

Translate this page into:
Probing the solvent effects and protonation sites of 1-phenyl-4-(phenyldiazenyl)-1H-pyrazol-5-amine (PDPA): DFT calculations meet pKa profiling
* Corresponding author E-mail address: nwazzan@kau.edu.sa (N. Wazzan)
-
Received: ,
Accepted: ,
Abstract
5-methyl-2-phenyl-4-(2-phenylhydrazono)-2,4-dihydro-3H-pyrazol-3-one (PDPA) was recently synthesized as a promising nuclear magnetic resonance (NMR) reference in studying the tautomeric structures of azo dyes. In this study, the interaction of PDPA derivatives with a single water molecule was studied using the density functional theory (DFT) method. The protonation reaction was proposed to identify the most basic center in the investigated species by computing the proton affinity (PA) and gas phase basicity (GB) values. The nature of the intermolecular hydrogen bond (IMHB) due to the interaction of the hydrogen atom of the water molecule and the nitrogen atom of the PDPA molecule was investigated using several aspects, including geometrical structures, quantum theory of atoms in molecules (QTAIM), natural bond orbital (NBO) analysis and the noncovalent interactions (NCIs) using the reduced density gradient (RDG) MAPS. Results revealed that the PDPA derivatives are medium potent and basic with average PA and GB values of 219.7 and 213.0 kcal/mol, respectively. The maximum PA and GB values of 228.5 and 221.7 were found for the PDPA-NH2 derivative, respectively, while the smallest ones of 213.5 and 206.7 for the PDPA-CN derivative, reflecting the effect of the nature of the substituted group. Our results revealed that IMHB has moderate strength and is strongly dependent on the nature of the substituted group. The results of QTAIM indicated that the average IMHB strength is 5.0 kcal/mol and that between the water molecule and thiazolidine-2,4-dione and its derivatives is 4.8 kcal/mol. Moreover, NBO analysis showed that the average IMHB strength is 6.7 kcal/mol. Results also revealed that substitution of the electron donating groups strengthens the IMHB, while that of the electron withdrawing groups weakens it.
Keywords
Gas-phase basicity (GB)
Intermolecular hydrogen bond (IMHB)
PDPA
Proton affinity (PA)
Quantum theory of atoms in molecules (QTAIM)

1. Introduction
Azo dyes, which constitute over 65% of the synthetic dyes, are a huge and significant type of dyes. These dyes are used in some foods, textiles, and leather articles (Correia et al., 1994). Some examples include water-soluble azo dyes based on H-acid, J-acid, and γ-acid. Azo dyes have various biological applications in molecular data processing, such as in ion sensors and optical materials (Geng et al., 2015). Although useful, azo dyes can impact the quality of ground water by blocking light transmission, affecting photosynthesis, and reducing dissolved oxygen in water (Meghwal et al., 2020). Chemically, azo dyes consist of an aminobenzene group, including Ar-N=N-P(NH2)-Ar, Ar-N-N=P(C=O)-Ar, and Ar-N=N-Ar, where Ar, P(C=O), P(NH2) (Zollinger 2003). The nature of the substituted groups on the phenyl ring plays a vital role in the discrepancy of the chemical and physical properties (solubility or color) of the azo dyes (Cerezo et al., 2015; Comez et al., 2016).
Among the numerous molecular properties that can be calculated for the molecular compounds are proton affinity (PA) and gas-phase basicity (GB), which are associated with the protonation reaction. PA and GB represent the negative values of the change in enthalpy (ΔH) and Gibbs free energy (ΔG), respectively. Furthermore, PA and GB properties are very useful in the investigation of proton donor and acceptor position. The determination of PAs using experimental methods such as mass spectrometry (Valadbeigi and Farrokhpour, 2013), kinetic methods, calorimetry, ion cyclotron resonance (Blake et al., 2009), and ion mobility spectrometry (Tabrizchi and Shooshtari, 2003) is often challenging (Dixon and Lias, 1987). The accuracy of the experimental PA and GB values, as well as the challenges, difficulties, and quantities of the available materials, leads to the use of computational methods as alternatives. Most studies used Density Functional Theory (DFT) methods to calculate PA and GB quantities, which tend to calculate these quantities as accurately as the high-level ab-initio methods such as MP2 and CCTD (Rebber et al., 2006; Safi and Wazzan, 2021).
There are various types of noncovalent solute-solvent interactions (NCI), which cover the solvation progression of soluble molecules in media of interest. In particular, intermolecular forces, which can include hydrogen bonds and electrostatic interactions that lead to changes in the physiochemical nature of the azo dyes (Bani-Yaseen 2017). The interaction of water molecules can occur through implicit or explicit interaction. For explicit interactions, it is important to predict the most basic site that with the capability to interact with water molecules via intermolecular hydrogen bonds (IMHBs), which can occur by a protonation reaction (Safi and Omar 2014; Zhao and Zhang 2004).
IMHB is usually known as a weak non-covalent interaction involving a donor-acceptor pair. Quantum Theory of Atoms In Molecules (QTAIM) of Bader (Bader 1991) and Natural Bond Orbital (NBO) analysis are repeatedly applied in the analysis of hydrogen bonds, including intermolecular and intramolecular H-bonds (Lamsabhi 2008; Safi 2016; Osman 2017; Bayoumy et al., 2020; El-Sayed et al., 2024). Analysis of the electron density at the bond critical point and its related topological parameters was compared with the results of traditional experiments such as X-ray measurements (Espinosa et al., 1996; Espinosa et al., 1998).
A recent study (Deneva et al., 2019) synthesized and characterized two series of azo dyes derived from pyrazolamine and pyrazolone, which can be used as NMR reference in the study of the tautomerics of azo dyes. Of particular interest, 3-methyl-1-phenyl-4-(phenyldiazenyl)-1H-pyrazol-5-amine (PDPA) was chosen for this study (Scheme 1). As can be seen in Scheme 1, PDPA exhibits an exceptional characteristic based on the presence of a variety of functional groups. These functional groups can theoretically facilitate the noncovalent solute-solvent interactions with components present within its molecular environments. In the current study, the methyl group at position 5 of the pyrazol ring has been replaced by several groups with different natures, including H, NH2, NO2, CHO, Cl, F, and CN. We aim to provide insights into the explicit molecular solute-water interactions of PDPA using the DFT method. Protonation reaction was used to predict the most basic nitrogen atom that could explicitly interact with the water molecule. The nature of the intermolecular interactions between the PDPA derivatives and water molecule were analyzed and discussed in depth in terms of geometrical structures, QTAIM, NCI, and NBO analysis.
2. Computational details
Computational calculations and visualization of the investigated species were conducted using the Gaussian 09 software package (Gaussian09 2009) and Gaussview (Dennington et al., 2016), respectively. Geometries of PDPA derivatives were optimized, followed by frequency calculations employing the DFT method with B3LYP (Becke 1996) functional and 6-311++g(d,p) level of theory in the gas phase. To improve the accuracy of the results, DFT-D3 and BJ-damping functional correction proposed by Grimme et al (S. Grimme 2010) was also used in geometry optimization. To achieve more reliable energy, single-point energy calculations were done using the B3LYP/6-311++g(3df,2p) level of theory. The selected level of theory was examined previously and found to compute accurate PA values (Nguyen and Chandra 1998; Safi 2016). The explicit solvent effect was computationally examined at the same level of theory by the complexation of the PDPA molecule and its derivatives with a single water molecule. The corresponding PA and GB of PDPA and its derivatives were calculated using the following expressions:
Here, ΔEelec, ΔZPE, and ΔEvib represent, correspondingly, the differences between the total electronic energy, zero-point vibrational energy and temperature-dependent vibrational energy of reactants and products at T=298 K, between the neutral and protonated forms of PDPA derivatives. The (5/2)RT and were taken as 1.48 and -6.28 kcal/mol, respectively (Safi and Omar 2014). A second-order perturbation method in the NBO framework (Reed et al., 1988) was used to analyze the interactions between water molecules and PDPA derivatives. QTAIM analysis (Bader 1991) was performed using the Multiwfn software (Lu and Chen 2012). The NCI analysis, which is known as reduced density gradient (RDG), was performed using the independent gradient model (IGM) analysis based on promolecular density in the framework of Multiwfn sotware. RDG maps were visualized under VMD (Humphrey et al., 1996) and Gnuplot (Kelley 2004-2021) programs.
3. Results and discussion
The PDPA molecule has five nitrogen atoms, labeled N1, N2, N3, N4, and N5, which underwent protonation and consequently interacted with a water molecule during the solvation process via IMHB formation. A preliminary study based on pKa calculation as implemented in the Marvin sketch program (Cherinka et al., 2019) indicates that the N1 atom is the most preferable center for protonation; see Fig. SM1 of the supplementary materials. It is also important to mention that protonation of PDPA-F and PDPA-NO2 has been excluded due to the reduction of the nitro group during the pKa calculations and high electronegativity of the F atom. Furthermore, the % protonated form (%P) increases and decreases as the power of the EDG increases and EWG decreases, respectively.
3.1 Analysis PA and GB results
The calculated PA and GB values of the investigated species using the DFT method have been displayed in Table 1. The corresponding total energies, zero-point energy (ZPE), and the thermal corrections for energies, enthalpies, and Gibbs free energies (TCE, TCH, and TCG) have been collated in Table SM1 of the supplementary materials. Our preliminary DFT calculations for the parent PDPA-H molecule show that N2 and N5 are the least basic atoms with PA values of 189.2 and 189.2 kcal/mol, respectively; Whereas the competition between N1, N3, and N4 has been clearly observed. Based on these results, the PA and GB values of N1, N3, and N4 atoms have been considered in our discussion. It was found that N1 is more basic than N3 and N4. For the parent molecule, the PA and GB values of the N1 atom are higher than the corresponding ones for N3 and N4. The average PA and GB values of N1 and N2 atoms are 219.7 and 213.1 kcal/mol, respectively, compared with those of N3 (219.2 and 212.2 kcal/mol) and N4 (218.3 and 211.2 kcal/mol) atoms. The average PA and GB values indicate that all atoms have almost the same basicity, with the N1 atom being the most basic. These results agree with those obtained by pKa calculation. These findings indicate that the PDPA molecule and its derivatives are moderate potent bases and their basicity is close to that of 5-metylhydantoin derivatives (PA=185-207 kcal/mol) (Safi and Frenking 2013), aniline (PA = 210 kcal/mol) (Jolly 1991), and triazepines derivatives (PA = 207 kcal/mol) (Safi and Lamsabhi 2007), with PA values of, 186.6-209.7 kcal/mol. It was also found that the basicity of the PDPA molecule is much higher than that of Helium (PA= 42.6 kcal/mol) and lower than that of methanide (PA = 416.8 kcal/mol) (Jolly 1991).
| PDPA derivatives | PA (kcal/mol) | GB (kcal/mol) | ||||
|---|---|---|---|---|---|---|
| N1 | N3 | N4 | N1 | N3 | N4 | |
| PDPA-H | 223.5 | 220.0 | 218.6 | 217.1 | 212.7 | 211.6 |
| PDPA-CH3 | 226.5 | 222.4 | 220.0 | 220.5 | 215.0 | 212.6 |
| PDPA-NH2 | 228.5 | 219.1 | 225.2 | 221.7 | 212.0 | 218.0 |
| PDPA-Cl | 219.5 | 217.7 | 218.0 | 212.7 | 210.4 | 211.1 |
| PDPA-F | 216.5 | 216.0 | 216.8 | 209.7 | 209.4 | 209.5 |
| PDPA-CHO | 215.7 | 225.2 | 224.9 | 209.3 | 218.1 | 217.7 |
| PDPA-CN | 213.5 | 213.3 | 212.1 | 206.7 | 207.1 | 204.8 |
| PDPA-NO2 | 213.5 | 220.1 | 210.9 | 206.8 | 212.9 | 203.9 |
| 219.7 | 219.2 | 218.3 | 213.0 | 212.2 | 211.2 | |
PDPA: 5-methyl-2-phenyl-4-(2-phenylhydrazono)-2,4-dihydro-3H-pyrazol-3-one.
In general, our results show that the stronger the EDG power is, the more basic the center is, while the stronger the EWG power is, the less basic the center is (Grabowski 2001; Safi and Frenking 2013; Safi and Omar 2014). For the N1 atom, results in Table 1 demonstrate that the maximum PA values of ∼229 and ∼ 227 kcal/mol correspond to -NH2 and -CH3 derivatives, respectively. These results can be attributed to the higher ability of the lone-pair electrons on the N atom of the NH2 group, as well as the inductive effect of the CH3 group, leading to an increase the electron density of the N1 atom, and consequently increasing the tendency of the N1 atom to interact with the incoming proton. On the other hand, the lowest PA value of 213.5 kcal/mol belongs to -CN and -NO2 derivatives, indicating that substitution of electron withdrawing groups decreases the electron density on the N1 atom, and consequently, decreases the propensity of the interaction of the N1 atom with the proton. A similar interpretation can also be used for GB results. These results can be confirmed by investigating the NBO results. Data in Table SM2 show that the maximum occupancies of the neutral and protonated N1 atom, (), of 1.9456 and 1.7682 a.u., respectively. Whereas the minimum occupancies for the same atom are 1.9386 and 1.6336 a.u., respectively. Therefore, the intrinsic basicity of N1 atoms of the investigated derivatives can be arranged as follows: PDPA-NH2 > PDPA-CH3 > PDPA-H > PDPA-Cl > PDPA-F > PDPA-CHO > PDPA -CN ≈ PDPA-NO2.
For the N3 atom, an irregular decrease was observed in the PA and GB values of PDPA-NH2 derivative compared to the parent molecule, see Table 1. In contrast, an unexpected increase in the PA and GB values of PDPA-CHO and PDPA-NO2 derivatives compared to the parent molecule, which might be explained in terms of the formation of a weak IMHB between the proton and the oxygen atom of both -CHO and -NO2 groups, resulting in reducing the potential of the positive charge of the incoming proton. For N4, the trends of PA and GB values are almost similar to N1, except for the PDPA-CHO molecule, which is 6.3 kcal/mol higher than that of PDPA-H. It is found that the PA of the PDPA-CHO derivative is higher than that of the parent molecule by 6.3 kcal/mol. For both N3 and N4 atoms, the presence of the phenyl group, which is directly bonded to the azo-dye group, has a crucial role in decreasing the basicity due to the delocalization effect.
The geometries of the neutral and protonated (at N1) forms of some investigated derivatives, including PDPA-H, PDPA-NH, PDPA-CHO, and PDPA-NO2, have been shown in Fig. 1, while the other derivatives have been shown in Fig. SM2 of the supplementary materials. Full geometries of the protonated forms, at N2 and N3 atoms, are available from authors upon request.
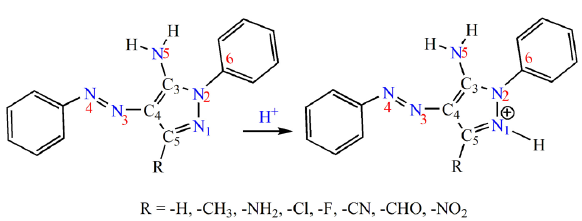
- Schematic representation of the studied PDPA molecule and its derivatives. PDPA: Poly (2,5-dialkyl-phenylene) amine.
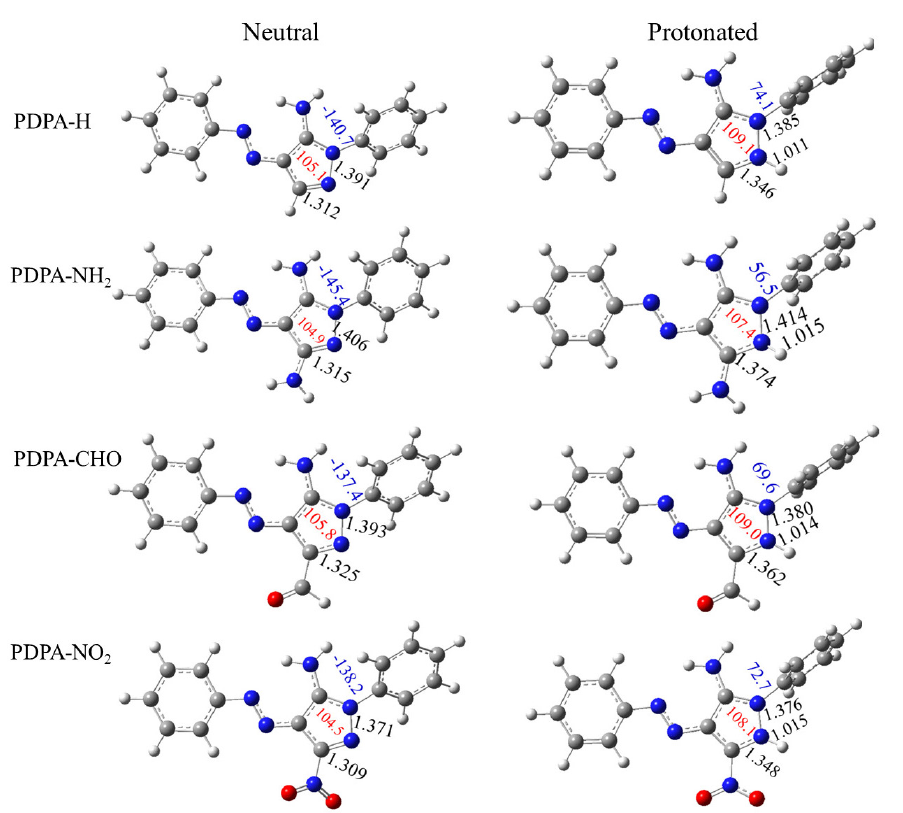
- Optimized structure of neutral and protonated forms at N1 atom of PDPA-H, PDPA-NH2, PDPA-CHO and PDPA-NO2 derivatives. Bond lengths, C5-N1, N1=N2 and N1-H+ in Å (black color), the ∠N2N1C5 bond angle in (°) (in red color) and the dihedral angle, φC5N1N2C6 in (°) are shown in blue color.
A general inspection of the results obtained shows that the bond lengths and bond angles of the investigated derivatives exhibit the expected variation upon protonation. It is found that the most effective changes are located in the bond lengths and the bond angles neighboring the protonated nitrogen atom, while slight variations are observed for other bonds. The C5=N1 and N1-N2 bond distances lengthen with averages of, correspondingly, 0.050 and 0.003 Å, while the ∠N2N1C3 angle increases with an average of 3.3°. In addition, the phenyl group attached to the pyrazole ring is twisted out of plane. It is also found that the average bond distances of , are 1.014, 1.027, and 1.023 Å, respectively, which agree with the PA results. This signifies that the interaction between the N1 atom and the incoming proton is the strongest one.
3.2 Analysis of single water-PDPA interactions
Frontier orbital theory (FOT) affirms that the most reactive molecular orbitals (MO) in the molecule are the highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO). The energy gaps of the individual water and PDPA derivatives (ΔE) and those of water-PDPA interactions (ΔE1 and ΔE2) can be calculated using the following expressions Eq. (2).
The corresponding EHOMO and ELUMO and HOMO-LUMO energy gaps (ΔE, ΔE1 and ΔE2) have been displayed in Table 2.
| Derivative | EHOMO | ELUMO | ΔE | ΔE1 | ΔE2 |
|---|---|---|---|---|---|
| H2O | -8.788 | -0.634 | 8.155 | - | - |
| PDPA-H | -5.836 | -2.172 | 3.664 | 6.616 | 5.202 |
| PDPA -CH3 | -5.766 | -2.127 | 3.639 | 6.661 | 5.132 |
| PDPA -NH2 | -5.727 | -2.114 | 3.613 | 6.674 | 5.093 |
| PDPA -Cl | -6.028 | -2.321 | 3.707 | 6.467 | 5.394 |
| PDPA -F | -6.053 | -2.321 | 3.732 | 6.467 | 5.419 |
| PDPA -CHO | -6.063 | -2.446 | 3.617 | 6.342 | 5.429 |
| PDPA -CN | -6.253 | -2.578 | 3.675 | 6.210 | 5.619 |
| PDPA -NO2 | -6.323 | -2.758 | 3.565 | 6.030 | 5.689 |
| Average | -6.006 | -2.355 | 3.652 | 6.434 | 5.372 |
A larger EHOMO value is known to accelerate the atom’s tendency to donate electrons, while a lower ELUMO increases the atom’s propensity to accept electrons. Examination of Table 2 indicates that the PDPA-NH2 derivative has the highest capability to donate electrons to water molecules, while the PDPA-NO2 derivative has the lowest. Contrarily, the PDPA-NO2 derivative has the maximum ability to accept electrons, while PDPA-NH2 has the minimum. These results clearly indicate that as the substitution of EDG increases, the electron density of the lone pair of electrons on N1 increases, consequently increasing its ability to donate electrons to the LUMO of the water molecule. In contrast, the substitution of EWG decreases the electron density on N1 therefore increasing it’s ability to accept electrons from water molecules. It is also found that the studied PDPA derivatives are characterized by small energy gaps, which lie in the range of 3.565-3.732 eV, signifying that they are chemically reactive and kinetically unstable.
Insight of the above findings, the question to be addressed is whether the electron’s flow is more favorable. The results in Table 2 clearly indicate that , signifying that the electrons flow can take place from the HOMO of PDPA derivatives to the LUMO of water. This means that the interaction between the H2O and PDPA molecules might have occurred by the interaction of the lone pair of electrons of the N1 atom of PDPA derivatives and the hydrogen water molecule. It is also found that as the power of EDG increases, the N1…H interaction power increases. Contrarily, EWG substitution decreases the strength of the N1…H interaction. Therefore, it can be predicted that the strength of N1…H interaction can be arranged as follows: PDPA -NH2 > PDPA -CH3 > PDPA -H > PDPA -Cl > PDPA -F > PDPA -CHO > PDPA -CN > PDPA -NO2.
The solvent effect was explicitly performed by the interaction of PDPA derivatives with single water molecules at the N1 atom through the formation of IMHB. Fig. 2 shows the geometrical structures of the PDPA-water complexes. The first inspection of Fig. 2 shows that the bond distance of N1…H in the PDPA-water complexes lies between 1.926 – 2.379 Å. Among the investigated complexes, the longest N1…H bond of 2.379 Å was found for the PDPA-NO2.H2O complex, which is attributed to the possible formation of another IMHB with 2.290 Å between the hydrogen water molecule and the oxygen NO2 group. On the other hand, the shortest IMHB of 1.953 Å, corresponds to the PDPA-NH2.H2O complex.
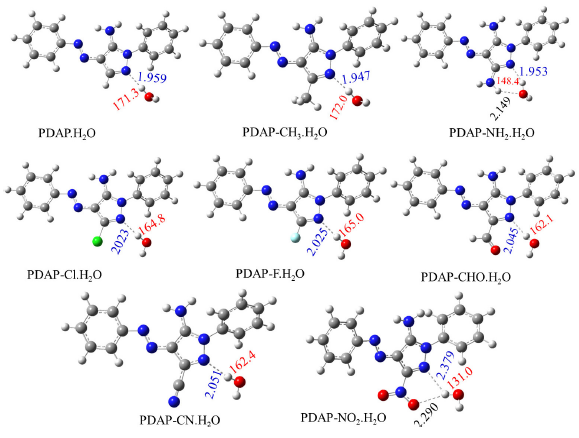
- Optimized structure of PDPA-H2O complexes and its derivatives as obtained using B3LY{/6-311++G(d,p) level of theory. The intermolecular hydrogen bond length and bond angle between PDPA and water N3…H as shown in blue and red colors, respectively. Units are in Å for N3…H bond length and in (°) for ∠N1HO bond angles.
In general, our results indicate that as the power of EWGs increases, the length of the IMHB decreases, which can be explained by the increase in electron density at the N1 atom and vice versa. It is also found that as the strength of EDG increases, the hydrogen bond angle, ∠N1HO, increases. Contrary, as the strength of EWG increases, the ∠N1HO decreases, see Fig. 2.
The intermolecular interactions between the water molecule and the PDPA derivatives can be represented by plotting the scatter plot of δ ginter/intra (a.u.) vs. sign(λ2)ρ (a.u.) and the corresponding RGD maps, see Fig. 3 and Fig. SM3 of the supplementary materials. As seen in Figs. 3 and SM3, the intermolecular region has been represented by red dots, while the intramolecular interactions have been represented by black dots. The density of the red region reflects the strength of the intermolecular interactions between water molecules and PDPA derivatives. The density of the red dot is directly proportional to the intermolecular interaction. It is obvious from Fig. 3 that minimum intermolecular interaction corresponds to the PDPA-NO2.H2O complex, which the maximum one belongs to, correspondingly, PDPA-NH2.H2O complex.
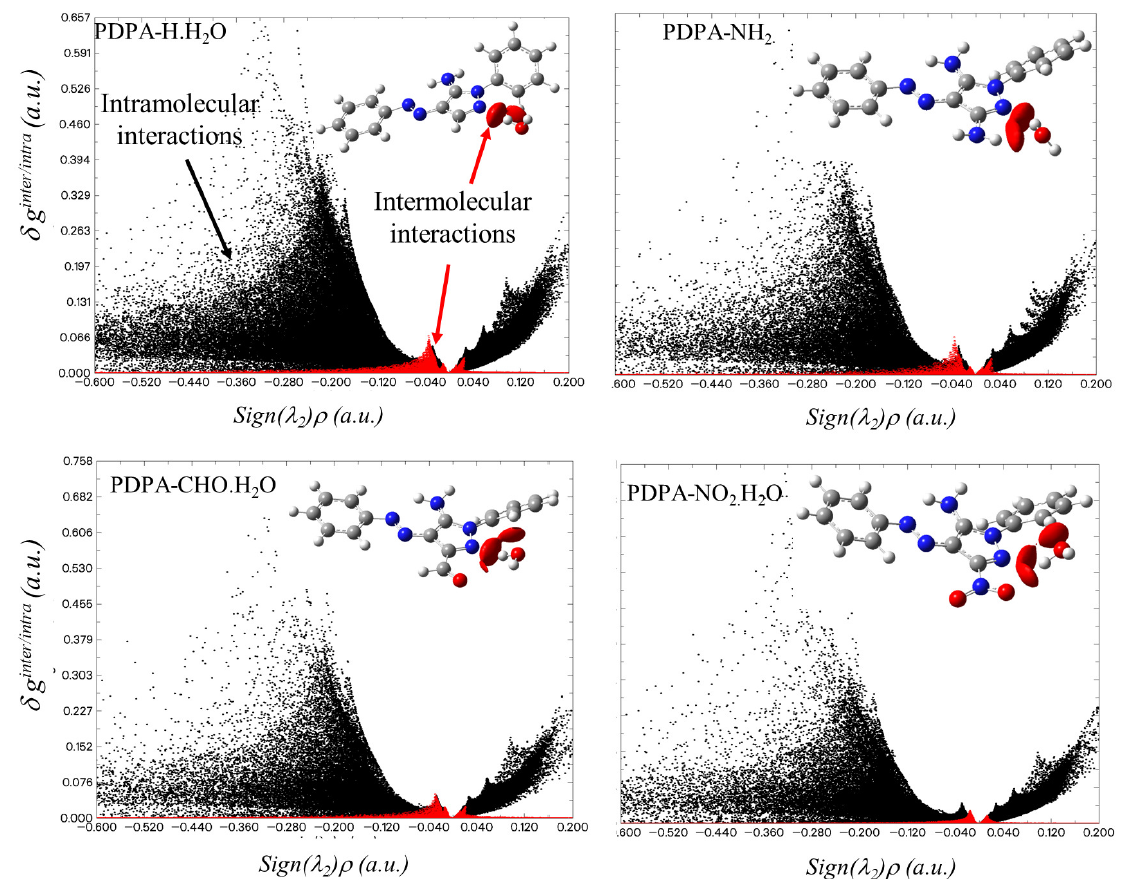
- Scatter plots of δginter/intra (a.u.) vs. Sign(λ2)ρ (a.u) of some PDPA-water complexes. The corresponding RGD maps are superimposed at the top right of each panel. The red dots correspond to the intermolecular interactions, while the black dots correspond to the intramolecular interactions. The corresponding plots of the other species have been shown in Figure SM3 of the supplementary materials
The above results have been confirmed by the estimation of the interaction energy () of the reaction using the following expression:
Here, , and are the single point energies with the corrected ZPE of PDPA-water complexes, PDPA derivatives and water molecule, respectively. The corresponding values have been listed in Table 3. The corresponding energies, ZPE and Thermal corrections have also been displayed in Table SM1.
| Species | Eint | ECT | b G(r) | H(r) | V(r) | G(r) | BO | EHB | ||
|---|---|---|---|---|---|---|---|---|---|---|
| PDPA-H.H2O | -4.8 | 9.0 | 0.0275 | 0.0867 | 0.0205 | -0.0012 | -0.0193 | 0.0205 | 0.94 | 6.1 |
| PDPA-CH3.H2O | -5.0 | 9.7 | 0.0285 | 0.0883 | 0.0212 | -0.0009 | -0.0202 | 0.0212 | 0.96 | 6.4 |
| PDPA-NH2.H2O | -6.6 | 9.5 (3.4) | 0.0295 | 0.0924 | 0.0225 | -0.0006 | -0.0219 | 0.0225 | 0.97 | 6.9 |
| PDPA -Cl.H2O | -4.5 | 6.4 | 0.0237 | 0.0775 | 0.0175 | -0.0019 | -0.0156 | 0.0175 | 0.89 | 4.9 |
| PDPA -F.H2O | -4.4 | 6.5 | 0.0235 | 0.0772 | 0.0173 | -0.002 | -0.0153 | 0.0173 | 0.89 | 4.8 |
| PDPA-CHO.H2O | -4.7 | 5.5 | 0.0223 | 0.0753 | 0.0167 | -0.0022 | -0.0145 | 0.0167 | 0.87 | 4.5 |
| PDPA-CN.H2O | -4.2 | 5.5 | 0.0223 | 0.074 | 0.0164 | -0.0021 | -0.0143 | 0.0164 | 0.87 | 4.5 |
| PDPA-NO2.H2O | -5.5 | 0.6 (1.6) | 0.011 | 0.0395 | 0.0082 | -0.0017 | -0.0065 | 0.0082 | 0.79 | 2.0 |
| Average values | -5.0 | 6.7 | 0.02354 | 0.0764 | 0.89750 | 0.01754 | -0.0016 | -0.0160 | 0.01754 | 5.0 |
Our results show that the average value is -5.0 kcal/mol, which belongs to moderate IMHB. The maximum value of -6.6 kcal/mol corresponds to PDPA-NH2.H2O, while the minimum one of -4.2 kcal/mol belongs to the PDPA-CN.H2O complex, illustrating the significant role of the nature of substituted groups in the strengthening or the weakening the IMHB. Furthermore, a second-order NBO perturbation analysis clearly depicts the interaction between the lone pairs of electrons on the N1 atom of PDPA derivatives, nonbonding orbital, and the antibonding orbitals of water molecule () → ), resultsing in a stabilization energy (ECT). This ECT enhances system stabilization by intermolecular charge transfer. According to Table 3, the ECT value ranges from 0.62 to 9.7 kcal/mol, ensuring a moderate strength IMHB. The existence of an additional electron transfer is also observed in the case of PDPA-NH2.H2O and PDPA-NO2.H2O complexes. The extra stabilization of the PDPA-NH2.H2O complex is due to the ((O) → ) interaction, which is associated with an ECT of 3.4 kcal/mol. On the other hand, the interaction between the nonbonding orbitals of oxygen nitro group with antibonding orbitals of the O-H of water molecule ((O) → ), which is associated with ECT of 1.6 kcal/mol, destabilizes the PDPA-NO2.H2O complexes.
Additional understanding of the nature of the IMHB in PDPA-water complexes can be obtained by analyzing the AIM results of Bader’s theory (Bader 1991). Analysis of the BCP’s properties was usually utilized to estimate the hydrogen bond strength (Nguyen and Chandra 1998; Lamsabhi 2008; Safi 2016). The most relevant results topological properties, including the electron charge, ρBCP(r), computed at the BCP (3,−1) for the IMHB and its related parameters, including Laplacian (∇ 2ρ(r)), Lagrangian and Hamiltonian kinetic energies (G(r) and H(r), and the potential electron density (V(r), have been displayed in Table 3. The corresponding molecular graphs have been shown in Fig. 3. It was discovered that ρBCP(r) and values fall within the hydrogen bond range with averages of 0.0253 and 0.0816 a.u., respectively, indicating that the investigated IMHB belongs to medium interaction (Nguyen and Chandra 1998; Lamsabhi 2008). The largest ρBCP(r) (0.0924 a.u.) was reported for the PDPA-NH2.H2O complex, while the smallest value (0.0395 a.u.) was for the PDPA-NO2.H2O complex. Our results in Table 3 strongly affirm that substitution of the EDGs increases the ρBCP(r) and its related topological parameters, while substitution of EWGs decreases the ρBCP(r) and its related topological parameters. Furthermore, the IMHB energy and its bond order (BO) were evaluated as EHB = -V(r)/2) (Espinosa et al., 1998), and BO= |V(r)|/G(r) and their results have been displayed in Table 3. Our results show that the average value of the BO is 0.91, with maximum and minimum BO values corresponding to PDPA-NH2.H2O and PDPA-NO2.H2O complexes, respectively. It is also found that EHB values are ranged from 1.6 to 6.9 kcal/mol, which confirm the results of Eint and ECT, in agreement with similar systems (Nguyen and Chandra 1998; Lamsabhi 2008; Safi 2016).
The NCI’s plot is useful in illustrating the correlation between strong directional attractions with the localized atom-atom contacts and the molecular division having weak interactions. The color-filled RDG isosurfaces and the scatter plots of the Hessian of the electron density, Sign(λ2)ρ, for some PDPA-water complexes have been shown in Fig. 4, while the other plots have been shown in Fig. SM4 of the supplementary materials. Examination of RDG isosurfaces indicate three different regions, which correspond to (i) weak interaction forces (green color) that occur between the atoms in the molecule, (ii) strong field interactions (red arrow) belong to steric effect of the aromatic rings and (iii) hydrogen bond interactions (blue color). A close examination of the 3D-Isosurface of color scaling of weak interactions diagram (NIC, Fig. 4 (right)) indicates that the interactions between LP(1)N1 (proton acceptor) and the proton donor (hydrogen water molecule) are stronger in case of NH2 and CH3 derivatives compared to the others. Except for the PDPA-NO2.H2O complex, the presence of another IMHB field is indicated by the presence of blue circle (assigned by a blue arrow). A weak IMHB field was also observed between the oxygen nitro group (-NO2 derivative) and the hydrogen water in case of PDPA-NO2. Also, another IMHB region was observed between the oxygen water molecule and the amino hydrogen atom in the case of PPDPA-NH2.H2O complex. These results agreed with NBO results as discussed above.
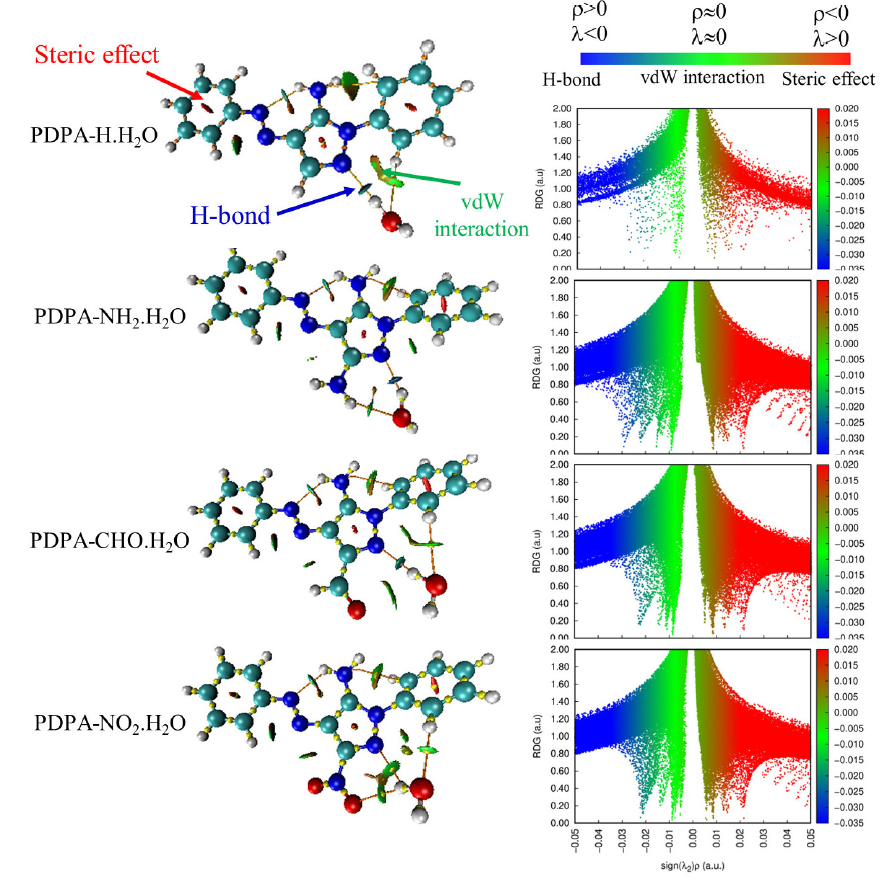
- RDG maps (left) and Scatter plots of RDG (a.u.) versus Sign(λ2)ρ (a.u) (right) of some PDPA-water complexes, including PDPA-H.H2O, PDPA-NH2.H2O, PDPA-CHO.H2O, and PDPA-NO2.H2O. Note: Molecular graphs based on QTAIM are superimposed (left images, orange points, and yellow lines). QTAIM: quantum theory of atoms and molecules.
Conclusions
Several derivatives of polyfunctional azo dye pyrazole-based (PDPA) were computationally studied for explicit solvent effects. The protonation sites were identified using two strategies based on DFT and pKa calculations. Results reveal that pKa, as a rapid calculation, is useful for identifying the protonation sites with some limitations, especially for -F and -NO2 groups. On the other hand, DFT/ B3LYP(6-311++G(3df,2p) predicts the appropriate site for protonation based on PA values. The PA and GB values suggest that PDPA and its derivatives are potent and basic, and the nature of the substituted group has a great impact on the intrinsic basicity. The stronger the EDGs, the higher the PA and GB values, while the stronger the EWGs, the lower the PA and GB values. The PDHP-water interaction shows a moderate-strength intermolecular hydrogen bond between the nitrogen atom and the water molecule. Interaction energies, NBO analysis, and topological properties within the framework of the QTAIM concluded that the PDPA-NH2.H2O complex has the strongest IMHB, while the PDPA-CN.H2O and PDPA-NO2.H2O complexes have the weakest one. Based on the results obtained in this article, the research can be extended to study the non-covalent interactions between the between the PDPA and its derivatives with different solvents, including protic and aprotic solvents.
Acknowledgments
Authors gratefully acknowledge King Abdulaziz University’s High-Performance Computing Centre (Aziz Supercomputer) (http://hpc.kau.edu.sa) for assisting with the calculations for the work of this paper.
CRediT authorship contribution statement
Nuha Wazzan: Methodology, Investigation, Resources, Writing - Review & Editing, Project administration; Zaki Safi: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data Curation, Writing - Review & Editing, Visualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of Generative AI and AI-assisted technologies in the writing process
The authors confirm that there was no use of Artificial Intelligence (AI)-Assisted Technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Supplementary material
Supplementary data to this article can be found online at https://dx.doi.org/10.25259/JKSUS_259_2024.
References
- A quantum theory of molecular structure and its applications. Chemical Reviews. 1991;91(5):893-928.
- [Google Scholar]
- Computational molecular perspectives on the interaction of propranolol with β-cyclodextrin in solution: Towards the drug-receptor mechanism of interaction. J. Mol. Liq.. 2017;227:280-290. https://doi.org/10.1016/j.molliq.2016.12.023
- [Google Scholar]
- Functionalization of graphene quantum dots (GQDs) with chitosan biopolymer for biophysical applications. Opt Quant Electron 2020:52. https://doi.org/10.1007/s11082-019-2134-z
- [Google Scholar]
- Density‐functional thermochemistry. IV. A new dynamical correlation functional and implications for exact‐exchange mixing. J. Chem. Phys.. 1996;104:1040-1046.
- [Google Scholar]
- Proton-transfer reaction mass spectrometry. Chem. Rev.. 2009;109:861-896. https://doi.org/10.1021/cr800364q
- [Google Scholar]
- Modeling solvent broadening on the vibronic spectra of a series of coumarin dyes From implicit to explicit solvent models. J. Chem. Theory Comput.. 2015;11:5810-5825. https://doi.org/10.1021/acs.jctc.5b00870
- [Google Scholar]
- Marvin: A tool kit for streamlined access and visualization of the SDSS-IV maNGA data set. AJ. 2019;158:74. https://doi.org/10.3847/1538-3881/ab2634
- [Google Scholar]
- Molecular properties of aqueous solutions: A focus on the collective dynamics of hydration water. Soft Matter. 2016;12:5501-5514. https://doi.org/10.1039/c5sm03119b
- [Google Scholar]
- Characterisation of textile wastewaters‐a review. Environ. Technol.. 1994;15:917-929.
- [Google Scholar]
- Tautomerism in azo dyes: Border cases of azo and hydrazo tautomers as possible NMR reference compounds. Dyes Pigm.. 2019;165:157-163. https://doi.org/10.1016/j.dyepig.2019.02.015
- [Google Scholar]
- GaussView, Version 6.1 GaussView, Version 61. Semichem Inc, Shawnee Mission; 2016.
- Molecular structure and energetics. In: Liebman JF, Greenberg A., eds. Physical Measurements. Deereld Beach, FL: VCH; 1987. p. :269.
- [Google Scholar]
- Investigating the electronic properties of edge glycine/biopolymer/graphene quantum dots. Sci. Rep.. 2024;14:21973. https://doi.org/10.1038/s41598-024-71655-1
- [Google Scholar]
- Hydrogen bonds: first quantitative agreement between electrostatic potential calculations from experimental X−(X+ N) and theoretical ab initio SCF models. J. Am. Chem. Soc.. 1996;118:2501-2502.
- [Google Scholar]
- Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett.. 1998;285:170-173. https://doi.org/10.1016/s0009-2614(98)00036-0
- [Google Scholar]
- 1, mj frisch, gw trucks, hb schlegel, ge scuseria, ma robb, jr cheeseman, g. Scalmani, v. Barone, b. Mennucci, ga petersson et al., gaussian. Inc., Wallingford CT. 2009;121:150-166.
- [Google Scholar]
- 2-amino-4-chloro-5-formylthiophene-3-carbonitrile derived azo dyes. Dyes Pigm.. 2015;117:133-140. https://doi.org/10.1016/j.dyepig.2015.02.010
- [Google Scholar]
- An estimation of strength of intramolecular hydrogen bonds—ab initio and AIM studies. J. Mol. Struct.. 2001;562:137-143.
- [Google Scholar]
- VMD: Visual molecular dynamics. J. Mol. Graph.. 1996;14:33-38. https://doi.org/10.1016/0263-7855(96)00018-5
- [Google Scholar]
- Modern inorganic chemistry.
- gnuplot 5.5 An Interactive Plotting Program. [accessed 2024 Oct 01]. Available from: http://sourceforge.net/projects/gnuplot.
- Specific hydration effects on oxo-thio triazepine derivatives. J. Phys. Chem. A. 2008;112:1791-1797. https://doi.org/10.1021/jp709960g
- [Google Scholar]
- Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem.. 2012;33:580-592. https://doi.org/10.1002/jcc.22885
- [Google Scholar]
- Effect of dyes on water chemistry, soil quality, and biological properties of water. Impact of textile dyes on public health and the environment. IGI Global Scientific Publishing; 2020. p. :90-114.
- Protonation and deprotonation energies of uracil implications for the uracil–water complex. Faraday Trans.. 1998;94:1277-1280. https://doi.org/10.1039/a708804c
- [Google Scholar]
- DFT study of the structure, reactivity, natural bond orbital and hyperpolarizability of thiazole azo dyes. Int. J. Mol. Sci.. 2017;18:239. https://doi.org/10.3390/ijms18020239
- [Google Scholar]
- Theoretical investigation of the proton affinity and gas-phase basicity of neutral x,y-dihydroxybenzoic acid and its derivatives. Eur J Mass Spectrom (Chichester). 2006;12:385-396. https://doi.org/10.1255/ejms.823
- [Google Scholar]
- Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev.. 1988;88:899-926. https://doi.org/10.1021/cr00088a005
- [Google Scholar]
- A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) J. Chemical. Physics. review. B. 2010;132:154104.
- [Google Scholar]
- Gas-phase reactivity of 2,7-dimethyl-[1,2,4]-triazepine thio derivatives toward cu+Cation: A DFT study. J. Phys. Chem. A. 2007;111:2213-2219. https://doi.org/10.1021/jp068642h
- [Google Scholar]
- A theoretical study on the structure of thiazolidine-2,4-dione and its 5-substituted derivatives in the gas phase Implications for the thiazolidine-2,4-dione -water complex. Arab. J. Chem.. 2016;9:616-625. https://doi.org/10.1016/j.arabjc.2015.03.016
- [Google Scholar]
- Protonation of 5‐methylhydantoin and its thio derivatives in the gas phase: A theoretical study. Int J of Quantum Chemistry. 2013;113:908-915. https://doi.org/10.1002/qua.24017
- [Google Scholar]
- Proton affinity and molecular basicity of m-and p-substituted benzamides in gas phase and in solution: a theoretical study. Chem. Phys. Lett.. 2014;610:321-330.
- [Google Scholar]
- Benchmark calculations of proton affinity and gas-phase basicity using multilevel (G4 and G3B3), B3LYP and MP2 computational methods of para-substituted benzaldehyde compounds. J. Comput. Chem.. 2021;42:1106-1117. https://doi.org/10.1002/jcc.26538
- [Google Scholar]
- Proton affinity measurements using ion mobility spectrometry. J. Chem. Thermodyn.. 2003;35:863-870. https://doi.org/10.1016/s0021-9614(02)00316-6
- [Google Scholar]
- DFT, CBS‐Q, W1BD and G4MP2 calculation of the proton and electron affinities, gas phase basicities and ionization energies of saturated and unsaturated carboxylic acids (C1–C4) Int. J. Quantum Chem.. 2013;113:1717-1721.
- [Google Scholar]
- Benchmark calculations of proton affinity and gas-phase basicity using multilevel (G4 and G3B3), B3LYP and MP2 computational methods of para-substituted benzaldehyde compounds. J. Comput. Chem.. 2021;42:1106-1117. https://doi.org/10.1002/jcc.26538
- [Google Scholar]
- Proton transfer reaction rate constants between hydronium ion (H3O+) and volatile organic compounds. Atmos. Environ.. 2004;38:2177-2185. https://doi.org/10.1016/j.atmosenv.2004.01.019
- [Google Scholar]
- Color chemistry: syntheses, properties, and applications of organic dyes and pigments. John Wiley & Sons; 2003.




