

Translate this page into:
New quinazoline-N-4-fluorophenyl derivatives as potential anticancer agents: Discovery of a promising dual EGFR/VEGFR-2 inhibitor
⁎Corresponding author. m.geesi@psau.edu.sa (Mohammed H. Geesi)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
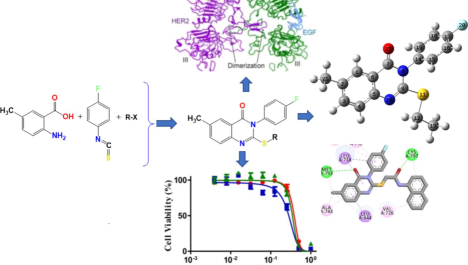
This research is dedicated to synthesizing a new group of quinazoline-N-4-fluorophenyl 4a–d structures and evaluating their anticancer efficacy across multiple cancer cell lines. The molecular design of these derivatives was based on the structural features required for dual inhibition of VEGFR-2 and EGFR. The new derivatives were structurally characterised by NMR analyses. Cytotoxicity was assessed in this study against various cancerous cell strains. Among these, the top three products were further assessed for their capacity to block the enzymatic activity of (VEGFR-2) and (EGFR). Product 4b, in particular, exhibited a strong cytotoxic profile, with IC50 values of 68.2 ± 1.54 nM against EGFR and 189 ± 5.66 nM against VEGFR-2. Molecular docking studies demonstrated that compound 4b effectively interacts with the active sites of both VEGFR-2 and EGFR, potentially influencing its action pathway as a powerful inhibitor.
Keywords
Quinazoline
Molecular docking
MTT assay
EGFR
VEGFR-2
Tyrosine kinase

1 Introduction
A broad category of critical and complicated diseases falls under the umbrella of cancer. According to statistics published by the WHO, nearly nine million cancer-related deaths were recorded in 2020. The same source predicts that the number of cancer cases could exceed 12 million by 2030 (Bray et al., 2021). Current research focuses on developing innovative approaches to target specific signalling pathways in order to eradicate the negative aspects of conventional chemotherapy in cancer treatment (Liu et al., 2018).
Heterocyclic derivatives that include at least one atom different from carbon particularly nitrogen, oxygen, or sulfur are often involved in the inhibition of key proteins associated with tumor growth and cellular proliferation (Kumar et al., 2023; Martins et al., 2015; Tilahun et al., 2025; Peerzada et al., 2021; Drakontaeidi et al., 2024; Papanotas and Pontiki, 2024; Obaid et al., 2022). Tyrosine kinase (TK) is fundamental to growth factor signalling, as it represents a key focus in anticancer therapy. Inhibition of angiogenesis, particularly through targeting Vascular Endothelial Growth Factor Receptor VEGFR, is an important strategy for impeding tumour progression (Zahran et al., 2023). Furthermore, VEGFR is pivotal in both normal and pathological angiogenesis, rendering it a prime target for novel cancer therapies (Ghorab et al., 2017).
The aberrant expression of Vascular Endothelial Growth Factor Receptor 2 (VEGFR-2) has been detected in several types of cancer cells (colorectal and lung). Therefore, targeting VEGFR-2 is a crucial strategy in anti-angiogenic therapy (Cook and Figg, 2010). Scientists are currently engaged in developing new Type II and III inhibitors to block VEGFR-2 following sustained TK inhibition (Traxler and Furet, 1999). On the other hand, Vascular Endothelial Growth Factor (VEGF) and Epidermal Growth Factor (EGF) can independently contribute to cancer progression and resistance to treatment through overlapping signalling pathways (Panigrahy et al., 2005). The emergence of resistance to VEGFR-2 inhibitors underscores the importance of exploring novel therapeutic options (Cao et al., 2011).
The Epidermal Growth Factor Receptor (EGFR) is a crucial protein in cellular signalling and is largely responsible for the accelerated multiplication of cancer cells (Gschwind et al., 2004; Li and Li, 2014). EGFR mutations are commonly associated with reduced responsiveness to TKIs, which often leads to resistance and cancer progression (Ayati et al., 2020). One notable example is the EGFRT790M mutation, which increases ATP binding affinity to levels similar to the non-mutated form, thus reducing the effectiveness of TKIs in the presence of ATP (Stockley et al., 2018). Targeting EGFR, along with the VEGF pathway, has emerged as a promising strategy for enhancing therapeutic responses, particularly in cases of non-small cell lung cancer (Byers and Heymach, 2007). Suppression of VEGF signalling is pivotal in cancer treatment (Panigrahy et al., 2002), and FDA-approved small molecule inhibitors like quinazoline derivatives (e.g., Vandetanib, Gefitinib) target both EGFR and VEGFR-2 pathways to inhibit tumour progression (Yu and Pao, 2013) (Fig. 1-A).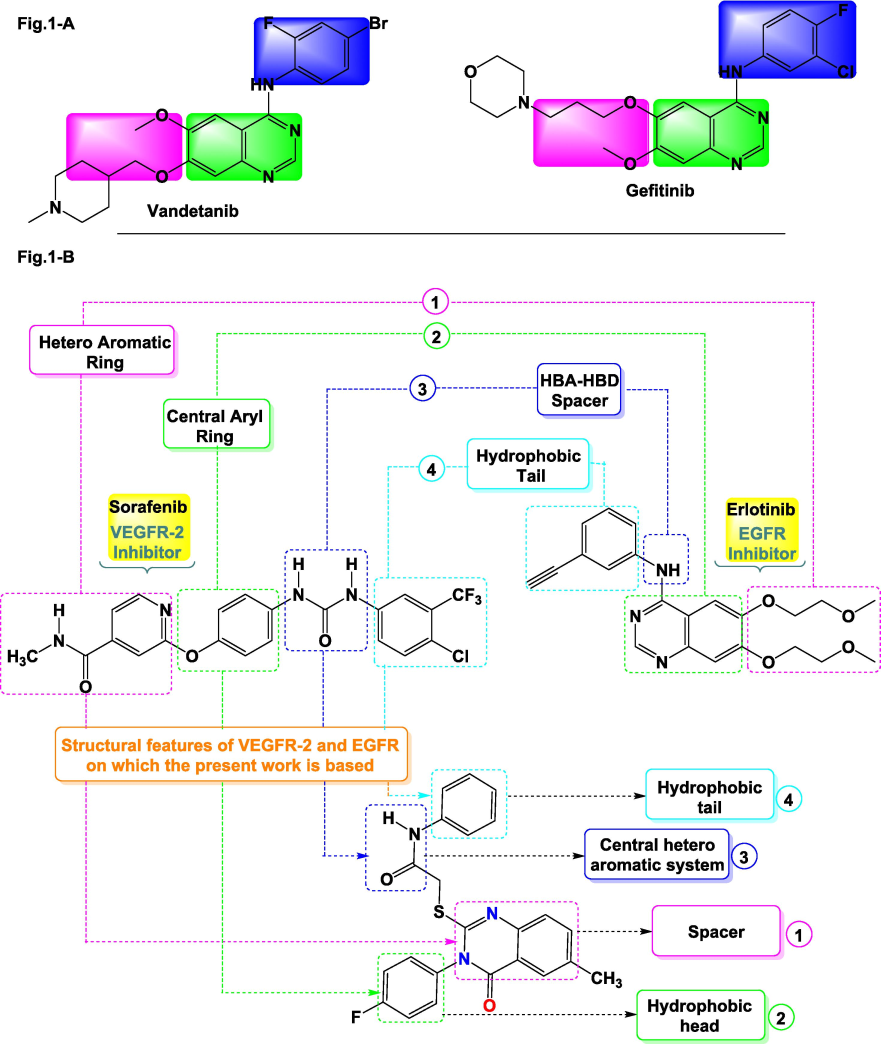
Compounds (Gefitinib & Vandetanib) active against TKIs (A) and highlights the fundamental structural characteristics of VEGFR-2 and EGFR inhibitors (B).
Quinazoline-based compounds, including WHI-P180, Erlotinib, Afatinib, Gefitinib, Lapatinib, and, Vandetanib are clinically significant for their ability to target EGFR and VEGFR-2 pathways and inhibit tumour growth (Abdullaziz et al., 2017). Moreover, quinazoline scaffolds are of particular interest in designing potential anticancer agents (Ghorab et al., 2018). Hybridisation strategies aimed at combining EGFR and VEGFR inhibition with other pharmacophores possessing potent antitumor activity, such as quinazolines, seem to be promising for enhancing compound efficacy (Ying et al., 2010) (Fig. 1-A).
Guided by existing literature and a thorough analysis of the proteins in question (refer to Fig. 1-B), this study centers on the insertion of new pharmacophoric groups derived from the literature on EGFR and VEGFR-2, taking into consideration their biological relationship with the quinazoline scaffold. We will develop new substituted quinazoline derivatives, using Erlotinib and Sorafenib (inhibitors of EGFR and VEGFR-2) as starting points to introduce structural modifications. The structural analysis of these derivatives has revealed the following information: a hetero-aromatic moiety to enhance affinity towards ATP (1), a central hydrophobic spacer (2), a binding site to facilitate hydrogen bonding with specific sites on the biological receptor such as Asp1046 and Glu885 (3), and finally, an allosteric hydrophobic group at the molecule's terminus (4). This design is illustrated in Fig. 1-B (Lee et al., 2010). Moreover, the quinazolinone derivatives 4a–d developed in this research exhibit the key pharmacophoric characteristics of EGFR and VEGFR-2. The rational analysis of the SAR of Erlotinib and Sorafenib towards EGFR and VEGFR-2 has led us to introduce various substituents onto the molecules to be synthesised, including (1) a hydrophobic group at the molecule's terminus, (2) a hetero-aromatic motif to enhance affinity towards Lys721 and Met769, (3) an N–H spacer to facilitate hydrogen bond formation with the biological target, and (4) a second hydrophobic group opposite the first one (Liu et al., 2006; Soliman et al., 2019) (Fig. 1-B).
In order to expand the SAR profile of the quinazoline derivatives, a fluorine atom was introduced at the aromatic ring in para position to improve the hydrogen bonding capabilities. Additionally, modifications were made to the sulfur substitution at the C2 position by incorporating various aromatic groups. The derivatives were then subjected to incubation with multiple cancer cell lines for a 24-hour period, followed by molecular modelling studies to explore their interactions and potential effectiveness.
2 Experimental section
The compounds involved in this research were procured from Aldrich and Acros (The United States). Physical properties, such as the melting point, were determined using a Kofler bench. NMR analysis was conducted at a resolution of 500 MHz using a Bruker Avance III spectrometer.
3 Synthesis of quinazolinone 3
A combination of 0.01 mol of 2-amino-5-methylbenzoic acid 1 and 5-fluorophenyl isothiocyanate 2, along with a few drops of Et3N in 30 mL of ethanol, was prepared. The mixture was refluxed for approximately 2 h. The formed precipitate was filtered and recrystallized in ethanol after treatment with HCl (Geesi et al., 2020).
4 Synthesis of quinazolinone-N-substituted compounds 4a-d
For 10 h of stirring in acetone as the solvent, an equimolar mixture of derivative 3 (5 mmol) and acetamide derivatives or alkyl halide (added separately) was refluxed in the presence of K2CO3. The precipitate formed after the reaction was recrystallised in ethanol (Geesi, 2020; Riadi et al., 2024).
4.1 2-((3-(4-Fluorophenyl)-6-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(naphthalen-1-yl)acetamide (4a)
(81 %, 173 ± 2 ◦C). 1H NMR (500 MHz, CDCl3): δ = 2.49 (s, 3H, CH3), 3.89 (s, 2H, CH2), 7.20 (m, 3H, HC-Ar), 7.30 (m, 2H, HC-Ar), 7.52 (d, 2H, HC-Ar), 7.58–7.67 (m, 3H, HC-Ar), 7.85 (m, 3H, HC-Ar), 8.04 (d, 1H, HC-Ar), 10.19 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δ = 21.08 (CH3); 36.49 (CH2); 116.77 (C); 116.95 (HC-Ar); 118.50 (C-Ar); 119.35 (C-Ar); 124.74 (C-Ar); 127.07 (C-Ar); 129.38 (C-Ar); 129.53 (C-Ar); 130.82 (C-Ar); 130.90 (C-Ar); 132.54 (C-Ar); 136.72 (C-Ar); 137.11 (C-Ar); 142.12 (C-Ar); 144.55 (C-Ar); 157.08 (C); 161.00 (C); 162.26 (C); 164.26 (C); 166.84 (C); 196.72 (C).
4.2 2-((2-(4-Bromophenyl)-2-oxoethyl)thio)-3-(4-fluorophenyl)-6-methylquinazolin-4(3H)-one (4b)
C23H16BrFN2O2S. (84 %, 164 ± 2 ◦C). 1H NMR (500 MHz, CDCl3): δ = 2.47 (s, 3H, CH3), 4.55 (s, 2H, CH2), 7.14 (m, 1H, CHAr), 7.30 (m, 2H, CHAr), 7.39 (m, 2H, CHAr), 7.49 (dd, 1H, CHAr), 7.71 (m, 2H, CHAr), 7.98 (m, 2H, CHAr), 8.02 (d, 1H, CHAr). 13C NMR (125 MHz, CDCl3): δ = 21.10 (CH3); 39.18 (CH2); 116.74 (HC-Ar); 116.92 (HC-Ar); 119.25 (HC-Ar); 125.60 (HC-Ar); 126.56 (HC-Ar); 128.68 (HC-Ar); 129.86 (HC-Ar); 131.02 (HC-Ar); 131.09 (C-Ar); 135.03 (C-Ar); 136.07 (C-Ar); 136.26 (C-Ar); 145.27 (C-Ar); 154.71 (C); 161.60 (C); 164.26 (C); 192.70 (C).
4.3 N-(4-Acetylphenyl)-2-((3-(4-fluorophenyl)-6-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetamide (4c)
C25H20FN3O3S. (83 %, 155 ± 2 ◦C). 1H NMR (500 MHz, DMSO‑d6): δ = 2.55 (3H, s, CH3), 2.66 (s, 3H, CH3), 4.25 (s, 2H, CH2), 7.57 (m, 3H, HC-Ar), 7.72 (dd, 2H, HC-Ar), 7.76 (dd, 1H, HC-Ar), 7.87 (d, 2H, HC-Ar), 7.99 (s, 1H, HC-Ar), 8.07 (d, 2H, HC-Ar), 10.87 (s, 1H, NH). 13C NMR (125 MHz, DMSO‑d6): δ = 20.57 (CH3); 26.28 (CH3); 37.17 (CH2); 116.29 (HC-Ar); 116.47 (HC-Ar); 118.18 (HC-Ar); 119.11 (HC-Ar); 125.61 (HC-Ar); 125.78 (HC-Ar); 129.40 (HC-Ar); 131.69 (HC-Ar); 131.76 (C-Ar); 135.61 (C-Ar); 136.04 (C-Ar); 143.12 (C-Ar); 145.03 (C-Ar); 155.61 (C); 160.52 (C); 166.32 (C); 196.35 (C).
4.4 2-(Ethylthio)-3-(4-fluorophenyl)-6-methylquinazolin-4(3H)-one (4d)
C17H15FN2OS. (78 %, 159 ± 2 ◦C). 1H NMR (500 MHz, CDCl3): δ = 1.35 (t, 3H, CH3), 2.46 (s, 3H, CH3), 3.16 (q, 2H, CH2), 7.22 (t, 2H, HC-Ar), 7.29 (dd, 2H, HC-Ar), 7.53 (q, 2H, HC-Ar), 8.01 (s, 1H, HC-Ar). 13C NMR (125 MHz, CDCl3): δ = 13.62 (CH3); 21.04 (CH3); 26.76 (CH2); 116.42 (CHAr); 116.61 (CHAr); 119.24 (HC-Ar); 125.92 (HC-Ar); 126.37 (HC-Ar); 130.93 (C-Ar); 131.00 (C-Ar); 131.78 (C-Ar); 135.69 (C-Ar); 135.91 (C-Ar); 145.76 (C-Ar); 155.96 (C); 161.80 (C); 163.98 (C).
5 Theoretical details
The DFT method at the B3LYP/6–311++G(d,p) theoretical level (Gaussian 16 package) is employed to optimise the ground state geometries of compounds 4a–d (Frisch et al., 2016). Additionally, solvent effects and NMR chemical shifts (1H and 13C) were computed using the IEFPCM model (Tomasi et al., 2005) and the GIAO approach (Wolinski et al., 1990), respectively.
6 Biological evaluation
6.1 Cell culture
In this study, cancer cell lines were obtained from the company Sigma Aldrich. They were cultured under the following conditions: 37 °C, 5 % CO2, and in a culture medium containing 1 % penicillin–streptomycin, 11 % fetal bovine serum, and MEM.
6.2 Molecular docking study
The bioactivity of compounds 4a–c was evaluated against VEGFR-2/EGFR tyrosine kinase enzymes. These compounds demonstrated relatively good activity compared to the reference drug. Molecular docking was performed using the AutoDock package. The redocking of the original ligands in the active sites of two biological targets (EGFR: PDB 4ZAU, Yosaatmadja et al., 2015) and (VEGFR-2: PDB 4ASD, McTigue et al., 2012) was successfully reproduced, with RMSD values of 0.71 (for EGFR) and 1.49 (for VEGFR-2).
7 Results and discussion
The reaction steps followed in this study for synthesising compounds 4a–d are presented in Scheme 1. Initially, 2-amino-5-methylbenzoic acid 1 underwent a substitution reaction with 4-fluorophenyl isothiocyanate 2 in the presence of base (triethylamine). The reflux process in ethanol yielded product 3 in substantial amounts. S-alkylation of product 3 with different acetamide derivatives or alkyl halide in acetone in the presence of K2CO3 constituted the second step of our synthesis, resulting in quinazolinone-based hybrids 4a–d.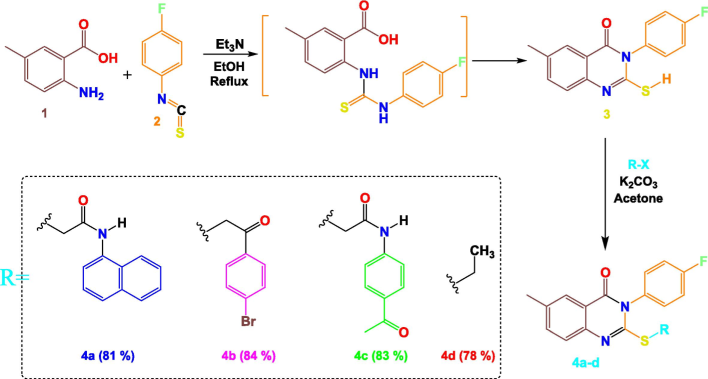
Synthetic pathway adopted in the present work.
The structures of derivatives 3 and 4a–d have been confirmed after performing several analytical techniques (1H, 13C NMR). For the quinazolinone products 4a–d, their 1H NMR spectra consistently show a singlet corresponding to the CH3 fragment attached to the quinazoline core, typically appearing around 2.50 ppm. In relation to the substituent added in the last stage of the synthesis process, singlet peaks have been detected at 10.19 and 10.87 ppm, corresponding to the protons carried on the NH atom in products 4a and 4c. Similarly, the 1H NMR spectrum of compound 4d reveals two distinct peaks: one appearing as a triplet at 1.35 ppm corresponding to the CH3 group, and the other as a quadruplet at 3.15 ppm, attributed to the methylene protons (CH2) directly attached to the sulfur atom. In addition, a multiplet is noted for the aromatic protons between 7.00 and 8.10 ppm. Further structural validation for products 4a–d is provided by their 13C NMR spectra, where a distinctive signal at 196.35 ppm relates to the carbonyl group (C = O) of the acetyl fragment located in the para position of product 4c. On the other hand, the linker CH2 in the case of products 4a–c is identified in the range of 36.49–39.18 ppm. Moreover, the 13C NMR spectrum for product 4d provides confirmation of the suggested structure by the presence of two unique peaks at 13.62 and 26.76 ppm who are associated with the CH3 and CH2 of the sulphide group. Moreover, the 13C NMR spectrum confirmed the absence of the thione (C = S) signal (for compound 3), typically observed around 175 ppm. Instead, the resonance of the −N = C- was detected at 155.80 ppm for compound 3 and at δ 154.71–157.08 ppm for quinazolinone derivatives 4a–d, corresponding to the chemical shift of the carbon atom (N = C), which suggests that compound 3 exists in its thiol form (Fig. 2) (Still et al., 1976).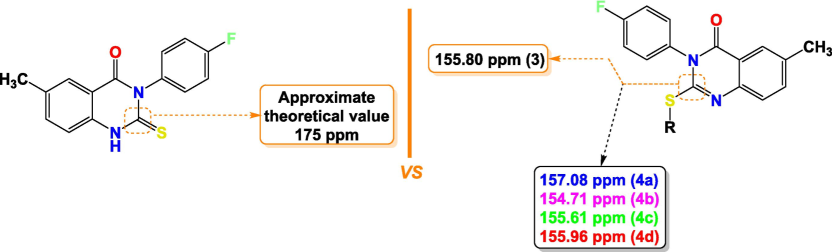
Comparison at the 13C NMR analysis level of the C2 of the starting compound 3 and the targeted compounds 4a–d.
7.1 NMR spectrum: Theoretical prediction
The chemical shifts found experimentally for NMR (1H & 13C) and those found theoretically are shown in Table 1. The optimised geometries with atomic numbering of 4a–d are shown in Fig. 3. It should be noted that the predicted NMR (1H & 13C) chemical shifts of 4a–d were obtained from their corresponding optimised geometries (Fig. 3). A: Prediected, B: Experimental.
4a
4b
4c
4d
A
B
A
B
A
B
A
B
H1
7.82
8.04
8.06
8.02
8.15
7.99
7.93
8.01
H3
7.40
7.85
7.59
7.71
7.95
7.57
7.67
7.53
H4
7.19
7.66
7.52
7.49
7.87
7.76
7.49
7.53
H12
4.06
3.89
4.61
4.55
4.71
4.25
3.01
3.16
H13
1.41
1.35
H15
7.25
7.85
7.30
7.39
7.40
7.57
7.28
7.29
H16
7.20
7.52
7.27
7.30
7.37
7.72
7.25
7.22
H18
7.23
7.52
7.27
7.30
7.42
7.72
7.25
7.22
H19
7.32
7.85
7.30
7.39
7.45
7.57
7.28
7.29
H21
2.63
2.49
2.45
2.47
3.03
2.66
2.51
2.46
H34
8.06
7.98
−
H35
7.52
7.14
−
H37
7.65
7.30
7.54
7.71
−
H38
7.41
7.20
7.93
7.98
8.60
8.07
−
H39
8.03
7.30
−
8.04
7.87
−
H40
7.93
7.60
−
−
H41
7.57
7.20
−
8.01
7.87
−
H42
7.52
7.20
−
6.91
8.07
−
H43
7.88
7.60
−
−
H45
−
2.34
2.55
−
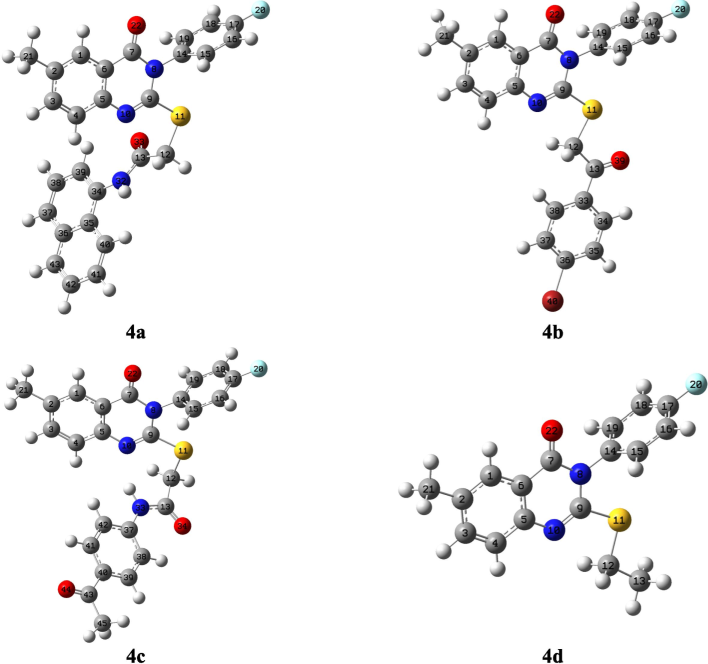
The targeted compounds’ geometric structures, following optimisation.
Based on the values in Tables 1 and 2, a very high correlation was observed between the values obtained experimentally and theoretically across both spectra (1H & 13C)-NMR, with percentages ranging from 93.30 to 99.9 % and 96.00 to 99.9 %, respectively. The comparison of values found for atoms H21 and H19 showed a difference of 0.1 ppm for compounds 4b and 4d. A: Prediected, B: Experimental.
4a
4b
4c
4d
A
B
A
B
A
B
A
B
C1
151.62
130.90
142.88
125.60
142.45
125.61
136.31
125.92
C2
165.12
161.00
158.93
136.26
155.54
135.61
148.07
135.91
C3
163.24
144.55
152.84
136.07
153.62
131.76
146.77
135.69
C4
151.33
130.82
141.94
119.25
140.89
119.11
136.44
126.37
C5
173.35
162.26
162.31
145.26
161.77
136.07
156.31
145.76
C6
143.83
119.35
135.01
116.92
134.77
118.18
129.33
119.24
C7
191.11
164.26
178.83
164.26
178.78
160.52
171.91
161.80
C9
189.62
157.08
177.55
154.71
179.81
155.61
171.96
155.96
C12
59.95
36.49
64.65
39.18
54.32
37.17
47.00
26.76
C13
195.48
166.84
211.69
192.69
186.22
196.35
21.02
13.62
C14
160.80
142.12
150.56
131.95
149.98
131.69
145.01
131.78
C15
159.07
136.72
149.08
131.95
148.86
129.40
143.13
130.93
C16
141.68
116.77
132.60
116.74
132.40
116.47
127.26
116.61
C17
196.28
196.72
150.90
135.02
184.00
166.32
176.31
163.98
C18
141.69
116.95
132.60
116.74
132.66
118.18
127.26
116.42
C19
159.20
136.72
149.07
131.08
148.86
129.40
143.13
131.00
C21
35.19
21.08
32.76
21.10
32.31
20.57
31.69
21.04
C33
148.94
131.02
−
−
−
−
C34
159.54
137.11
145.36
126.56
−
−
−
−
C35
151.13
129.38
148.36
129.86
−
−
−
−
C36
160.58
137.11
166.11
161.60
−
−
−
−
C37
150.30
124.74
148.03
129.86
161.07
143.12
−
−
C38
150.72
127.07
147.28
128.68
132.01
116.29
−
−
C39
143.49
116.95
−
−
148.75
129.40
−
−
C40
144.30
118.50
−
−
146.62
129.40
−
−
C41
151.98
132.54
−
−
144.26
125.78
−
−
C42
151.58
129.53
−
−
132.83
118.18
−
−
C43
154.94
132.45
−
−
169.72
145.03
−
−
C45
−
−
−
38.08
26.28
−
−
Conversely, for hydrogen atom H42 in compound 4c, a difference of 1.16 ppm was observed (Table 1). In the 13C NMR spectrum, deviations between the observed chemical shifts and predicted ones are in ranges of 0–33.00, 4.00–22.4, 11.00–23.00, and 7.00–21.00 ppm for 4a, 4b, 4c, and 4d, respectively (Table 2).
8 Biological evaluation
8.1 Determining the cytotoxic potential
To evaluate the effect of quinazolinone products 4a–d on the development of multiple cancer cell lines (A-549, MDA, and HeLa), a cytotoxicity assessment was carried out. The cytotoxic characteristics was analyzed through the MTT assay. The Docetaxel (a chemotherapy drug for treating different cancer types) serving as a positive control.
Products 4a–c exhibited the strongest cytotoxic effects across all cell lines, with IC50 values between 0.49 ± 0.01 and 4.36 ± 0.09 µM. Product 4b manifested an exceptionally strong cytotoxic activity against Hela, A-549, and MDA cell lines (ten times more active than the reference ''Docetaxel''), with IC50 values of 1.24 ± 0.03, 0.49 ± 0.01, and 1.07 ± 0.02 µM, respectively. Conversely, product 4d displayed low potential to inhibit cancer cells, with elevated IC50 values of 48.5 ± 1.02 µM against HeLa cells and 46.3 ± 0.97 µM against A-549 cells. Nevertheless, derivative 4d exhibited greater cytotoxicity against MDA cells, achieving an IC50 value of 5.75 ± 0.12 µM. Evaluation against A-549 cancer cells revealed excellent anticancer activity for derivatives 4a and 4c, with IC50 values of 1.31 ± 0.03 µM and 3.94 ± 0.08 µM, respectively. Conversely, these derivatives exhibited weaker anticancer effects against Hela cancer cells, with IC50 values of 3.48 ± 0.07 µM (4a) and 2.59 ± 0.05 µM (4c). Notably, significant cytotoxicity was observed against MDA cancer cells for both compounds.
8.2 In-vitro VEGFR-2 and EGFR kinase inhibitory assay
For the purpose of assessing the inhibitory activity of our synthesised products against EGFR, we specifically selected three compounds (4a, 4b and 4c) based on their potent cytotoxic profiles. The evaluation was conducted using an HTRF assay (El-Sattar et al., 2021). Docetaxel served as the standard with an IC50 of 56.1 ± 1.17 nM (Table 3). Compound 4c exhibited significant EGFR inhibitory activity, closely resembling Docetaxel with an IC50 of 62.3 ± 1.47 nM. In comparison, Docetaxel had an IC50 of 56.1 ± 1.17 nM. Compound 4b, a quinazoline derivative, also demonstrated notable EGFR inhibition with an IC50 of 68.2 ± 1.54 nM, showing a 12.1 nM difference compared to the reference. Additionally, compound 4a, which features a naphthalene group substitution on the amide function, displayed moderate efficacy in EGFR inhibition, with an IC50 measured at 104 ± 2.18 nM, half as potent as Docetaxel in terms of cytotoxicity. In this assessment, we selected compounds 4a–c (with high cytotoxic activities) tested against VEGFR-2 and EGFR according to the mechanism applied by Sogabe (Sogabe et al., 2013). Table 3 presents the IC50 values in nM (50 % inhibitory concentration). The tested derivatives exhibited excellent to good inhibitory activity, with IC50 values ranging from 90.1 ± 2.69 to 189 ± 5.66 nM for compounds 4a–c. Compounds 4a and 4c demonstrated VEGFR-2 inhibition comparable to Docetaxel, with IC50 values of 92.2 ± 2.71 and 90.1 ± 2.69 nM, respectively. Conversely, compound 4b exhibited moderate activity in front of VEGFR-2, with an IC50 of 189 ± 5.66 nM (approximately half as active as the reference).
Compound
IC50 a,b
Helaa
A-549a
MDAa
EGFRb
VEGFR-2b
4a
4b
4c
4d
Docetaxel3.48 ± 0.07
1.24 ± 0.03
2.59 ± 0.05
48.5 ± 1.02
9.65 ± 0.21.31 ± 0.03
0.49 ± 0.01
3.94 ± 0.08
46.3 ± 0.97
10.8 ± 0.234.36 ± 0.09
1.07 ± 0.02
3.51 ± 0.07
5.75 ± 0.12
3.98 ± 0.08104 ± 2.18
68.2 ± 1.54
62.3 ± 1.47
NT
56.1 ± 1.1792.2 ± 2.71
189 ± 5.66
90.1 ± 2.69
NT
89.3 ± 2.67
8.3 Molecular docking
To illustrate and understand the interesting cytotoxic profile observed for compounds 4a–c against the targeted enzymes, a molecular docking analysis was conducted to identify the fragments of our products responsible for this activity, as well as assess the binding mode with the biological targets. Table 4 includes several parameters, such as the number of amino acids that can be incorporated into the complex formation between our products and the biological target, the binding energy of these complexes, and the number of hydrogen bonds. * Number of amino acids in the active site that show binding interaction with 4a-c.
Compound
Free binding energy
H-Bonds
(HBs)Number of
amino acids *IC50 ± SEM
(nM)
VEGFR-2 kinase
4a
−9.21
2
13
92.2 ± 2.71
4b
−7.75
3
14
189 ± 5.66
4c
−7.14
0
15
90.1 ± 2.69
Docetaxel
89.3 ± 2.67
EGFR
4a
−7.54
1
9
104 ± 2.18
4b
−8.10
2
7
68.2 ± 1.54
4c
−7.11
1
7
62.3 ± 1.47
Docetaxel
56.1 ± 1.17
The complexes formed between 4a–c into the binding sites of the target enzymes show negative binding energies (BEs) in −7.11 and −9.21 kcal/mol (Table 4). The negative binding affinities of the stable complexes may explain the inhibitory efficiency of compounds 4a–c towards the target enzymes (EGFR and VEGFR-2 kinase). However, BEs show slight differences. Thus, this parameter may not be used as a strong descriptor to explain the observed inhibition. Our focus will be on the type and number of interactions that may appear in the presence of the substituted groups of Sulphur atoms. Figs. 4 and 5 show the binding modes of derivatives 4a–c into the binding sites of VEGFR2 and EGFR tyrosine kinases.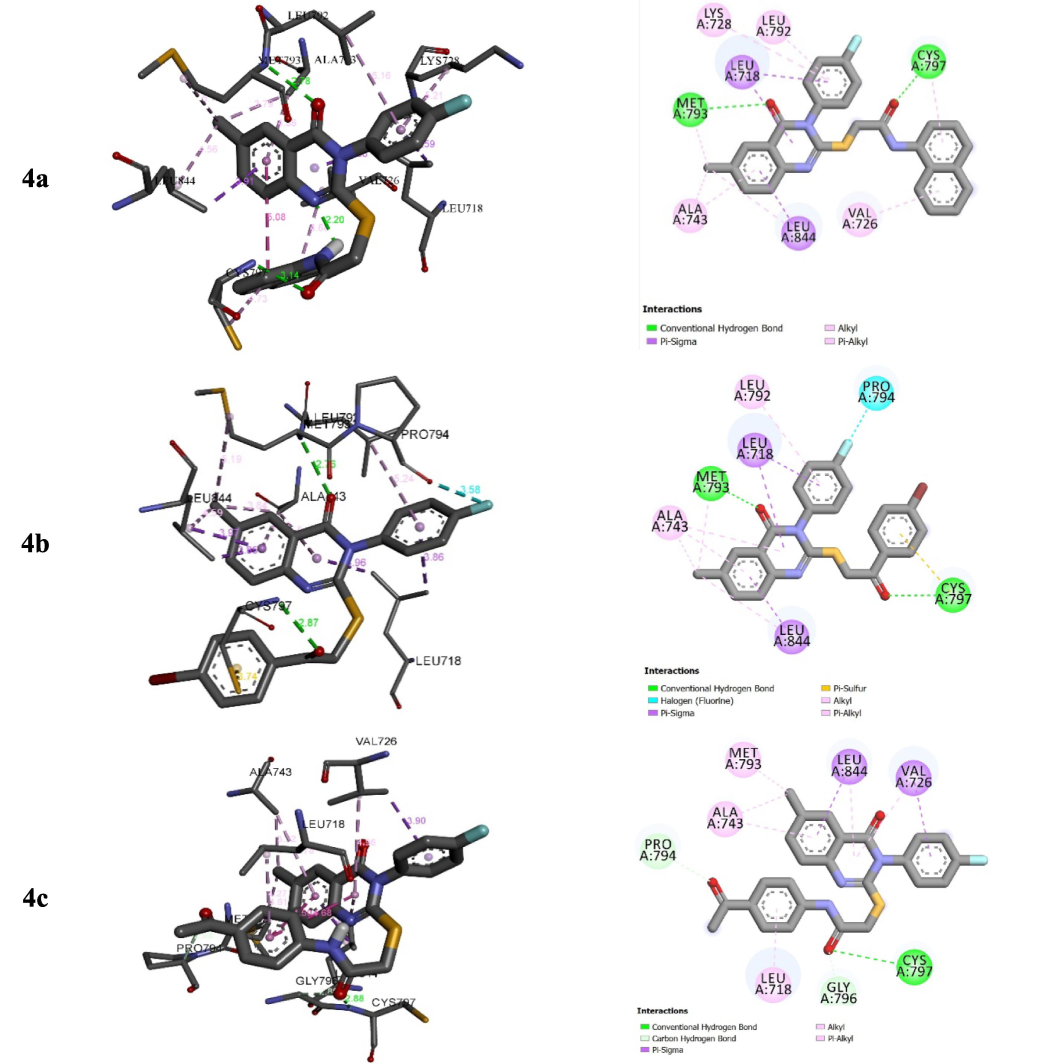
Interactions between compounds 4a–c and EGFR.
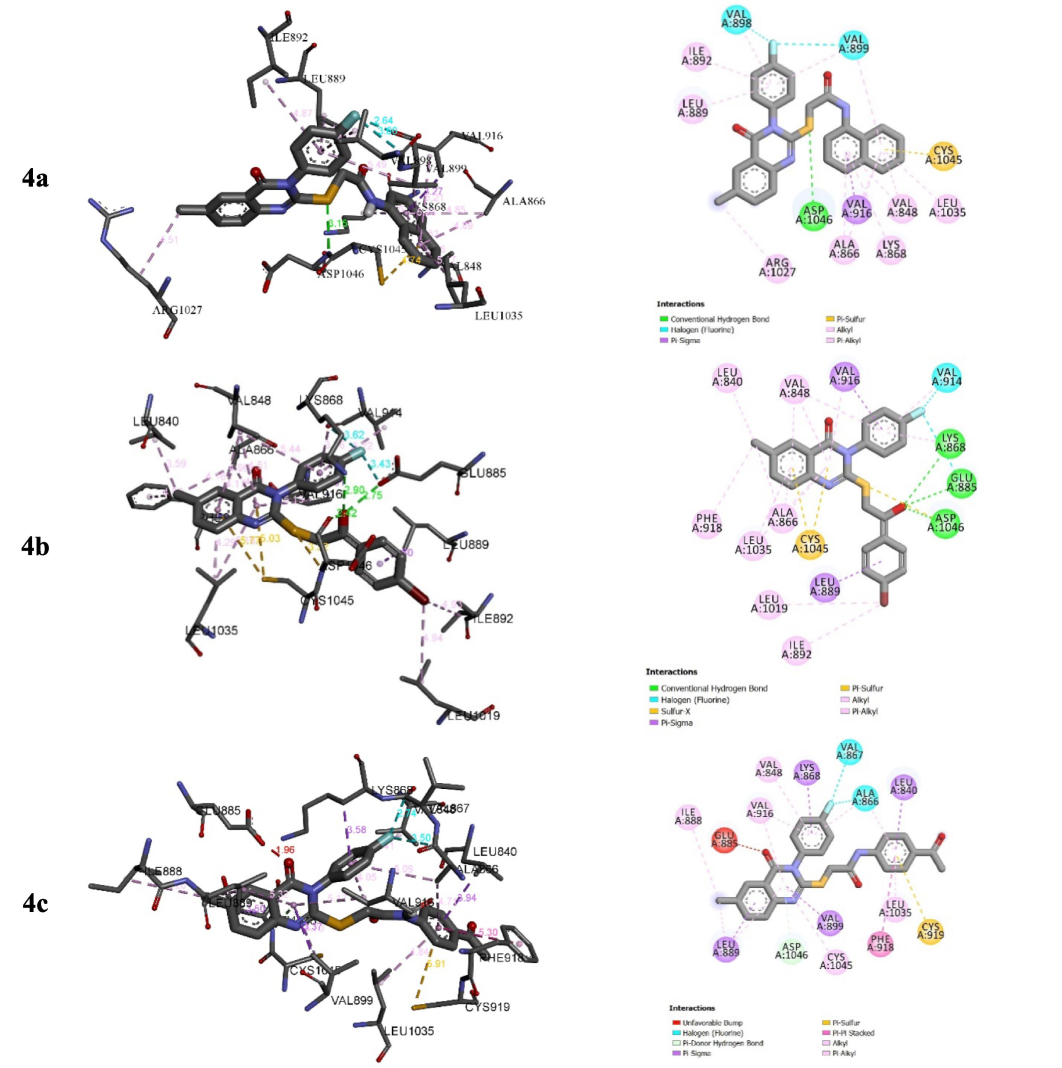
Interactions between compounds 4a–c and VEGFR-2.
For EGFR, 4c shows the highest inhibition efficiency followed by 4a and 4b with the lowest inhibition (in-vitro). The highest inhibition of 4c is in good accordance with the number of interactions that form the N-(4-acetyl phenyl)acetamide fragment of compound 4c into the binding site of EGFR tyrosine kinase compared to the ones established with N-(naphthalene-1-yl)acetylamide substituted group of 4a and the substituted group 1-(4-bromophenyl)ethan-1-one of 4b (Fig. 4). Indeed, N-(4-acetylphenyl)acetamide fragment of compound 4c interacts with CYS A79 via hydrogen bond of GLY A796 and PRO A794 via carbon-hydrogen bond, and LEU A78 via π-σ interaction (Fig. 5). For VEGFR2 tyrosine kinase inhibition, the high inhibition efficiency of 4c and 4a compared to 4b may return to the π-sulphur interactions that formed with groups N-(4-acetylphenyl)acetamide and N-(naphthalen-1-yl)acetamide substituted groups in derivatives 4c and 4a, respectively (Fig. 5).
9 Conclusion
Four novel quinazoline derivatives were synthesised as dual VEGFR-2/EGFR inhibitors. Newly synthesised quinazolines were structurally characterised by NMR analyses. The cytotoxicity test showed that compound 4b demonstrated the highest efficacy against A-549 cells, with an IC50 value of 0.49 ± 0.01 µM. Based on their cytotoxic performance, the inhibitory activity of products 4a–c against EGFR and VEGFR-2 was assessed in subsequent evaluations. Notably, product 4c proved to be the most potent, exhibiting an IC50 of 62.3 ± 1.47 nM against EGFR and 90.1 ± 2.69 nM against VEGFR-2. Studies involving molecular docking of product 4c uncovered substantial interactions inside the active sites of VEGFR-2 and EGFR offering insights into its remarkable anticancer efficacy.
CRediT authorship contribution statement
Mohammed H. Geesi: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Software, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization.
Funding
“This study is supported via funding from Prince Sattam bin Abdulaziz University project number (PSAU/2024/R/1446) ”
Acknowledgement
“This study is supported via funding from Prince Sattam bin Abdulaziz University project number (PSAU/2024/R/1446) ”
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Design, synthesis, molecular docking and cytotoxic evaluation of novel 2-furybenzimidazoles as VEGFR-2 inhibitors. Eur. J. Med. Chem.. 2017;136:315-329.
- [CrossRef] [Google Scholar]
- A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem.. 2020;99:103811
- [CrossRef] [Google Scholar]
- Soerjomataram I., The ever-increasing importance of cancer as a leading cause of premature death worldwide. CancerCancer. 2021;127(16):3029-3030.
- [CrossRef] [Google Scholar]
- Dual targeting of the vascular endothelial growth factor and epidermal growth factor receptor pathways: rationale and clinical applications for non-small-cell lung cancer. Clin. Lung. Cancer.. 2007;8:S79-S85.
- [CrossRef] [Google Scholar]
- Forty-year journey of angiogenesis translational research. Sci. Transl. Med.. 2011;3:114rv113.
- [CrossRef] [Google Scholar]
- Angiogenesis inhibitors: current strategies and future prospects. CA Cancer J. Clin.. 2010;60(4):222-243.
- [CrossRef] [Google Scholar]
- Multitarget pharmacology of sulfur-nitrogen heterocycles: anticancer and antioxidant perspectives. Antioxidants. 2024;13(8):898.
- [CrossRef] [Google Scholar]
- Design, synthesis, molecular docking and in silico ADMET profile of pyrano [2,3-d] pyrimidine derivatives as antimicrobial and anticancer agents. Bioorg. Chem.. 2021;115:105186
- [CrossRef] [Google Scholar]
- Frisch, M J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Petersson G.A., Nakatsuji H., Li X., Caricato M., Marenich A.V., Bloino J., Janesko B.G., Gomperts R., Mennucci B., Hratchian H.P, Ortiz J.V., Izmaylov A.F., Sonnenberg J.L., Williams., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V.G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J.r.. J A., Peralta J.E., Ogliaro F., Bearpark M.J., Heyd J.J., Brothers E.N., Kudin K.N., Staroverov V.N., Keith T.A., Kobayashi R., Normand J., Raghavachari K., Rendell A.P., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Millam J.M., Klene M., Adamo C., Cammi R., Ochterski J.W., Martin R.L., Morokuma K., Farkas O., Foresman J.B., Fox D.J., Gaussian 16 Rev. C.01, Wallingford, CT, 2016.
- Synthesis, antibacterial evaluation, Crystal Structure and Hirshfeld surface analysis of a new 2-Benzylsulfanyl-3-(4-fluoro-phenyl)-6-methyl-3H-quinazolin-4-one. J. Mol. Str.. 2020;1208:127894
- [CrossRef] [Google Scholar]
- Synthesis, antibacterial evaluation, raman, crystal structure and hirshfeld surface analysis of a new 3-(4-fluorophenyl)-6-methyl-2-(propylthio) quinazolin-4 (3H)-one. J. Mol. Str.. 2020;1215:128265
- [CrossRef] [Google Scholar]
- VEGFR-2 inhibitors and apoptosis inducers: synthesis and molecular design of new benzo[g]quinazolin bearing benzenesulfonamide moiety. J. Enzyme. Inhib. Med. Chem.. 2017;32:893-907.
- [CrossRef] [Google Scholar]
- Dual EGFR/HER2 inhibitors and apoptosis inducers: New benzo-quinazoline derivatives bearing benzenesulfonamide as anticancer and radiosensitizers. Bioorg. Chem.. 2018;80:611-620.
- [CrossRef] [Google Scholar]
- The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer. 2004;4(5):361-370.
- [CrossRef] [Google Scholar]
- Nitrogen containing heterocycles as anticancer agents: a medicinal chemistry perspective. Pharmaceuticals. 2023;16(2):299.
- [CrossRef] [Google Scholar]
- Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur. J. Med. Chem.. 2010;45:5420-5427.
- [CrossRef] [Google Scholar]
- Epidermal growth factor receptor inhibitors: a patent review (2010–present) Expert Opin. Ther. Pat.. 2014;24(3):309-321.
- [CrossRef] [Google Scholar]
- Liu, Y.i., Gray, N.S., 2006. Rational design of inhibitors that bind to inactive kinase conformations, Nat. Chem. Biol. 2 (7), 358-364.doi: 10.1038/nchembio799.
- EGFR-TKIs resistance via EGFRindependent signaling pathways. Mol. Cancer.. 2018;17:1-9.
- [CrossRef] [Google Scholar]
- Heterocyclic anticancer compounds: recent advances and the paradigm shift towards the use of nanomedicine’s tool box. Molecules. 2015;20(9):16852-16891.
- [CrossRef] [Google Scholar]
- McTigue M, Murray B W, Chen J H, Deng Y-L, Solowiej J, Kania R S., 2012. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proceedings of the National Academy of Sciences. 109:18281-18289. doi: 10.1073/pnas.1207759109.
- Inhibitory potential of nitrogen, oxygen and sulfur containing heterocyclic scaffolds against acetylcholinesterase and butyrylcholinesterase. RSC Adv.. 2022;12(31):19764-19855.
- [CrossRef] [Google Scholar]
- PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J. Clin. Investig.. 2002;110:923-932.
- [CrossRef] [Google Scholar]
- PPARγ as a therapeutic target for tumor angiogenesis and metastasis. Cancer Biol. Ther.. 2005;4(7):687-693.
- [CrossRef] [Google Scholar]
- Multitarget Pharmacology of Sulfur-Nitrogen Heterocycles: Anticancer and Antioxidant Perspectives. Antioxidants. 2024;13(8):898.
- [CrossRef] [Google Scholar]
- Deciphering the key heterocyclic scaffolds in targeting microtubules, kinases and carbonic anhydrases for cancer drug development. Pharmacol. Ther.. 2021;225:107860
- [CrossRef] [Google Scholar]
- Design, characterization, and DFT exploration of new quinazoline-N-substituted analogs: Anti-cancer activity and molecular docking insights. J. Mol. Str.. 2024;140420
- [CrossRef] [Google Scholar]
- Structure-based approach for the discovery of pyrrolo [3, 2-d] pyrimidine-based EGFR T790M/L858R mutant inhibitors. ACS Med. Chem. Lett.. 2013;4(2):201-205.
- [CrossRef] [Google Scholar]
- Novel sulfonamide benzoquinazolinones as dual EGFR/HER2 inhibitors, apoptosis inducers and radiosensitizers. J. Enzyme Inhib. Med. Chem.. 2019;34(1):1030-1040.
- [CrossRef] [Google Scholar]
- Carbon-13 nuclear magnetic resonance spectra of organic sulfur compounds. Substituent chemical shift (scs) effects in the 4-thiazoline-2-thione series. Can. J. Chem.. 1976;54(10):1660-1664.
- [CrossRef] [Google Scholar]
- Evidence-based best practices for EGFR T790M testing in lung cancer in Canada. Curr. Oncol.. 2018;25(2):163-169.
- [CrossRef] [Google Scholar]
- New isoxazoline-linked 1,3,4-thiadiazole derivatives: synthesis, antiproliferative activity, molecular docking, molecular dynamics and DFT. J. Mole. Str.. 2025;1319:139368
- [CrossRef] [Google Scholar]
- Quantum mechanical continuum solvation models. Chem. Rev.. 2005;105:2999-3094.
- [CrossRef] [Google Scholar]
- Traxler, P., Furet, P., 1999. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors, Pharmacol. Ther. 82, 195-206. doi: 10.1016/s0163-7258(98)00044-8.
- Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc.. 1990;112:8251-8260.
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of quinoline amide derivatives as novel VEGFR-2 inhibitors. Bioorg. Med. Chem. Lett.. 2010;20(22):6653-6656.
- [CrossRef] [Google Scholar]
- Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J. Stru. Bio.. 2015;192:539-544.
- [CrossRef] [Google Scholar]
- Afatinib-new therapy option for EGFR-mutant lung cancer. Nat. Rev. Clin. Oncol.. 2013;10(10):551-552.
- [CrossRef] [Google Scholar]
- Antiproliferative, antiangiogenic and apoptotic effect of new hybrids of quinazoline-4(3H)-ones and sulfachloropyridazine. Eur. J. Med. Chem.. 2023;245:114912
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2024.103518.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




