

Translate this page into:
Anti-inflammatory and antioxidant activity, toxicity prediction, computational investigation, and molecular docking studies of 2-thiophenecarbonitrile
⁎Corresponding authors at: Department of Physics, Arignar Anna Government Arts College (Affiliated to Bharathidasan University), Musri, Tiruchirappalli 621 211, India (V. Balachandran). Department of Biomedical Science, Faculty of Science, University Tunku Abdul Rahman, Jalan University, Bandar Barat, Kampar 31900, Malaysia (S. Djearamane). brsbala@rediffmail.com (Vadivelu Balachandran), sinouvassane@utar.edu.my (Sinouvassane Djearamane)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Density Functional Theory (DFT) and Molecular docking are pivotal computational techniques in modern chemistry and drug design. This work investigates the electronic structure and reactivity of 2-thiophenecarbonitrile (2TCN) with an emphasis on important factors such as HOMO-LUMO energy gap, MEP, Mulliken atomic charges, natural population analysis, and Mutiwfn (ELF, LOL, ALIE, and RDG) analysis. The MEP and FMO studies were calculated in various solvents like acetonitrile, water, gas, and methanol. The anti-inflammatory and antioxidant investigations revealed substantial activities by 2TCN. Additionally, molecular docking studies are performed to elucidate the binding interaction between the compound and target proteins, providing insights into its potential therapeutic mechanisms. The results demonstrate the binding energies, interaction residues, and the most favorable docking poses. This approach underscores the integration of theoretical and computational methods in advancing molecule design and therapeutic discovery.
Keywords
Solvation effect
DFT
Docking
Antioxidant
2-thiophenecarbonitrile

1 Introduction
Among the organic molecules that are classified as heterocyclic compounds, 2-thiophenecarbonitrile is one of the compounds that can be found. Specifically, it possesses a thiophene ring, which is a five-membered ring consisting of four carbon atoms and one sulfur atom. Additionally, it possesses a cyano group (−C≡N) that is connected to the second carbon position on the ring. The aromatic nature of the thiophene ring makes it a useful building block in a variety of organic synthesis reactions. This property also contributes to the fact that it is stable. The cyano group results in the introduction of reactivity and polarity. Depending on the purity of the substance and the conditions under which it is found, it can manifest as a colorless to light yellow liquid or solid. Several different medicinal molecules are synthesized with the help of 2-thiophenecarbonitrile, which serves as an intermediate (Molvi et al., 2008). Due to its reactivity, it is an extremely valuable starting material for the development of pharmaceuticals that possess antifungal, antibacterial and other bioactive qualities. It is utilized in the manufacturing of conductive polymers and other materials, particularly those that call for heterocyclic frameworks, such as polymers based on thiophene (Alomar et al., 2013). In addition to its role as a precursor in the synthesis of various thiophene derivatives, 2-thiophenecarbonitrile is also engaged in the construction of heterocyclic compounds that consist of more complex structures. As a result of its ability to participate in nucleophilic addition processes, the cyano group (−C≡N) is frequently utilized in the production of a wide range of amines and amidines. The thiophene ring contains the ability to undergo electrophilic substitution reactions, which enables it to be utilized in the synthesis of a wide variety of substituted thiophene derivatives (Baildya et al., 2021). On account of its versatility in organic and medicinal chemistry, 2-thiophenecarbonitrile has garnered a lot of attention in the field of scientific research. Through modification, it is possible to create derivatives that possess potentially beneficial biological activities, or it can be utilized in the development of material science advances such as semiconductors and sensors. Thiophene derivatives have gained interest in medicinal chemistry for their diverse biological activities, including antidiabetic, antimicrobial, and anti-inflammatory properties (Ruan and Lodish, 2003) (Ruan and Lodish, 2003) (Chaudhury et al., 2017). This study presents a comprehensive vibrational analysis of 2-thiophenecarbonitrile (2TCN) by integrating actual IR and Raman spectrum data with theoretical information utilizing a scaled quantum chemistry approach based on DFT. The calculation of stabilization energies E(2) is performed using NBO, which provides definitive proof of stabilization resulting from hyperconjugation in different intramolecular interactions. The analysis of the HOMO and LUMO has been employed to clarify details about the transmission of electric charge within the molecule, in various solvents. The topology analyses were derived using the Multiwfn software, which functions as a wave function analyzer. In addition, biological experiments were conducted, including investigations on anti-inflammatory, and antioxidant properties, as well as molecular docking.
2 Experimental
2.1 Materials and methods
The 2TCN chemical was obtained from Sigma-Aldrich with purity of 99 percent. The Infrared spectrum was obtained by utilizing the FTIR spectrophotometer with KBr pellets, whereas the FT-Raman spectral analysis was conducted using the Brucker Spectrometer. The PerkinElmer Lamda-35 spectrophotometer was utilized to capture the emission spectra, utilizing ethanol as the solvent.
2.2 Computational analysis
The computational analysis of 2TCN was carried out using Gaassview and Gaussian 09 W software(Frisch et al., 2009). Using B3LYP functional, the 2TCN molecule geometry was calculated. Fig S1 shows the optimization structure of the 2TCN molecule. The interactions between orbitals within and between molecules were examined using NBO computations. Table S1 presents the optimized geometrical parameters for both methods. The HOMO-LUMO and MEP analyses were performed in the gas phase(Domínguez-Flores and Melander, 2022). Using Atomistic Online, a web tool for creating input files for common RDG diagrams, and Gauss-sum software, the DOS plot was constructed. The investigations of ELF, LOL, and ALIE were carried out by Multiwfn (Lu and Chen, 2012). The output data obtained from Multiwfn were utilized along with the VMD 1.9.1 software for generating the isosurface maps. The software Autodock-4.2.6 and Discovery Studio were used to do the molecular docking study(Lalpara et al., 2022).
2.3 Antioxidant (DPPH) activity
It had 0.2 mL of DPPH radical solution 0.5 mM in ethanol, 0.1 mL of sample at different concentrations, and 2 mL of pure ethanol in it. When DPPH mixes with an antioxidant that can gives off hydrogen (Ezati et al., 2022; Elangovan et al., 2023). After 30 min of reaction, the color changed from deep violet to bright yellow and the change in the absorbance was measured using UV–VIS spectrophotometer at 517 nm. The blank was the mixture of ethanol and sample. Combining the DPPH radical solution with ethanol produced the control solution.
2.4 Anti-inflammatory activity
A 0.2 mL of a 2 % egg albumin solution, 0.1 mL of sample extract at different strengths, and 2.7 mL of phosphate-buffered saline (pH 7.4) were mixed to make a 3 mL reaction mixture(Arulmurugan et al., 2024; Elangovan et al., 2023). The control was made by mixing 3 mL of triple-distilled water, 0.1 mL of 2 % egg albumin solution, and 2.7 mL of phosphate-buffered saline. Next, the mixtures were kept at 37 ± 2°C for 30 min. After that, the mixtures were heated in a water bath at 70 ± 2°C for 15 min. UV/Vis spectrophotometer was used to measure the absorbance at 280 nm.
3 Results and Discussion
3.1 Vibrational assignments
The investigation of the IR and Raman vibrational spectra can yield valuable insights into the vibrational modes of the 2TCN molecule. Experimental spectral analyses using IR and Raman techniques have been conducted(Elangovan et al., 2023). The results are presented in Fig. 1&2. The B3LYP/6–311 ++G(d,p) and B3LYP/6–311 + G(d,p) basis sets were used in corresponding theoretical calculations. The assignments for each spectrum are found in Table 1. Typically, the C–H stretching vibration modes are not affected by substituent groups(Arulmurugan et al., 2024). However, there are specific vibrations that are sensitive to substituents and can offer important structural information because of their mechanical interaction with the substituent groups. Typically, the spectral range of 3100–3000 cm−1 is where the asymmetric stretching vibrations C–H are commonly found. The current work on 2-thiophenecarbonitrile detected the stretching C–H vibration in Raman at 3110 cm−1, and in IR at 3108 cm−1 and 2908 cm−1. Stretching vibrations at 3116 cm−1, 3108 cm−1, and 2914 cm−1 were anticipated by theoretical calculations employing B3LYP/6–311 ++ G(d,p). B3LYP/6–311 + G(d,p) computations also projected vibrations at 3112 cm−1, 3103 cm-1, and 2910 cm-1.Observations of C–H bending vibrational modes were found within the 1530–1000 cm−1 frequency range(Elangovan et al., 2023). Whereas the Raman spectrum features peaks at 1513 and 1079 cm−1, the IR spectrum comprises peaks at 1672, 1576, 1131, and 1079 cm−1 were noticed. While with the B3LYP/6–311 ++ G(d,p) approach the values are 1673, 1575, 1514, 1475, and 1078 cm−1, the predicted values for the B3LYP/6–311 + G(d,p) method are 1677, 1578, 1519, 1480, and 1083 cm−1. Within the frequency range 900–625 cm−1, the C–H bending vibration in the out-of-plane direction exists. Note: ν-stretching, δ-in-plane bending, γ-out -of -plane bending, δring -ring vibration.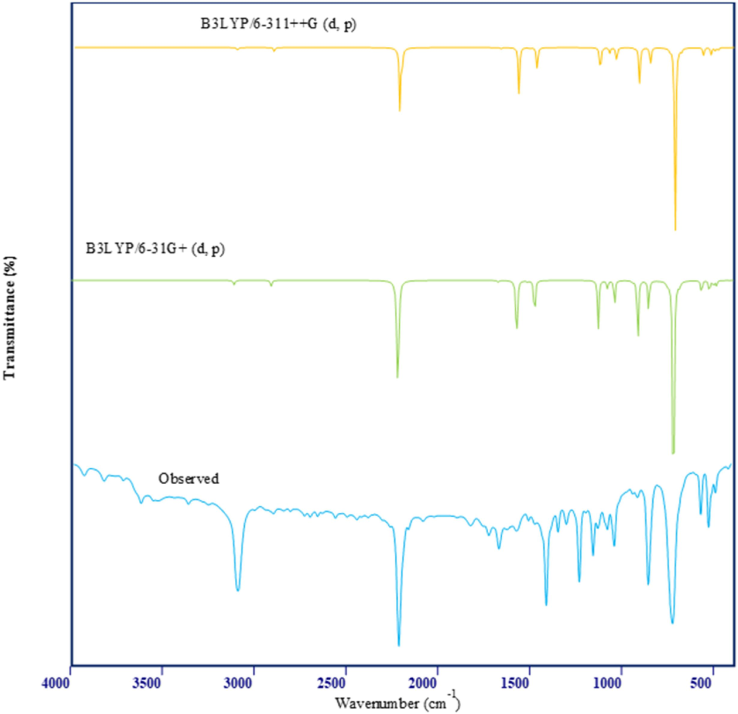
Observed and simulated FT-IR spectrum of 2TCN.
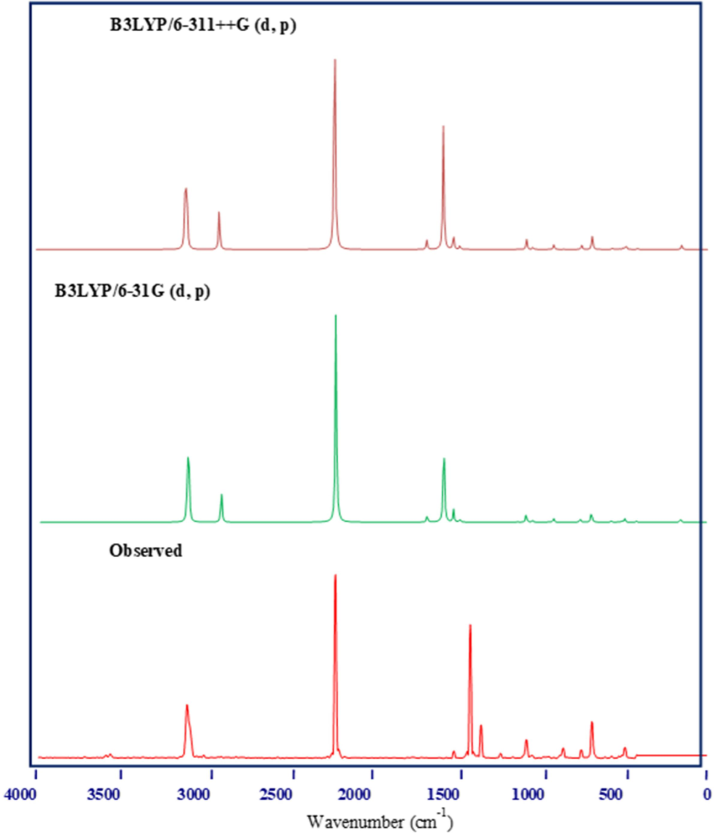
Observed and simulated Raman spectrum of 2TCN.
No
FT-IR
FT-RAMAN
B3LYP/ 6–311 + G(p,d)
B3LYP/ 6–311++G(p,d)
Vibrational assignments
1
3116
3112
νCH(99)
2
3102
3108
3103
νCH(98)
3
2908
2914
2910
νCH(99)
4
2221
2222
2224
2221
νCN(88)
5
1672
1677
1673
νCC(71)
6
1576
1578
1575
νCC(70),
7
1513
1519
1514
νCH(70),
8
1475
1480
1475
νCH(68), νCC(16), νCN(10)
9
1131
1133
1130
νCC(68), CH(15)
10
1079
1079
1083
1078
νCH(70), νCC(18), νCN(10)
11
1041
1044
1040
νCN(68)
12
940
947
941
νCS(72), CH(16)
13
914
918
915
νCS(70), CH(17)
14
856
859
859
855
νCC(67), CH(12)
15
751
758
750
γCH(62), γring (21)
16
722
724
721
νCC(58),νCH(17)
17
686
690
687
γCH(60), γring (19)
18
567
571
565
γCH(60), γ CN(15)
19
524
528
524
γCN(55)
20
506
502
γCC(49)
21
487
488
490
485
γring (52)
22
416
419
415
γring (50)
23
156
153
γring (49)
24
146
142
γring (49)
The recorded IR spectrum at 567 and Raman spectrum at 751 and 686 have equivalent theoretical values of 758, 690, and 571 cm−1 for B3LYP/6–311 ++G(d,p), and 750, 687, and 565 cm−1 for B3LYP/6–311 + G(d,p). Nitrogen compounds containing double and triple bonds, such as nitrile and cyanate, exhibit a distinct spectrum characterized by a single, typically prominent peak at 2280–2200 cm−1. B3LYP/6–311 ++G(d,p) and B3LYP/6–311 + G(d,p) have comparable theoretical values of 2224 and 2221 cm−1, respectively, for the stretching vibration of C-N in IR and 2222 cm−1 in FT-Raman (Elangovan et al., 2023). The bending vibration of B3LYP/6–311 ++G(d,p) and B3LYP/6–311 + G(d,p) was observed at 1041 cm−1 in the IR spectrum, while B3LYP/6–311 + G(d,p) has a frequency of 1044 cm−1 and 1040 cm−1, respectively. The C–C stretching vibrations commonly occur within the range of 1635 to 1080 cm−1. The stretching vibration of the C–C modes was detected at frequencies of 1672, 1576, and 1131 cm−1 in IR. The theoretical values for these vibrations are 1677, 1578, and 1133 cm−1 for B3LYP/6–311 ++G(d,p), and 1673, 1575, and 1130 cm−1 for B3LYP/6–311 + G(d,p).
3.2 Electronic spectra
Electronic spectra, often referred to as UV–Vis (ultraviolet–visible) spectra, are used to study the absorption of ultraviolet (UV) and visible light by molecules. The absorption of light in these regions of the electromagnetic spectrum causes electronic transitions in molecules, typically between different energy levels of electrons. The σ to σ* transitions occur when light excites an electron from a bonding sigma orbital (σ) to an antibonding sigma orbital (σ*). These transitions are very high in energy and typically occur in the far UV region (below 200 nm). The n to σ* transition involves the excitation of a non-bonding electron (n) to an antibonding sigma orbital (σ*). These transitions occur in the UV region (around 150–250 nm). The π to π* transitions occur when electrons in a pi bonding orbital (π) are excited to an antibonding pi orbital (π*). These transitions usually occur in the UV to visible range (180–400 nm), especially in compounds with conjugated systems like alkenes and aromatic rings. The n to π* transition involves the excitation of a non-bonding electron to an antibonding pi orbital (π*). These transitions typically occur in the near-UV region (200–400 nm). The absorption spectroscopic properties of the title molecule were forecasted using the TD-DFT technique, specifically with the B3LYP/CPCM solvation method. Ethanol was used as the solvent in both the experimental and theoretical analysis(Elangovan et al., 2023). Table S2 displays the UV–visible data analysis(Elangovan et al., 2024). From Fig. 3; the molecule exhibits absorption peaks at 279.20 nm in the experimental section and at 236.27 nm in the theoretical technique with 80 % of the contribution coming from the HOMO and LUMO.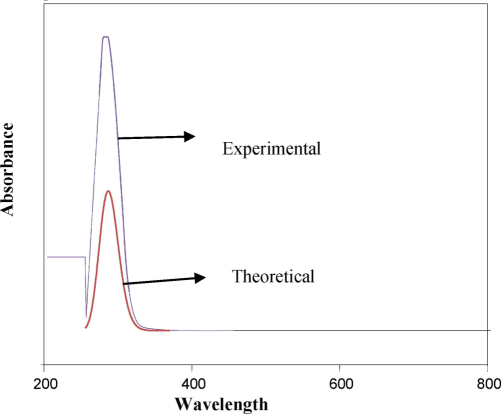
Absorption spectra of 2TCN.
3.3 Donor acceptor interaction
The HOMO and LUMO play a pivotal role in assessing the chemical stability of a molecule. To comprehend the behavior of molecules, one must possess knowledge regarding electronic transitions and the corresponding disparities in energy, which are denoted by the energy gap(Priya et al., 2023). Moreover, many chemical reactivity descriptors are crucial in comprehending this matter. Fig. 4 graphically depicts the energies of the HOMO and LUMO, along with the band gap, which is the difference between them. The color green signifies the unfavorable stage, whilst the color red signifies the favorable stage. The HOMO is connected to the nucleophilic part of the molecule, while the LUMO tells us about the electrophilicity of the molecule(Gobinath et al., 2024). The energy difference of 2TCN is determined to be 5.21 (eV) in solvents methanol, water, and acetonitrile and the gas phase is 5.23 (eV) respectively. A high-energy HOMO indicates strong nucleophilic behavior, making thiophene derivatives more reactive towards nucleophilic. The global reactivity description for the specified chemical has been calculated and condensed in Table S3, for both gas phase solvent phases. The diagram illustrates that both the HOMO and the LUMO are localized inside the entire molecule. The smaller energy gap means that the molecule can carry electrons more effectively, which makes it more biologically active.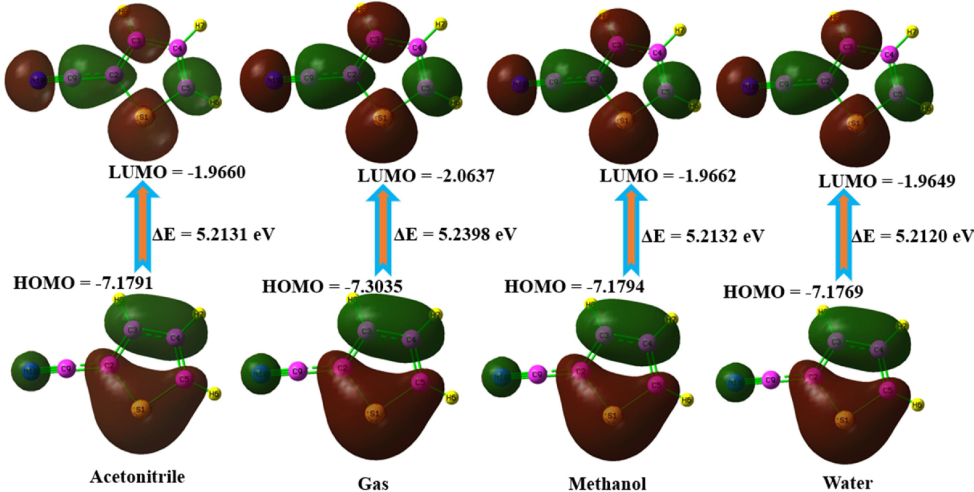
HOMO-LUMO surface map of 2TCN in various solvents and gas phase.
3.4 Study of DOS
The Density of States (DOS) is a concept used in physics and chemistry to describe the number of states that are available for occupation by electrons (or other particles like phonons) at each energy level in a material. It's particularly important in solid-state physics, quantum mechanics, and materials science for understanding electronic structure and how materials interact with light, electricity, and other external factors. Fig S2 shows the DOS spectrum, which shows how many energy levels there are per unit of energy increase and what kinds of levels and Fig S2 displays the HOMO and LUMO energy levels. The HOMO by the green line and the LUMO by the red line(Elangovan et al., 2021). When looking at the plot's energy axis, the range from −20 eV to −5 eV is called “filled orbitals,” and the range from 0 eV to 20 eV is called “virtual orbitals.” The empty orbitals are called recipient orbitals, and the full orbitals are called donor orbitals.
3.5 MEP map analysis
The MEP (molecular electrostatic potential) is utilized to forecast the positions of nucleophilic and electrophilic reactive sites within a molecule, offering a valuable understanding of their comparative reactivity. An MEP analysis was performed on the optimized structure of the title compounds(Diya et al., 2023).This method is valuable for data verification, demonstrating that these chemicals interact as inhibitors. The MEP method utilizes color categorization to precisely determine a molecule's dimensions, configuration, and positive, negative, and neutral regions. While the red color signifies areas with low electrostatic potential, indicating a high concentration of electrons, the blue hue represents places with very high electrostatic potential. This designation means the absence of electrons in this area, making it a favorable location for nucleophilic assault. The MEP surface is seen in Fig. 5 for the gas phase and solvent phases. The depicted surface range spans from −9.003 to + 9.003 atomic units in acetonitrile and water(Elangovan et al., 2023). The gas phase water ranges from −8.929 to + 8.829 and −9.005 to + 9.005 a.u respectively. The red region corresponds to the site of the electrophilic attack, located around N10. On the other hand, the blue hue reflects the region of nucleophilic assault, which is situated around the sulfur atom for both the gas phase and solvent phases.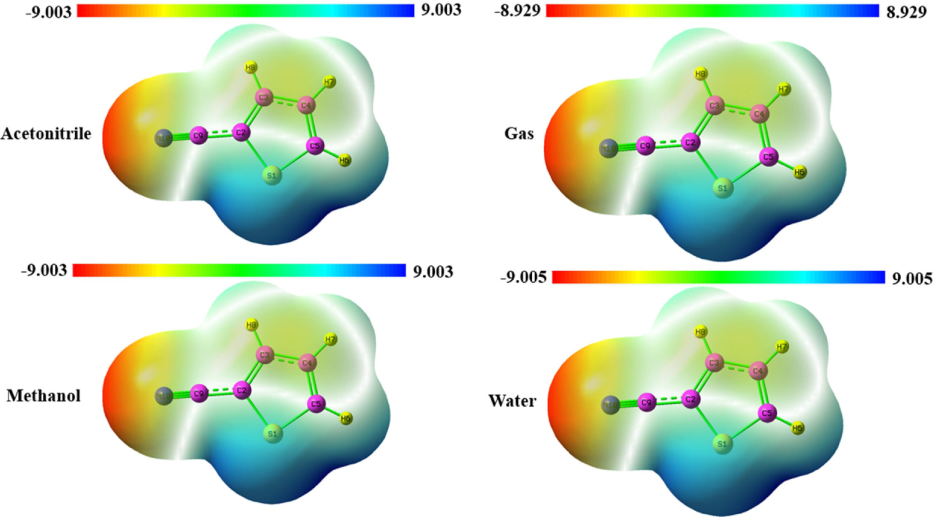
MEP surface map of 2TCN in various solvents and gas phase.
3.6 Mulliken atomic analysis
The distribution of Mulliken's atomic charges is crucial in quantum chemical calculations. It represents the electrostatic potential that ultimately persists beyond the molecule's surface(Geethapriya et al., 2023). Charge transfer between donor and acceptor pairs will occur within the molecule. Table S4 shows the MPA study results. The hydrogen atom as a whole has a net positive charge ranging from 0.142 to 0.263. The carbon atoms C2, C3, and C5 carry a positive charge, whereas the atoms C4, C9, and S1 carry negative charges. Atoms with a high positive Mullikan charge might be more electrophilic while atoms with high negative charge might be more nucleophilic.
3.7 NPA analysis
The natural population analysis provides the electron density distribution and charge transfer between atoms or within molecules. The NPA was employed to illustrate the distribution of electrons within the subshells of atomic orbitals. Table S5 presents the inherent energies of each atom, together with the number of electrons in the nucleus, valence, and Rydberg subshells. The carbon atom displays both electronegativity and electronegativity. The carbon atom at position C5 (−0.415) exhibits higher electronegativity, while the carbon atom at position C3 (0.295) exhibits higher electro-positivity(Muthukumar et al., 2022). All hydrogen atoms have electropositive charges. The elements with atomic numbers 6, 7, and 8 exhibit electropositive charges of 0.227, 0.233, and 0.241, respectively. S1(0.547) displayed the highest electropositive charge. Based on the electrostatic perspective, most electropositive atoms can take an electron, while most electronegative atoms can readily donate their electrons. The whole natural population analysis of the 2TCN molecule has been computed as
Core: 21.99346 (99.9703 % of 22).
Valence:33.81282 (99.4495 % of 34).
Rydberg: 0.19372 (0.3459 % of 56).
3.8 NCI analysis
The Reduced Gradient Density (RDG) is employed to analyze non-covalent interactions, which can be attracting (noncovalent), repulsive (steric), or neutral (van der Waals). The Fig. 6 displays a 2D scatter plot and a 2D RDG analysis(Kazachenko et al., 2022). The scatter plot reveals that the Red flaky has a range of 0.02 to 0.005 a.u., indicating stronger repulsive interactions and demonstrating the steric effect. The green patches indicate the presence of van der Waals interactions, which occur within the range of the negative area from −0.005 to −0.015. The presence of a blue color patch within the range of −0.020 to −0.035 a.u indicates a significant hydrogen bond interaction.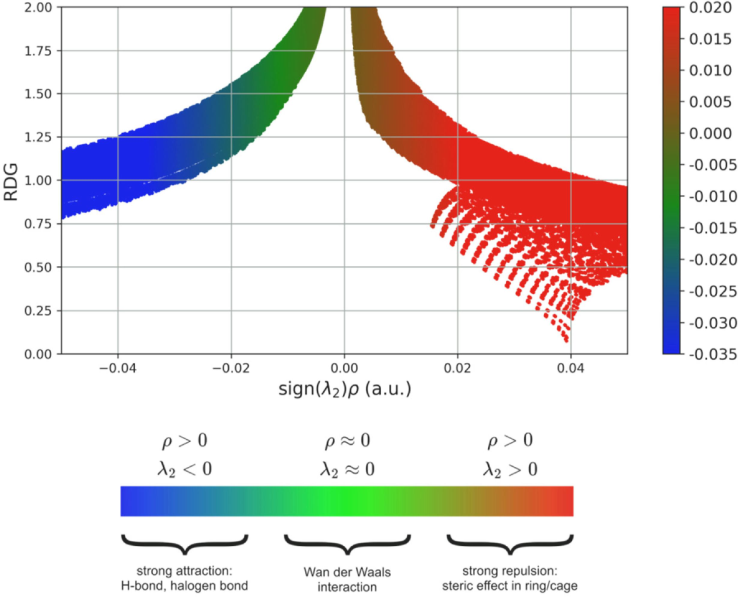
RDG scatter graph of 2TCN.
3.9 ELF and LOL
The Electron Localization Function (ELF) is a powerful tool used in computational chemistry and materials science to analyze and visualize the degree of electron localization in molecules and solids. ELF helps to provide insight into the nature of chemical bonding, stability, and reactivity by mapping regions of space where electrons are likely to be found. It allows chemists to study the distribution of electron density in a way that reveals the behavior of electrons beyond what traditional bond descriptions offer. We utilized the software MULTIWFN version 3.8 to do a topological study of ELF and LOL. Fig S3 displays the three-dimensional mapping of the ELF and LOL. Within the range of 0.001 to 1.000, there exist various color codes. The red zone indicates higher levels of (ELF) values, whereas the blue region represents lower ELF values. ELF studies aim mostly to understand the quantitative behavior of electrons in a system(Arulaabaranam et al., 2021). The Pauli repulsion between two electrons with the same spin arises from the disparity in their kinetic energy density and is connected to the behavior of the electron. The presence of Pauli repulsion is denoted by the red and blue zones, respectively. A red zone including hydrogen atoms surrounds the higher ELF region, suggesting the presence of highly localized bonding and nonbonding electrons. Carbon, sulfur, and nitrogen atoms are represented by blue, indicating the electron-depleted region. On the LOL map, the white center region of the hydrogen atom (H6, H7, and H8) indicates that the electron density surpasses the upper limit of the color scale. The presence of the blue color in atoms C2, C3, C4, C5, C9, and N10 signifies the phenomenon of electron delocalization.
3.10 Average localization energy (ALIE)
It is possible to get rid of the electrons in a point system by ionizing the average local energy. The favored reaction sites for electrophiles or radicals are those with the lowest energy, indicating the position of the most tightly bound electrons. The compound's average localization energy is depicted in Fig S4. The study demonstrated that electrons can contribute to the stability and bioactivity of molecules, whether they are localized or delocalized, depending on the specific context of the molecule(Vincy et al., 2022). The Bohr scale for these sites runs from −8.65 to 8.65 and is represented by the colors blue (0.000) to red (2.000). The hue red represents core electrons, the color blue represents localized bonded electrons, and the color greenish-yellow represents delocalized electrons in molecules.
3.11 Toxicity prediction
Toxicity prediction methods are widely recognized for their safety, but cell-based toxicity studies do not comprehensively grasp their potential negative impacts. The terms “active” and “inactive” describe whether a substance is expected to have a hazardous activity or not based on specific biological endpoints or assays when toxicity prediction is done utilizing web servers. When a substance is designated as “active” in a toxicity prediction, it indicates that the model being employed predicts that the compound will demonstrate hazardous activity (Kolahalam et al., 2021). To put it another way, the substance is probably going to have a hurtful impact on biological systems, such as mutagenesis consequences, organ toxicity, or cell destruction. When a substance is classified as “inactive,” it indicates that the prediction model indicates that it is unlikely to be hazardous under the tested conditions (Pillai et al., 2020). Based on the supplied data, it does not exhibit any noteworthy negative interactions or consequences. To forecast the organ toxicity (OC toxicity), toxicological parameters (TEMs), and LD50 (Median LD50) for the 2TCN chemical, we utilized the web server pro-tox II program (Fnfoon and Al-Adilee, 2023). The LD50 value and pFPP of the 2TCN molecule are displayed in the Table S6. Based on the results, the title compound was classified as having acute toxicity class IV. The acute toxicity prediction results encompass the classification of toxicity classes. Class IV generally indicates a low level of toxicity compared to more hazardous classes. The results indicate that the title molecule does not possess any carcinogenic, mutagenic, or cytotoxic effects.
3.12 Antioxidant activity
The 2.2 diphenyl-1-picrylhydrazyl (DPPH) assay was employed to evaluate the antioxidant capacity of the 2TCN molecule. The DPPH free radical scavenging method is a well-established technique used to assess the antioxidant capacity of a sample. In the DPPH experiment, the sample causes the violet DPPH solution to undergo a reduction in an amount that depends on its concentration, resulting in the formation of a yellow by-product known as diphenyl picryl hydrazine (Lalpara et al., 2022). After a 30-minute incubation period, the samples' absorbance was determined at 517 nm. The percentage of inhibition determined using the formula OD of sample – OD of blank / OD of control × 100 is illustrated in Table S7. The scavenging activity percentage (AA%) was obtained, and the inhibition percentage of ascorbic acid was found to be 78 % at 100 µg, while 2TCN exhibited an inhibition percentage of 72 % at 200 µg.
3.13 Anti-inflammatory activity
The anti-inflammatory effect of 2TCN compounds at four doses (25, 50, 100 and 200 µg) was assessed using albumin denaturation. The OD of control − OD of sample / OD of control x 100 equals % of albumin denaturation inhibition. The corresponding percentage of inhibition for each concentration was found to be 32, 38, 46, and 60 %, respectively. The reference drug, diclofenac sodium, exhibited a percentage of inhibition of 72 %. Diclofenac sodium is used as a reference drug because it has a well-established efficacy and safety profile(Souza et al., 237 (2022)). Albumin denaturation inhibition may signify an anti-inflammatory impact, as it plays a crucial function in regulating inflammatory reactions and sustaining oncotic pressure in the bloodstream. Denaturation of proteins such as albumin can result in alterations to their structure and function, potentially indicating underlying physiological abnormalities (Bihari et al., 2020). The values are presented in Table S8.
3.14 Ramachandran plot of 2TCN
It helps in assessing the quality and validity of protein structure by showing whether the ɸ and ψ angles fall within allowed regions for stable conformation. Deviations from these regions indicate potential structure issues in protein models. The Ramachandran plot consists of four quadrants, with the phi and psi angles plotted on the x and y axes, respectively, ranging from −180 to + 180 degrees(Shntaif et al., 2021). The red color represents the allowed zone of the core, whereas the blue color denotes the preferred regions with no steric hindrance. The protein's Ramachandran plots are depicted in Fig S5. The majority of residues fall within acceptable ranges that indicate the stability of proteins suited for computer-aided drug design docking.
3.15 Molecular docking study
After downloading the 2IEG, 3S9Y, 4PF7, and 6R9W receptor macromolecules from the Protein Data Bank at https://www.rcsb.org/pdb, they were converted to pdb format. Finding the receptor's cavity is the first step in locating the residues. It was from https://www.cgl.ucsf.edu/chimera/ that the offline Auto dock application was downloaded (Kerkour et al., 2023). Macromolecular receptors might be separated from ligands, solvents, and unusual residues. Macromolecular and non-molecular components were isolated using the Discovery Studio software. The PDB format was used to save the output of the separation. The ligand structure of the produced compound can be downloaded from the PubChem website (https://pubchem.ncbi.nlm.nih.gov). A change to the mol file format and an adjustment to the number of action torsions were used to optimize this using Auto-dock tools. While preparing the receptor by adding hydrogen polar, the grid box was built up to know the binding site location, and the format was converted to pdbqt form(Izuchukwu et al., 2022). The docking of molecules was done with the help of Auto-dock vina. The ligands and receptors that were previously on drive C: were copied and saved in a notepad file called conf.txt, and Auto-dock Vina was executed using the command prompt tool. An analysis of the study findings was made possible by extracting the binding docking data free energy value from the log.txt output. The docking structure in Fig. 7 for 2IEG, 3S9Y, 4PF7, and 6R9W macromolecules(Raza et al., 2023). Dashed lines in the image represent a hydrogen bond developing between the target protein and the ligand. The protein–ligand complex hydrogen bonds are Tyr185, Tyr260, Gly331, and Val65 for 2IEG, 3S9Y, 4PF7, and 6R9W with the binding energies are −5.10, −4.72, −4.73, and −4.55 kcal/mol respectively. The docking energies are presented in Table 2. The protein 6R9W showed the best binding affinity score when compared to other proteins.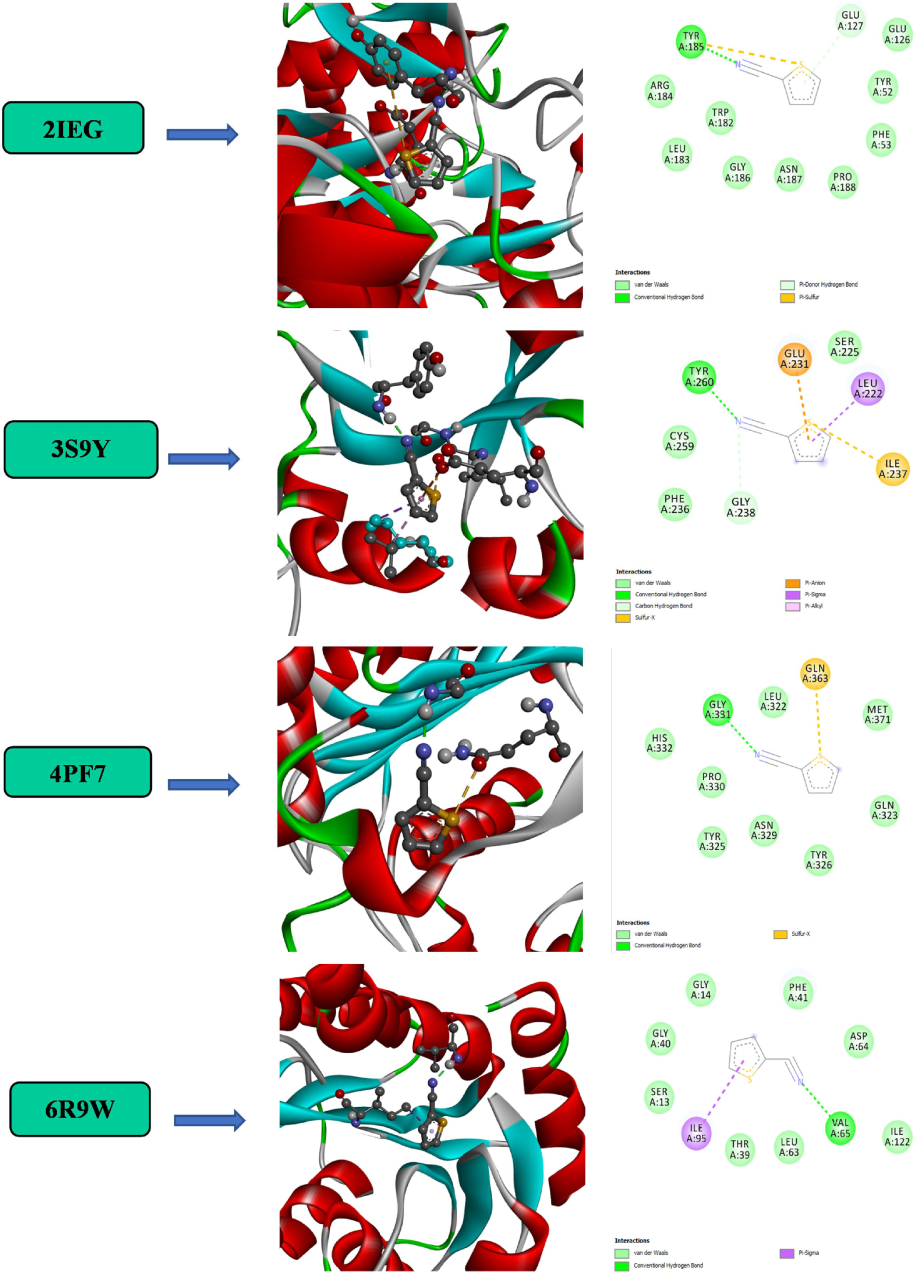
Molecular docking analysis of 2TCN.
Ligand
Activity
Protein ID
Binding energy
(kcal/mol)
Inhibition constant
RMSD
(Å)
2-Thiophenecarbonitrile
Antidiabetic
2IEG
−5.10
182.20
96.02
Antiviral
3S9Y
−4.72
348.42
49.56
Insulin inhibitor
4PF7
−4.73
339.06
14.79
Antituberculosis
6R9W
−4.55
463.22
84.73
4 Conclusion
The compound experienced characterization using infrared (IR), Raman, and ultraviolet (UV) spectroscopy. The UV–Vis spectra showed absorption bands below 400 nm in both methods (experimental and TD-DFT). The MEP elucidated the location of the nucleophilic site on the N atom and identified an electrophilic site in the vicinity of the sulfur within the molecule in both solvents (acetonitrile, water, and methanol) and gas phases. The 2TCN molecule exhibits a HOMO-LUMO gap of 5.21 (eV) in solvents methanol, water, and acetonitrile and the gas phase is 5.23 (eV), suggesting a high level of chemical stability. The localization and delocalization of electrons can be determined by topological investigations such as ELF and LOL. The compound 2TCN exhibited anti-inflammatory, and antioxidant effectiveness. According to molecular docking studies, 2TCN forms an interaction with 2IEG, 3S9Y, 4PF7, and 6R9W, and binding energies are −5.10, −4.72, −4.73, and −4.55 kcal/mol.
CRediT authorship contribution statement
Vaithilingam Sasikala: Methodology, Investigation, Formal analysis, Data curation. Vadivelu Balachandran: Visualization, Validation, Methodology, Investigation. Natarajan Elangovan: Validation, Supervision, Software, Resources, Project administration, Methodology. Sinouvassane Djearamane: Resources, Project administration, Methodology. Natarajan Arumugam: Validation, Supervision, Software, Resources, Project administration, Methodology. Ling Shing Wong: Software, Resources, Project administration, Methodology, Investigation. Saminathan Kayarohanam: Methodology, Investigation, Funding acquisition, Formal analysis.
Acknowledgment
The project was funded by Researchers Supporting Project number (RSP2024R143), King Saud University, Riyadh, Saudi Arabia.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis, structure and antifungal activity of thiophene-2,3-dicarboxaldehyde bis(thiosemicarbazone) and nickel(II), copper(II) and cadmium(II) complexes: Unsymmetrical coordination mode of nickel complex. J. Inorg. Biochem.. 2013;126:76-83.
- [CrossRef] [Google Scholar]
- K. Arulaabaranam, S. Muthu, G. Mani, A.S. Ben Geoffrey, Speculative assessment, molecular composition, PDOS, topology exploration (ELF, LOL, RDG), ligand-protein interactions, on 5-bromo-3-nitropyridine-2-carbonitrile, Heliyon 7 (2021) e07061. 10.1016/j.heliyon.2021.e07061.
- Synthesis, solvent role in TD-DFT (IEFPCM model), fluorescence and reactivity properties, topology and molecular docking studies on sulfathiazole derivative. J. Mol. Liq.. 2024;400:124570
- [CrossRef] [Google Scholar]
- Screening of potential drug from Azadirachta Indica (Neem) extracts for SARS-CoV-2: An insight from molecular docking and MD-simulation studies. J. Mol. Struct.. 2021;1227:129390
- [CrossRef] [Google Scholar]
- Albumin as a drug: its biological effects beyond volume expansion. Crit. Care Resusc. J. Australas. Acad. Crit. Care Med.. 2020;22:257-265.
- [CrossRef] [Google Scholar]
- Clinical review of antidiabetic drugs: implications for type 2 diabetes mellitus management. Front. Endocrinol. (lausanne). 2017;8:6.
- [CrossRef] [Google Scholar]
- Synthesis, spectral features, electronic structure studies, and molecular docking analysis of a Schiffbase (E)-1-(4-chlorophenyl)-N-(nitrophenyl)methanimine from 4-chloroaniline and 2-nitrobenzaldehyde. Vietnam J. Chem.. 2023;61:577-593.
- [CrossRef] [Google Scholar]
- Electrocatalytic rate constants from DFT simulations and theoretical models: Learning from each other. Curr. Opin. Electrochem.. 2022;36:101110
- [CrossRef] [Google Scholar]
- Synthesis, structural investigation, computational study, antimicrobial activity and molecular docking studies of novel synthesized (E)-4-((pyridine-4-ylmethylene)amino)-N-(pyrimidin-2-yl)benzenesulfonamide from pyridine-4-carboxaldehyde and sulfadiazine. J. Mol. Struct.. 2021;1241
- [CrossRef] [Google Scholar]
- Photoluminescence property and solvation studies on sulfonamide; Synthesis, structural, topological analysis, antimicrobial activity and molecular docking studies. Inorg. Chem. Commun.. 2023;155:111019
- [CrossRef] [Google Scholar]
- Solid-state Synthesis, electronic Structure Studies, Solvent Interaction through Hydrogen Bonding, and Molecular Docking Studies of 2,2’-((1,2-Phenylenebis(Azaneylylidene))Bis (Methaneylylidene))Diphenol from o-Phenylenediamine and Salicylaldehyde. Polycycl. Aromat. Compd. 2023
- [CrossRef] [Google Scholar]
- Theoretical Investigation on Solvents Effect in Molecular Structure (TD-DFT, MEP, HOMO-LUMO), Topological Analysis and Molecular Docking Studies of N-(5-((4-Ethylpiperazin-1-yl)Methyl)Pyridin-2-yl)-5-Fluoro-4-(4-Fluoro-1-Isopropyl-2-Methyl-1H-Benzo[d] Imi. Polycycl. Aromat. Compd. 2023:1-24.
- [CrossRef] [Google Scholar]
- Synthesis, solvent role (water and DMSO), antimicrobial activity, reactivity analysis, inter and intramolecular charge transfer, topology, and molecular docking studies on adenine derivative. J. Mol. Liq.. 2023;391:123250
- [CrossRef] [Google Scholar]
- Fluorescent Azomethine by the Condensation of Sulfadiazine and 4-Chlorobenzaldehyde in Solution: Synthesis, Characterization, Solvent Interactions, Electronic Structure, and Biological Activity Prediction. Polycycl. Aromat. Compd. 2023:1-22.
- [CrossRef] [Google Scholar]
- Synthesis, vibrational analysis, absorption and emission spectral studies, topology and molecular docking studies on sulfadiazine derivative. ChemistrySelect. 2024;9
- [CrossRef] [Google Scholar]
- Preparation and characterization of B, S, and N-doped glucose carbon dots: Antibacterial, antifungal, and antioxidant activity. Sustain. Mater. Technol.. 2022;32:e00397.
- [Google Scholar]
- Synthesis and spectral characterization of some metal complexes with new heterocyclic azo imidazole dye ligand and study biological activity as anticancer. J. Mol. Struct.. 2023;1271:134089
- [CrossRef] [Google Scholar]
- Gaussian 09, Revision B.01, Gaussian 09, Revis. B.01. Wallingford CT: Gaussian, Inc.; 2009. p. :1-20. citeulike-article-id:9096580
- Solid state synthesis of a fluorescent Schiff base (E)-1-(perfluorophenyl)-N-(o-toly)methanimine followed by computational, quantum mechanical and molecular docking studies. Results Chem.. 2023;5:100819
- [CrossRef] [Google Scholar]
- Design, synthesis of new 4,5-dibenzylidene-9,10-diphenyl-1,2,7,8,9,10 hexahydroacridine-3,6-dione derivatives using extract of Vitexnegundo: Cytotoxic activity & molecular docking study. Chem. Phys. Impact. 2024;8:100483
- [CrossRef] [Google Scholar]
- Synthesis, vibrational analysis, molecular property investigation, and molecular docking of new benzenesulphonamide-based carboxamide derivatives against Plasmodium falciparum. J. Mol. Struct.. 2022;1269:133796
- [CrossRef] [Google Scholar]
- Hydrogen bonds interactions in biuret-water clusters: FTIR, X-ray diffraction, AIM, DFT, RDG, ELF, NLO analysis. J. King Saud Univ. - Sci.. 2022;34:102350
- [CrossRef] [Google Scholar]
- Novel α-aminophosphonate derivates synthesis, theoretical calculation, Molecular docking, and in silico prediction of potential inhibition of SARS-CoV-2. J. Mol. Struct.. 2023;1272 134196
- [CrossRef] [Google Scholar]
- Saussurea lappa plant rhizome extract-based zinc oxide nanoparticles: synthesis, characterization and its antibacterial, antifungal activities and cytotoxic studies against Chinese Hamster Ovary (CHO) cell lines. Heliyon. 2021;7:e07265.
- [CrossRef] [Google Scholar]
- Water Promoted One Pot Synthesis of Sesamol Derivatives as Potent Antioxidants: DFT, Molecular Docking, SAR and Single Crystal Studies. Polycycl. Aromat. Compd. 2022
- [CrossRef] [Google Scholar]
- Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem.. 2012;33:580-592.
- [CrossRef] [Google Scholar]
- Synthesis, anti-inflammatory, analgesic and antioxidant activities of some tetrasubstituted thiophenes. J. Enzyme Inhib. Med. Chem.. 2008;23:829-838.
- [CrossRef] [Google Scholar]
- Synthesis, spectral analysis, antibacterial activity, quantum chemical studies and supporting molecular docking of Schiff base (E)-4-((4-bromobenzylidene) amino)benzenesulfonamide. J. Indian Chem. Soc.. 2022;99:100405
- [CrossRef] [Google Scholar]
- Green synthesis and characterization of zinc oxide nanoparticles with antibacterial and antifungal activity. J. Mol. Struct.. 2020;1211:128107
- [CrossRef] [Google Scholar]
- Absorption studies on serotonin neurotransmitter with the platinum metal cluster using the gas phase and different solvents, topological and non-covalent interaction: A DFT approach. Chem. Phys. Impact. 2023;7:100295
- [CrossRef] [Google Scholar]
- One Pot Efficient Synthesis of 1,3-di(Naphthalen-1-yl)Thiourea; X-Ray Structure, Hirshfeld Surface Analysis, Density Functional Theory, Molecular Docking and In-Vitro Biological Assessment. J. Mol. Struct.. 2023;1271:133989
- [CrossRef] [Google Scholar]
- Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev.. 2003;14:447-455.
- [CrossRef] [Google Scholar]
- Rational drug design, synthesis, and biological evaluation of novel N-(2-arylaminophenyl)-2,3-diphenylquinoxaline-6-sulfonamides as potential antimalarial, antifungal, and antibacterial agents. Digit. Chinese Med.. 2021;4:290-304.
- [CrossRef] [Google Scholar]
- Zinc(II) complexes bearing N, N, S ligands: Synthesis, crystal structure, spectroscopic analysis, molecular docking and biological investigations about its antifungal activity. J. Inorg. Biochem.. 2022;237 111995
- [CrossRef] [Google Scholar]
- Exploring the antibacterial activity of 1, 2 diaminoethane hexanedionic acid by spectroscopic, electronic, ELF, LOL, RDG analysis and molecular docking studies using DFT method. J. Mol. Struct.. 2022;1247:131388
- [CrossRef] [Google Scholar]
Further reading
- Synthesis, spectral analysis, antibacterial, antifungal, antioxidant and hemolytic activity studies of some new 2,5-disubstituted-1,3,4-oxadiazoles. J. Mol. Struct.. 2022;1253:132216
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2024.103526.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




