5-methylpyridine structure (Hemamalini and Fun, 2010). The assessment of the organic geometrical features reveals the presence of two types of cations: type I are planar layered cations (holds N1 and N2), with neighboring cations separated by a centroid–centroid span of 3.804 Å, as shown in (Fig. 2b), the type II (holds N3 and N4), form inclined rows of cations with neighboring cations separated with a distance of 3.728 Å suggesting the presence of two types of π…π interactions. In all adjacent layers, the cations are staggered from each other and exhibit an alternating orientation, likely to reduce electrostatic and steric interactions and consequently build a local inversion center. Additionally, the 2-Amino-5-methylpyridinium cation displays an evenly distributed volume, due to the para arrangement of the methyl and amino groups.
The interaction between anions and cations is regulated by three types of hydrogen bonds, as outlined in Table S2. These include hydrogen bonds involving an aminic hydrogen atom and a chloride ion, where donor–acceptor distances vary between 3.286(6) and 3.433(6) Å, hydrogen bonding connecting a pyridinic hydrogen atom and a chloride ion as for N1—H1…Cl2 and N3—H3A…Cl4, the collaboration of pyridinic and aminic hydrogen to form hydrogen bonds to a halide ion is commonly observed in other 2-Amino-5-methylpyridinium salts. The C—H…Cl hydrogen bonds, in conjunction with those aforementioned, generate a (8) ring motif.
4.3 Hirshfeld surface analysis
The normalized contact distance dnorm, mapped along the c-axis of (C6H9N2)2[CdCl4] is portrayed in Fig. 3a. The results discussed in the structural elucidation part, particularly concerning hydrogen bonds (HB), are efficiently summarized on the dnorm map, where red circular spots indicate close-contact interactions, including N—H…Cl and C—H…Cl HB. Beyond HB which contributes to the 3D architecture of the crystalline stack, this latter is further stabilized by other interactions. The Curvedness and Shape-index plots of the HS are both means to portray the C—H…π or the π…π stacking by the presence of wide flat areas with a blue outline around the antiparallel and neighboring pyridinium rings on the Curvedness graph and by the presence of red and blue triangles in the shape-index. Fig. 3b and c support the existence of π…π interactions in (C6H9N2)2[CdCl4].![Hirshfeld surfaces in 3d, mapped with dnorm(a) in the range [–0.354 –1.267] (red dotted lines represent hydrogen bonds), view of the curvedness (b) and the shape index (c) plots of (C6H9N2)2[CdCl4].](/content/185/2024/36/5/img/10.1016_j.jksus.2024.103178-fig4.png)
Hirshfeld surfaces in 3d, mapped with dnorm(a) in the range [–0.354 –1.267] (red dotted lines represent hydrogen bonds), view of the curvedness (b) and the shape index (c) plots of (C6H9N2)2[CdCl4].
The full 2D fingerprint plots (Fig. S3), generated by resolving the 3D dnorm surface, display the intermolecular contacts involved within the structure. These plots can be further decomposed to quantify individual contributions and emphasize pairs of atoms in close contact. The plots are generated based on de and di distance scales, shown on the graph axes. Additionally, the calculated values of the enrichment ratios for (C6H9N2)2[CdCl4] are given in Table 2. The enrichment ratio was computed to accentuate the contacts statistically favored within the crystal packing. The examination of the 2D fingerprint plots reveals that H…Cl/Cl…H contacts are the most prevalent interactions, constituting a relative contribution of 44.8 %, due to the abundant presence of chloride and hydrogen on the HS (SCl = 23.6 % and SH = 62.1 %). These contacts exhibit a de + di ∼ 2.39 Å less than the combined VdW radii of (Cl:1.75 Å + H:1.09 Å = 2.84 Å) classifying them as close contacts. The enrichment ratio EHCl = 1.53 clearly elicits the tendency to assemble N(C)—H…Cl HB.
Enrichment
H
Cl
C
N
Cd
H
0.83
1.53
0.84
0.83
1.61
Cl
–
0.12
0.09
0.39
–
C
–
–
3.33
–
–
N
–
–
–
–
–
Cd
–
–
–
–
–
% surface
62.10
23.60
9.65
3.30
0.85
H…H contacts constitute 32.1 % of the HS and correspond to an ERHH = 0.83. Additionally, H…C/C…H (10.1 %) and H…N/N…H (3.4 %) contacts are discernible on the HS, enriched with similar ratios (ER = 0.84; 0.83). These contacts reveal on their 2D digital plots the existence of two wings, each centered on a sum of de + di ∼ 3 Å and 2.8 Å, respectively. However, only the H…N/N…H contacts are considered close when compared to the combined VDW radii of the involved atoms. Among the contacts, C…N/N…C contacts exhibit the highest enrichment value, ECN = 4.08, followed by C…C contacts with an enrichment of ECC = 3.33. This may be attributed to the accumulation of aromatic pyridinium rings in the crystalline packing, and the latter may be related to π…π interactions, with SN = 3.3 % and SC = 9.65 %. Other contacts, such as H…Cd/Cd…H 1.7 %, Cl…Cl 0.7 %, N…Cl/Cl…N 0.6 %, Cl…C/C…Cl 0.4 %, contribute less to the HS.
4.4 Vibrational analysis
The vibrational behavior of the (C6H9N2)2[CdCl4] was discussed through a combination of observed and computed infrared spectra. The experimental IR spectrum recorded in the range [4000 – 400] cm−1 and modeled using the B3LYP/LANL2DZ basis set are illustrated in Fig. S4. The experimental infrared wavenumber and the calculated one obtained through the DFT method are detailed in Table S3. To ensure a closer match with experimental frequencies, uniform scaling factors, as recommended by Rauhut and Pulay (Daghar et al., 2022; Jomaa et al., 2023; Rauhut and Pulay, 1995; Bilgili et al., 2022) were applied. This adjustment was introduced to enhance the concordance with the experimental data, the minor differences noted are ascribed to the calculations carried out in the gas phase. Brief discussions of the assignments for the key functional groups are presented in the following sections.
4.4.1 [CdCl42−] vibrations
Based on theoretical calculations and comparisons with other materials containing the chlorocadmate anion (Jomaa et al., 2020; Jomma et al., 2021); it is evident that the inorganic component displays vibrations less than 500 cm−1. The asymmetric stretching of Cd—Cl appears at 174 cm−1, while the symmetric stretching is observed at 149 cm−1.
4.4.2 (C6H9N2+) vibrations
The νa and νs vibrations of N—H are detected, between 3421 and 3140 cm−1. The predicted frequency for these bands, based on DFT calculations, falls between 3506 and 3107 cm−1.
The bands identified at 1667 and 1662 cm−1 are attributed to the in-plane and out-of-plane deformation mode of N—H, respectively, with corresponding calculated frequencies of 1648 and 1634 cm−1. Medium-intensity bands between 1552 and 1350 cm−1 are ascribed to the δa and δs vibrations of —NH2 and —CH3, with predicted frequencies of 1554 and 1350 cm−1.
In the range between 3042 and 2907 cm−1, the observed bands are attributed to the ν(C—H) valence vibrations of the pyridine ring (Kazachenko et al., 2023). The computed values of these vibrations are νas = 3053 cm−1 and νs = 2910 cm−1. The in-plane and out-of-plane deformation modes of C—H in the pyridine ring are attributed to the bands detected at 1222 and 1151 cm−1, respectively. DFT simulations show that these modes have frequencies between 1221 and 1157 cm−1. (Ghatfaoui et al., 2021)
In the experimental spectrum, bands that range between 989 and 907 cm−1 correspond to ν(C—N). These vibrations are computed between 1061 and 963 cm−1. Furthermore, the bands observed at approximately 763 and 503 cm−1 are assigned to the out-of-plane deformation vibration modes δ(C—C) and δ(C⚌C). Theoretical calculations place these vibrations within the range of 760–519 cm−1. (Daghar et al., 2021)
4.5 Uv–vis absorption of (C6H9N2)2[CdCl4]
The predicted UV–Visible spectra of (C6H9N2)2[CdCl4], obtained via the TD–DFT/ B3LYP -LANL2DZ and TD–DFT/BLYP-LANL2DZ model chemistries, are compared in Figure S5. The experimental UV–Visible spectrum is given in Figure S6.a, it presents a single band with a maximum absorption at 330 nm. This band corresponds to the π → π* transition, related to aromatic conjugation in the organic 2-amino-5-methylpyridinium group, as supported by existing literature (Jin et al., 2005; Carnevale et al., 2010) and to the Ligand-Metal-Charge Transfer.
The identified band appears at 345 nm in the spectrum calculated at the BLYP level and at 283 nm for the B3LYP level. The determined band gap of (C6H9N2)2[CdCl4] is 3.43 eV, as revealed by the Tauc plot model shown in Figure S6.b.
4.6 Frontier molecular orbital and chemical quantum descriptors
To assess the energetic behaviors and reactivity of (C6H9N2)2[CdCl4], a thorough examination of the frontier orbitals was conducted. The 3D tracings of the HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) in both the gas phase and various solvents, including water, acetone, methanol, and DMSO are illustrated in Fig. S7. In these visual representations, green defines the negative phase; and red represents the positive phase. Analysis of the gas phase results indicated that HOMO components are distributed across both the organic cation and chlorine atoms, while LUMO components are situated only on the organic cation.
The energy gap between the HOMO-LUMO in the investigated compound is notably wide, measuring ΔE = 4.21 eV in the gas phase and almost the same value in the solvents which is 4.76 eV, indicating low chemical reactivity and heightened kinetic stability (Mhadhbi et al., 2022). Thus, we can conclude that the solvents increase the HOMO-LUMO energy gap (Kazachenko et al., 2022).
Furthermore, through the computed HOMO and LUMO energies and applying established formulas from the literature (Parr and Chattaraj, 1991), we can determine the global descriptors of chemical reactivity, as summarized in Table S4, for the different phases. All energies determined in the different phases of (C6H9N2)2[CdCl4], as well as the chemical potential, exhibit negative values. Therefore, we can infer that the crystal arrangement remains steady and doesn't undergo spontaneous decomposition into its elemental components. The compound exhibits in the gas phase a chemical hardness (η) of 2.10 eV and softness (S) of 0.23 eV, while in different solvents, it demonstrates a chemical hardness (η) and softness (S) of approximately 2.38 eV and 0.21 eV, respectively. These consistent results collectively point to the high kinetic stability of (C6H9N2)2[CdCl4] (Ahmed et al., 2017; Asif et al., 2022).
4.7 Molecular electrostatic potential (MEP)
The three-dimensional charge distributions of molecules can be insightfully revealed by the investigation of MEP. This analytical approach helps identify a molecule's most reactive electrophilic and nucleophilic sites, which are crucial for promoting the formation of HB (Kosar and Albayrak, 2011; Sajid et al., 2021). In the current study, MEP surfaces of (C6H9N2)2[CdCl4] were generated, considering both the gas phase and a diverse range of solvents.
The mapped MEP surfaces, illustrated in Figure S8, showcase the total electron density’s nuanced variation from –8.336 e–2 a.u. (dark red) to 8.336 e–2 a.u. (dark blue). The red color reveals regions of strong repulsion, whereas the blue color indicates areas of pronounced attraction. Consequently, the negative potential region is associated with the [CdCl4]2– group serves as an ideal site for nucleophilic attack. Conversely, the most positive potential is observed near the hydrogen atoms, particularly in proximity to the —NH+ group of the organic cation (C6H9N2)+. These outcomes indicate the existence of electrostatic attraction between the cations and the anion [CdCl4]2–. The presence of a yellow region, denoting a slightly inert area, confirms the existence of intermolecular interactions of the N—H…Cl type.
4.8 Topological study of non-covalent interactions: AIM‑RDG analysis
The visual representation of the Atoms-In-Molecule (AIM) analysis of (C6H9N2)2[CdCl4], is shown in Figure S9, while the corresponding geometric, topological, and energetic parameters are collected in Table 3. The AIM study discloses that the BCPs are situated across Cl…H and Cl…N bonds, and that Cl2…H9, Cl2…N23, and Cl3…H7 interactions exhibit topological properties similar to Cl4…H26, Cl4… N6, Cl5…H24 interactions (Kazachenko et al., 2022).
BCP
Density of all electrons
(r)
Laplacian of electron density
2
(r)
Potential energy density V(r)
Interaction
energy (Eint) kJ/mol
Lagrangian kinetic energy G(r)
Energy densityH(r)
Cl2…H9
0.025301
0.06430
–0.01561
–20.49
0.015846
0.00023
Cl2…N23
0.004178
0.01234
–0.00198
–2.59
0.002535
0.00055
Cl3…H7
0.035046
0.07207
–0.02498
–32.79
0.021500
–0.0035
Cl4…H26
0.025306
0.06431
–0.01562
–20.50
0.01585
0.00023
Cl4…N6
0.004112
0.01214
–0.00194
–2.55
0.002492
0.00054
Cl5…H24
0.035026
0.07206
–0.02496
–32.77
0.02148
–0.00347
Among these interactions, Cl3…H7 or Cl5…H24 particularly stand out with the highest electron density (0.035046–0.035026 a.u.), attributed to their shorter interatomic distance. Consequently, these two interactions are considered the strongest by the computed HB energy value, which is approximately –32.79 kJ.mol−1. Conversely, the remaining hydrogen bonds are regarded as weak, given their positive Laplacian and energy density values. The positive Laplacian values vary between 0.07207 and 0.01214 a.u., additionally, signifying a reduction of electronic charge across the bond track (Hammami et al., 2021).
Existing non-covalent interactions (NCIs) in (C6H9N2)2[CdCl4], were further evaluated using the NCIs descriptor named the Reduced Density Gradient (RDG), an extension of AIM theory for visual analysis. The outcomes are depicted in Fig. 4. a and 4. b. In these figures, the RDG isosurface is set at 0.5 a.u. with a color scale ranging from –0.035 to 0.02.![Plots of the reduced density gradient scatter (a), and color-filled isosurface of the (C6H9N2)2[CdCl4] compound illustrating the non-bonded interactions (b).](/content/185/2024/36/5/img/10.1016_j.jksus.2024.103178-fig5.png)
Plots of the reduced density gradient scatter (a), and color-filled isosurface of the (C6H9N2)2[CdCl4] compound illustrating the non-bonded interactions (b).
According to Fig. 4.a attractive interactions, VdW interactions, and repulsive interactions in (C6H9N2)2[CdCl4] are represented by blue, green, and red regions, respectively. Each data point on the graph corresponds to a coordinate in 3D space. The points of the six peaks approximate the positions of the critical points of AIM theory. The horizontal line at RDG = 0.5 a.u. corresponds to the points that the RDG isosurfaces were constructed from. Fig. 4.b, provides a clear visualization of different interaction types based on color. Light blue dots observed between the hydrogen and chlorine atoms indicate a notable attractive N—H…Cl interaction. Green spots surrounding the anion are indicative of VdW interactions, while the oval red plaque in the middle of the ring is due to repulsive interactions, highlighting a significant steric impact.
4.9 Thermal analysis
The thermal stability of (C6H9N2)2[CdCl4] was assessed simultaneously by Thermogravimetry (TG) and Differential Scanning Calorimetry (DSC) analyses. The observed thermal behavior is depicted in Fig. 5. The initial narrow endothermic peak, observed at 378 K without any mass loss, is attributed to the melting process. Subsequently, the second endothermic peak at 540 K is accompanied, as indicated on the TG curve, by a mass loss corresponding to pyrolysis and the release of 4HCl. It’s worth noting that the calculated and experimental mass loss percentages exhibit good agreement, with values of 76.14 % and 74.98 %, respectively.![TG–DSC curves of (C6H9N2)2[CdCl4].](/content/185/2024/36/5/img/10.1016_j.jksus.2024.103178-fig6.png)
TG–DSC curves of (C6H9N2)2[CdCl4].
4.10 Molecular docking
Molecular docking analysis was conducted to evaluate the binding characteristic of (C6H9N2)2[CdCl4] with known inhibitory ligands; H4B, HBI, and FC1 targeting the “iNOS” enzyme. The inducible Nitric Oxide Synthase (iNOS) protein is one of the three pivotal enzymes responsible for nitric oxide production, playing a crucial role in diverse physiological and pathophysiological states. Excessive production of nitric oxide can contribute to the development of specific degenerative neurological disorders, including Alzheimer's, schizophrenia (Midtvedt, 2009), and Parkinson's condition. iNOS is directly associated with neurodegenerative diseases, leading to the development of specific inhibitors (ligands) designed to target this protein. Inhibitors are substances designed to block the action of enzymes, to prevent or at least slow the progression of diseases.
Docking simulations generated ten possible positions (conformations) for each ligand in the active site of the enzyme. The most favorable binding positions for the selected ligands are depicted in Fig. S10. An intriguing observation arises as H4B, HBI, FC1, and (C6H9N2)2[CdCl4] occupy nearly identical positions. Fig. 6, illustrates the various interactions present in the ligand–protein ((C6H9N2)2[CdCl4]–iNOS). These interactions include pi–alkyl, pi–anion, pi–donor, conventional hydrogen bonding, and hydrogen donor bonding interaction, etc., underscoring the complexity of the binding.![Mode of interaction of (C6H9N2)2[CdCl4] with the active sites of iNOS (a), H-Bond surface (b), and aromatic surface map (c).](/content/185/2024/36/5/img/10.1016_j.jksus.2024.103178-fig7.png)
Mode of interaction of (C6H9N2)2[CdCl4] with the active sites of iNOS (a), H-Bond surface (b), and aromatic surface map (c).
The energy outcomes of the docking computations are presented in Table 4. All ligands exhibit favorable total energy scores upon binding to the target protein, spanning from –97.36 to –78.98 kcal.mol−1. It is important to emphasize that the predominant interactions between the ligands and the enzyme are of the van der Waals type and that electronic interactions are not exhibited by all ligands (Romani et al., 2020). Among the ligands, FC1 is the most potent inhibitor, with the highest total energy value of –97.36 kcal.mol−1 and the strongest VDW interaction. Figure S11 represents the diverse intermolecular interactions between the ligands (inhibitors) and the iNOS protein (receptor).
Protein
Ligand
Total energy
VDW
H bond
Electronic
iNOS
FC1
–97.36
–97.36
0
0
H4B
–81.51
–81.51
0
0
HBI
–86.97
–86.97
0
0
(C6H9N2)2[CdCl4]
–78.98
–64.99
–13.99
0
Furthermore, the molecular docking of (C6H9N2)2[CdCl4] in the iNOS active site results in a significant binding energy of approximately –78.98 kcal.mol−1, comprising contributions from van der Waals forces (EVDW = –63.292 kcal.mol−1) and hydrogen bond interactions (EH = –13.99 kcal.mol−1). Consequently, (C6H9N2)2[CdCl4] exhibited a superior VDW interaction compared to (C6H9N2)2[CuCl4] [–58.2 kcal.mol−1], as reported in the reference study (Gatfaoui et al., 2021). The enhanced performance of (C6H9N2)2[CdCl4] in our study suggests its potential as a potent inhibitor, showcasing improved binding capabilities in the iNOS enzyme's active site.“
These findings provide valuable insights into potential therapeutic strategies for addressing conditions like Alzheimer's, schizophrenia, and Parkinson's condition.
5 Conclusion
This contribution provides valuable insights into the weak non-covalent interactions and the inhibitory properties of (C6H9N2)2[CdCl4]. A crystallographic investigation was conducted on single crystals obtained through slow evaporation at ambient temperature. Vibrational bands were accurately assigned by comparing the vibration mode frequencies with those of similar compounds and theoretical predictions. This demonstrated excellent alignment between theoretical and experimental results. The molecular assembly and cooperative effects of weak non-covalent interactions were meticulously examined through detailed analyses. The computed HOMO and LUMO energies under gaseous and several solvent conditions, reveal the material’s hardness and great stability. UV–Vis spectra, obtained experimentally and through computations, indicated the existence of π → π* transitions. Lastly, the results from molecular docking suggest a substantial interaction of the compound with the target receptors thus, an excellent pharmacokinetic profile.
CRediT authorship contribution statement
Ikram Jomaa: Conceptualization, Methodology, Software, Formal analysis, Writing – original draft. Fehmi Bardak: Conceptualization, Methodology, Software. Noureddine Issaoui: Conceptualization, Methodology, Software, Writing – review & editing, Supervision, Validation. Aurelio Cabeza: Conceptualization, Software, Validation. Duane Choquesillo-Lazarte: Conceptualization, Software, Validation. Ahmet Atac: Conceptualization, Formal analysis, Validation. Houda Marouani: Conceptualization, Validation, Formal analysis, Writing – review & editing, Supervision. Omar M. Al-Dossary: Conceptualization, Formal analysis, Validation.
Acknowledgments
We acknowledge the assistance provided by the Tunisian Ministry of Higher Education and Scientific Research. This study was supported by the Researchers Supporting Project no. RSP2024R61 of King Saud University, Riyadh, Saudi Arabia.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis, structural studies and biological activities of three new 2-(pentadecylthio)-5-aryl-1,3,4-oxadiazoles. J. Mol. Struc. 2017;1129:50-59.
- [Google Scholar]
- Intermolecular interactions involved in the crystalstructure of 2-amino-5-methylpyridinium hexabromostannate (IV), (C6H9N2)2[SnBr 6] J. Chem. Crystallogr.. 2007;37:265-273.
- [Google Scholar]
- A first principles study on electrochemical sensing of highly toxic pesticides by using porous C4N nanoflake. J. Phy. Chem. Solids. 2022;160:110345.
- [Google Scholar]
- Evaluation of crystallographic data with the program DIAMOND. J. Res. Natl. Inst. Stand. Technol.. 1996;101:221-225.
- [Google Scholar]
- Investigation of the interionic interactions and spectroscopic features of 1-Octyl-3-methylimidazolium chloride, tetrafluoroborate, and hexafluorophosphate ionic liquids: an experimental survey and DFT modeling. J. Mol. Struct.. 2022;1261:132912
- [Google Scholar]
- The 2-aminopyridinium-carboxylate supramolecular heterosynthon: a robust motif for generation of multiple-component crystals. Cryst. Growth Des.. 2005;5:1169-1179.
- [Google Scholar]
- CO(II) halide complexes with 2-amino-3-methylpyridinium and 2-amino-5-methylpyridinium: synthesis, crystal structures, and magnetic properties. J. Coord. Chem.. 2010;63:2223-2238.
- [Google Scholar]
- Computational evaluation on molecular stability and binding affinity of methyldopa against lysine-specific demethylase 4D enzyme through quantum chemical computations and molecular docking analysis. J. Mol. Struct.. 2023;1286:135518
- [Google Scholar]
- Molecular structure, spectroscopy, quantum chemical and antibacterial activity investigations of 2-methylbenzylammonium perchlorate. J. Mol. Struct.. 2022;1247:131311
- [Google Scholar]
- Dennington, R., Keith, T.A., Millam, J.M., GaussView Version 6, 2016.
- Empirical and computational studies on newly synthesis cyclohexylammonium perchlorate. J. Mol. Struc. 2021;1230:129820.
- [Google Scholar]
- Experimental spectroscopic, structural (monomer and dimer), molecular docking, molecular dynamics simulation and hirshfeld surface analysis of 2-Amino-6-methylpyridine. Polycyclic. Aromat. Compd.. 2022;43:3910-3940.
- [Google Scholar]
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A. Jr., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J., Gaussian 16 Revision A.03 (Gaussian, Inc., Wallingford CT, (2016).
- Self assembly of a novel Cu(II) complex, (C6H9N2)2[CuCl4]: experimental, computational, and molecular docking survey. J. Iran. Chem. Soc.. 2021;18:2331-2343.
- [Google Scholar]
- Synthesis, experimental and computational study of a non-centrosymmetric material 3-methylbenzylammonium trioxonitrate. J. Mol. Struc. 2021;1225:129132.
- [Google Scholar]
- Solvation impact on the geometry and electrical properties of flufenoxuron: a topological and spectral insight by DFT approach. J. Mol. Liq.. 2023;391:123231
- [Google Scholar]
- F. Hammami, N. Issaoui,S. Nasr, Investigation of hydrogen bonded structure of urea-water mixtures through Infra-red spectroscopy and non-covalent interaction (NCI) theoretical approach, Computational and Theoretical Chemistry, 1199 (2021) 113218.
- A novel chlorocadmate hybrid cocrystal delivering intermolecular charge transfer enhanced nonlinear optical properties and optical limiting. Opt. Mater: X. 2021;117:111194
- [Google Scholar]
- 2-Amino-5-methylpyridinium 3-carboxy-4-hydroxybenzenesulfonate. Acta crystallogr Sect. e: Struct. Rep. Online. 2010;66:o2153-o2154.
- [Google Scholar]
- Jamróz, M.H., 2004. Vibrational Energy Distribution Analysis: VEDA 4.
- 2-Amino-5-methylpyridinium (2-amino-5-methylpyridine) trichloro-zincate(II) acta Crystallogr Sect. c: Cryst. Struct. Commun.. 2005;61:m197-m199.
- [Google Scholar]
- Synthesis, structure, NBO, Hirshfeld surface, NMR, HOMO-LUMO, UV, photoluminescence, z scan, vibrational and thermal analysis of piperazinedi-ium tetrakis (μ2-chloro)-diaqua-dichloro-di-cadmium single crystal. J. Mol. Struct.. 2022;1258:132685
- [Google Scholar]
- Exper- imental, computational, and in silico analysis of (C8H14N2)2[CdCl6] compound. J. Mol. Struct.. 2020;1213:128-186.
- [Google Scholar]
- Supramolecular association of (1,4-phenylenedimethanaminium) bis(perchlorate) monohydrate: a combined experimental and theoretical study. J. Mol. Struct.. 2023;1272:134212
- [Google Scholar]
- Insight into non-covalent interactions in a tetrachlorocadmate salt with promising NLO properties: Experimental and computational analysis. J. Mol. Struc. 2021;124215:130730.
- [Google Scholar]
- New semiconductor halocadmate [CdnXm](2n–m) crystal structure, molecular conformation and theoretical investigations. J. Solid State Chem.. 2023;322:123954.
- [Google Scholar]
- The computational study, 3D-QSAR, and molecular docking study of 2-amino 5-methyl pyridine. Polycyclic. Aromat. Compd.. 2023;43:1495-1508.
- [Google Scholar]
- Optimization of guar gum galactomannan sulfation process with sulfamic acid. Biomass Conversion and Biorefinery. 2023;13:10041-10050.
- [Google Scholar]
- Kazachenko A.S.,N. Issaoui,Medimagh M., Yu. Fetisova O., Berezhnaya Y.D., Elsuf’ev E.V., Al-Dossary O.M., Wojcik M.J., Xiang Z., Bousiakou L.G.; Experimental and theoretical study of the sulfamic acid-urea deep eutectic solvent, (2022) J. Mol. Liq. , 363, art. no. 119859.
- A. S. Kazachenko, M. Medimagh, N. Issaoui, O. Al-Dossary, M.J. Wojcik, S. A. Kazachenko, A. V. Miroshnokova, Angelina V. Y. N. Malyar, Sulfamic acid/water complexes (SAA-H2O(1-8)) intermolecular hydrogen bond interactions: FTIR,X-ray, DFT and AIM analysis, Journal of Molecular Structure, 1265, (2022) 133394.
- Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol. Spectrochim. Acta, Part A. 2011;78:160-167.
- [Google Scholar]
- The structural and spectroscopic investigation of 2-chloro-3-methylquinoline by DFT method and UV–VIS, NMR and vibrational spectral techniques combined with molecular docking analysis. J. Mol. Struct.. 2018;1163:147-160.
- [Google Scholar]
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785-789.
- [Google Scholar]
- Multiwfn: a multifunctional wave function analyzer. J. Comput. Chem.. 2012;33:580-592.
- [Google Scholar]
- DFT and molecular docking study of the effect of a green solvent (water and DMSO) on the structure, MEP, and FMOs of the 1-ethylpiperazine-1,4-diium bis(hydrogenoxalate) compound. J. Mol. Liq.. 2023;369:120851
- [Google Scholar]
- N. Mhadhbi, N. Issaoui, H. Walid S., A. Jahoor M., E. Abdelmonein S., A. Mohd, N. Houcine, R. Badraoui, Physico-Chemical Properties, Pharmacokinetics, Molecular Docking and In-Vitro Pharmacological Study of a Cobalt (II) Complex Based on 2-Aminopyridine, ChemistrySelect, 7, 321 (2022) e202103592.
- Midtvedt, T., 2009. Chapter 25 - Penicillins, cephalosporins, other beta-lactam antibiotics, and tetracyclines, Side Eff. Drugs Annu. 31, 413-425.
- Transferable scaling factors for density functional derived vibrational force fields. J. Phys. Chem.. 1995;99:3093-3100.
- [Google Scholar]
- Synthesis, crystal structures, Hirshfeld surface analysis and physico-chemical characterization of two new ZnII and CdII halidometallates. J. Mater. Sci: Mater. Electron.. 2021;32:10890-10905.
- [Google Scholar]
- Schrödinger, LLC, The {PyMOL} Molecular Graphics System, Version∼1.8, 2015.
- Properties and reactivities of niclosamide in different media, a potential antiviral to treatment of COVID-19 by using DFT calculations and molecular docking. Biointerface Res. Appl. Chem.. 2020;10(6):7295-7328.
- [Google Scholar]
- Remarkable static and dynamic NLO response of alkali and superalkali doped macrocyclic [hexa-]thiophene complexes; a DFT approach. RCS Adv.. 2021;7:4118-4128.
- [Google Scholar]
- Spectroscopic (FT-IR, FT-Raman, UV-visible), quantum mechanical based computational studies and molecular docking analysis of 2-Amino-3,5-dichloropyridine. Anal. Chem. Lett.. 2021;11:848-861.
- [Google Scholar]
- Donor acceptor groups effect, polar protic solvents influence on electronic properties and reactivity of 2-Chloropyridine-4-carboxylic acid. J. Indian Chem. Soc.. 2022;99:100478
- [Google Scholar]
- Synthesis, characterization, and computational survey of a novel material template o-xylylenediamine. J. Iran. Chem. Soc.. 2022;19:1499-1514.
- [Google Scholar]
- Organic–inorganic hybrid tetrachlorocadmates as promising fluorescent agents for cross-linked polyurethanes: synthesis, crystal structures and extended performance analysis. RSC Adv.. 2021;11:7713-7722.
- [Google Scholar]
- Wolff, S.K., Grimwood, D.J., McKinnon, J.J., Jayatilaka, D., Spackamn, M.A., 2013. Crystal- Explorer 3.1, University of Westren Australia, Perth, 2013 3.1.
- GEMDOCK: a generic evolutionary method for molecular docking. Proteins: Struct., Funct., Bioinf.. 2004;55:288-304.
- [Google Scholar]
- Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four coordinate geometry index, τ4. Dalton Trans.. 2007;9:955-964.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2024.103178.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




