

Translate this page into:
Next generation sequencing shows diversity of Omicron sub-lineages of SARS-COV2 circulating in Jeddah, Saudi Arabia
⁎Corresponding authors at: Centre of Excellence in Bionanoscience Research, King Abdulaziz University, Jeddah, Saudi Arabia. ammm@kau.edu.sa (Irfan A. Rather), jsabir@kau.edu.sa (Jamal S.M. Sabir)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The ever-evolving Omicron variant of the SARS-CoV-2 and its sub-lineages have prompted Saudi Arabia to continuously track circulating lineages. We focused on the presence of diverse SARS-CoV-2 circulation in Saudi Arabia and presented the whole genome sequencing study of 94 positive SARS-CoV-2 specimens procured between February and April 2022 in the city of Jeddah, Saudi Arabia. Following whole-genome sequencing, bioinformatics analysis was undertaken. The SARS-CoV-2 variant Omicron clades 21K and 21L constituted the entirety of sequenced specimens, belonging to BA.2 (n = 56) and BA.1.1 (n = 20), respectively, and low-frequency sub-lineages were BA.2.3 (n = 6), BA.1 (n = 4), BA.2.40.1 (n = 2), BA.1.14 (n = 1), BA.2.10 (n = 1), BA2.32 (n = 1), BA.2.57 (n = 1), BA2.64 (n = 1), and BA2.5 (n = 1). Mutational patterns were identified, as well as possible consequences for the spread of the virus. Comparative molecular docking of Omicron-specific Nucleocapsid protein harboring the mutations P13L, R203K, G204R, as well as S413R, and the deletions E31-, R32-, and S33- showed reduced interaction with human RIG-I protein with 8 interacting amino acid residues and 10 polar interactions, while the SARS-CoV-2 Nucleocapsid protein exhibited 15 interacting amino acid residues and 26 polar interactions. Ongoing monitoring is essential for assessing the genomic epidemiological consequences of tourist travel and pilgrimage in Jeddah and across Saudi Arabia, as well as the prompt identification of emerging variants for further investigation.
Keywords
SARS-CoV-2
Genome
Jeddah
Mutation

1 Introduction
Coronavirus disease 2019 (COVID-19) was recognized as a worldwide pandemic in early 2020 (Alzahrani et al., 2022; Nguyen et al., 2022; Rather et al., 2022; Zhu et al., 2020). The new coronavirus strain was discovered in late 2019 in central China. The patients had an unidentified form of respiratory viral infection. Using cell cultures, molecular detection tools and whole genome sequencing led to the isolation of a virus from the betacoronavirus genus indicating its close relatedness to other coronaviruses that cause severe respiratory infection, such as middle east respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV), (Muñoz-Fontela et al., 2020). The occurrence of the SARS epidemic resulted in a different version of beta-coronavirus that was first identified in Southern China in late 2002. By the time outbreak was contained in 2004, more than 8000 cases of SARS were reported in China and 774 deaths with a 7 % case-fatality rate (Anderson et al., 2004). Less than a decade later in 2012, another version of coronavirus MERS was first reported in Saudi Arabia. The outbreak of 2014–2016 resulted in 667 cases of MERS and a mortality rate of 32.97 % (Al-Omari et al., 2019).
The current COVID-19 pandemic caused by SARS-CoV-2 dwarfs both previous coronavirus outbreaks in terms of the scale of spread, number of infected cases, number of fatalities, and severe disruption of economic activity and daily life (Anderson et al., 2020). Initially, a major bottleneck in identifying infected patients (both symptomatic and non-symptomatic) was reliably scalable and widespread testing, however, that hurdle was overcome with widespread testing using validated methods (Ravi et al., 2020). Another critical milestone was developing, approving, and administering vaccines against SARS-CoV-2 (Creech et al., 2021).
Since SARS-CoV-2 first appeared, consecutive variants of concern (VOC) facilitated rapid spread globally. A VOC is defined as a variation that increases transmission, severity, or changes in illness presentation, or that reduces the potency of vaccines, diagnostic modalities, and therapeutic approaches. Lengthy infection and concurrent infections with certain SARS-CoV-2 variants may result in genomic recombination, which is a crucial driver in the continuing development of SARS-CoV-2 variants. Uniquely to the Omicron variant, the spike region of its genome contained 35 mutations leading to 30 instances of amino acid changes, three in-frame deletions, and an insertion of three amino acids at position 214 (ins214EPE). Of particular importance is that 15 of these changes are located in the RBD region. Furthermore, the nucleocapsid protein had 6 mutations while the membrane protein exhibited 3 mutations (Shrestha et al., 2022).
While the root of the Omicron is still unknown. Phylogenetic analyses of worldwide SARS-CoV-2 genomes have not shown any near transition sequences connecting Omicron and its preceding variants; thus, the question of how Omicron evolved is still subject to speculation. Interestingly, several evolutionary analyses could not uncover any unique mutational profile spectrum or frameshifting episode that would indicate its descent from earlier variants of SARS-CoV-2. A complex evolutionary history may be inferred by the presence of the unusually lengthy branch of the Omicron lineage in the time-calibrated phylogenetic tree (Fig. 1). This has led to speculation that it may have evolved in an animal host followed by an episode of zoonotic spread from the supposed host to humans. A study has found that Omicron has 5 mutations that are mouse-adapted, even though the initial SARS-CoV-2 was not compatible with mice (Wei et al., 2021). Significant overlap existed between alterations in the Omicron spike protein and earlier variants increasing the affinity of spike protein binding with the mouse cellular entrance receptor, thus showing adaptation to mice. Another structural biology study further corroborated the hypothesis since it demonstrated that the Omicron RBD region mutations are structurally adapted to ACE2 (Zhang et al., 2022).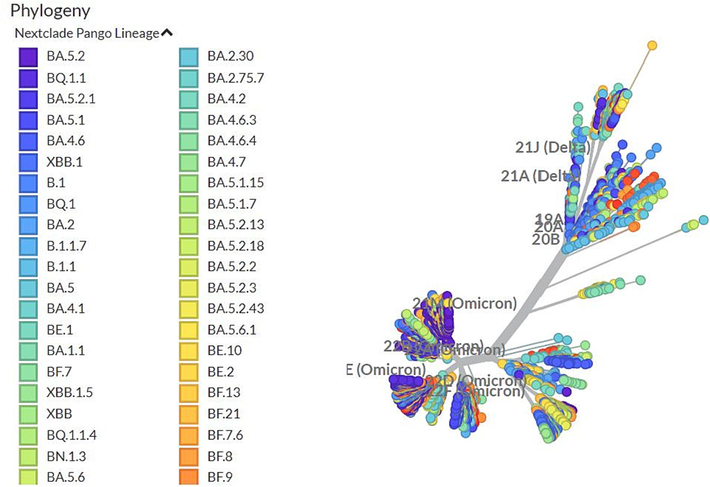
A time-stamped worldwide representative sample of 2816 SARS-CoV2 genomes sampled between Dec 2019 and Feb 2023 shown in a phylogenetic tree, with the rounded tips colored as per Nextstrain clades that pertain largely to variations of concern. The Omicron branches away from previous variants.
The onset of numerous consecutive variants presents the greatest threat to pandemic containment. As a result, strong comprehensive monitoring programs are required to uncover variations that may increase infectivity, treatment efficacy, and vaccination effectiveness. (Shrestha et al., 2022).
Jeddah city in Saudi Arabia holds a unique and crucial position in infectious disease surveillance. Its significance stems from a confluence of factors that make it a potential hotspot for disease outbreaks and a critical node in global disease transmission networks. It serves as the primary gateway for millions of pilgrims annually during the Hajj and Umrah pilgrimages, creating an ideal environment for the introduction and spread of infectious diseases. The city's status as a major commercial hub, with its international airport and seaport, facilitates the movement of goods and people worldwide, thereby increasing the risk of importing and exporting infectious diseases. Despite these challenges, Jeddah boasts a well-developed healthcare infrastructure, including hospitals, primary care centers, and specialized laboratories, providing the foundation for effective disease surveillance and outbreak response. The city's proximity to major research institutions and universities fosters collaboration and knowledge exchange in infectious disease epidemiology. Given these factors, Jeddah city plays a pivotal role in global infectious disease surveillance. The city's strategic location, high population turnover, and diverse population make it a critical node for detecting and responding to emerging infectious disease threats. Therefore, strengthening surveillance systems, promoting public health education, and enhancing collaboration between local, national, and international agencies are essential to safeguard Jeddah's population and protect global health security. (Badur et al., 2022).
2 Material and methods
2.1 Sample collection
Procurement of samples for whole genome sequencing was conducted at Alborg Laboratories, Jeddah, Saudi Arabia between February and April 2022. The inclusion criterion was dependent on RT-PCR positive specimens for SARS-CoV-2 with a Ct value less than 30. A Sum of 96 specimens was chosen of which residual nasopharyngeal swab specimens were used for downstream viral RNA extraction. The Illumina CovidSeq sequencing kit was used for the sequencing. (Bhoyar et al., 2021).
2.2 RNA extraction and sequencing
The automated Abbott M1000 nucleic acid extraction instrument employing the QIAamp Nucleic Acid Extraction Kit (Qiagen) was used. The amplicon-based Illumina CovidSeq standard protocol was utilized. The Illumina MiSeq instrument was used to sequence libraries using the 600-cycle v3 MiSeq Reagent kit.
2.3 Genome assembly, phylogenetic clustering, classification of lineage and mutation analysis
Raw fastq paired-end reads were submitted for assembly using Exatype pipeline version 1.7.12 for NGS output SARS-CoV-2 pipeline v1.7.12 that applies quality control and mapping of read to reference genome of isolate Wuhan-Hu-1 NC_045512.2 ( https://www.sars-cov-2.exatype.com). All assembled reads were then saved in the GISAID reference database ( https://www.gisaid.org/), and corresponding accession numbers were provided in supplementary Table S1. Consensus sequences were then submitted to Nextclade web tool v2.5 ( https://clades.nextstrain.org/) for dynamic monitoring of SARS-CoV-2 evolution for phylogenetic analysis, clade, and lineage assignment (Aksamentov et al., 2021). Utilizing Nextclade v2.5 and comparing against the Wuhan-Hu-1 wild type, mutation analysis was performed. The phylogenetic tree for the sequences (n-95) was generated and visualized on Auspice v2.39.0.
2.4 3D modeling of Omicron Nucleocapsid protein
The Uniprot database (Uniprot ID: P0DTC9) was used to obtain the reference amino acid sequence. Omicron-specific mutations in the generated sequencing data were introduced to the sequence as follows amino acid substitution S413R, G204R, R203K, and P13L along with the deletions E31-, R32-, and S33-. The i-TASSER server was used for structural prediction of the Omicron Nucleocapsid (Yang and Zhang, 2015). I-TASSER is a protein structure prediction method that uses a combination of threading, fragment assembly, and refinement techniques to generate high-quality 3D models of proteins from their amino acid sequences. It first identifies structural templates from the Protein Data Bank (PDB) and then assembles fragments of known protein structures to construct a full-length model of the query protein. The assembled model is then refined using various techniques to improve its overall structure and energy. The predicted structure of the mutant with the best C-score, TM-score, and RMSD was aligned with the SARS-COV2 Nucleocapsid protein (PDB ID: 8f5d) using PyMol. Protein structural alignment using PyMOL involves loading protein structures (PDB files), selecting reference and mobile structures, performing sequence and structural alignment (align command), refining the alignment (align command), visualizing the results (color and transparency adjustments), analyzing alignment metrics (RMSD, TM-score, GDT-TS), and saving the aligned structures (PDB format) (“The PyMOL Molecular Graphics System, Version 2.0, Schrödinger, LLC.,” n.d).
2.5 Molecular docking study of the interaction of SARS-COV2 nucleocapsid protein with human RIG-I protein
The open-access server of Patchdock (Schneidman-Duhovny et al., 2005) was utilized to predict and assess the interaction between SARS-COV2 Nucleocapsid protein and human RIG-I protein. The electron microscopic structure of the SARS-COV2 Nucleocapsid protein was retrieved from the RSCB Protein Data Bank (PDB ID: 8f5d) along with the crystal structure of the human RIG-I protein (PDB ID: 2qfd). Two molecular docking studies were run in parallel; the human RIG-I protein with non-mutated Nucleocapsid protein, and RIG-I protein with the predicted structure of the mutated Nucleocapsid protein harboring Omicron-specific mutations. The best poses were determined based on weighted scores. PyMol was used to analyze docked structures to identify interacting residues of human RIG-I protein with SARS-COV2 Nucleocapsid protein, and the predicted Omicron Nucleocapsid protein respectively as well as the number of polar interactions.
2.6 Ethical approval.
This study was approved by the National Committee of Biomedical Ethics at King Abdul-Aziz City for Science and Technology, Saudi Arabia (HAP-02-J-099) on 13 December 2022.
3 Results
This study was undertaken with dual primary objectives. The initial goal involved the comprehensive whole-genome sequencing of SARS-CoV-2 positive samples (n = 96) collected within Jeddah city, aimed at identifying prevailing viral variants. Subsequently, the study sought to elucidate the potential consequences of sequence modifications within the Nucleocapsid on its interaction dynamics with the interferon signaling pathway, particularly through the intermediary of the RIG-I protein. Firstly, all samples were collected and processed in Alborg Laboratories in Jeddah in the period between the 3rd of February 2022 and the 10th of April 2022 and included 49 females (52.1 %), 34 males (36.2 %), and 11 unknown (11.7 %) ranging from 2 to 70 years old, while 7 % of samples were of unknown age (samples metadata are provided in supplementary Table S3). The sequencing output yielded 95 sequences, while one sample produced no product. Consensus sequences were further filtered based on coverage; with low coverage sequence (n = 1) at 41.6 % (Ns more than 50 %) excluded. The remaining 94 samples had an average coverage of 97.9 % and a median of 99.6 % and were submitted to the GISAID database.
Secondly, the Omicron variant of SARS-CoV-2 carries several mutations in its Nucleocapsid protein, which plays a crucial role in viral replication and immune evasion. Predictive structural modeling revealed that these mutations alter the folding of the Nucleocapsid protein, particularly in the loop regions. Additionally, molecular docking studies demonstrated that the Omicron Nucleocapsid protein interacts with the human RIG-I protein, an innate immune sensor, in a distinct manner compared to the original SARS-CoV-2 Nucleocapsid protein. These findings suggest that the mutations in the Omicron Nucleocapsid protein may have implications for its ability to evade the immune system and contribute to the observed transmissibility of the Omicron variant.
Dynamics of the Omicron Clades 21K 26.3 % and 21L 73.7 % in Jeddah are shown in Fig. 1. Results refer to a period of transition between the two major clades and their sub-lineages (Omicron BA.1, BA.1.1, and BA.1.14) and (Omicron BA.2, BA.2.10, BA.2.3, BA.2.32, BA.2.40.1, BA.2.5, BA.2.57, and BA.2.64). The surveillance shows the earliest detection of BA.2 and its sub-lineages in Saudi Arabia. Our genomic surveillance showed co-circulation of both omicron variants (BA.1 and BA.2) and their respective sub-lineages. However, BA.2 exhibited higher frequency throughout the study (Fig. 2) (see Fig. 3 and Fig. 4).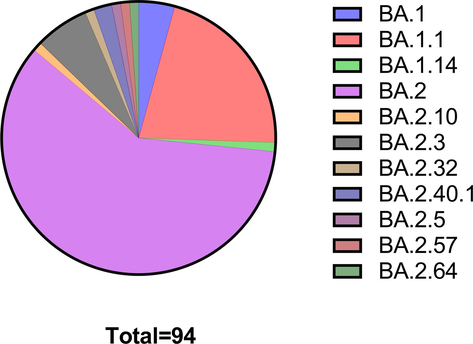
Fractional distribution of Omicron lineages and sub-lineages in the study population.
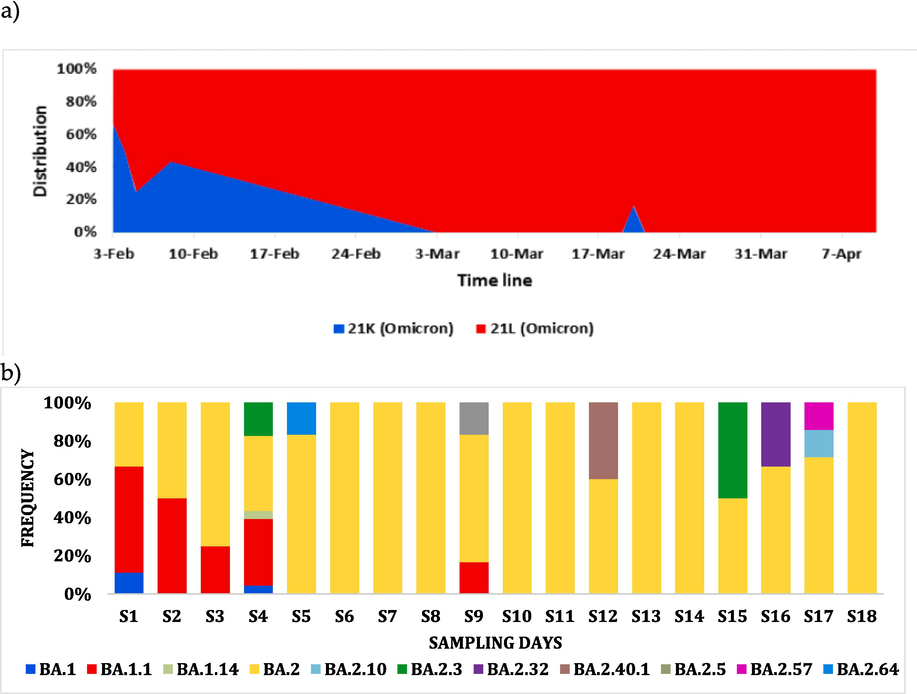
Omicron SARS-COV2 dynamics in Jeddah City describing frequency (a) and distribution (b) of SARS-COV2 Omicron sub-lineages detected in this study across time (18 sampling dates).
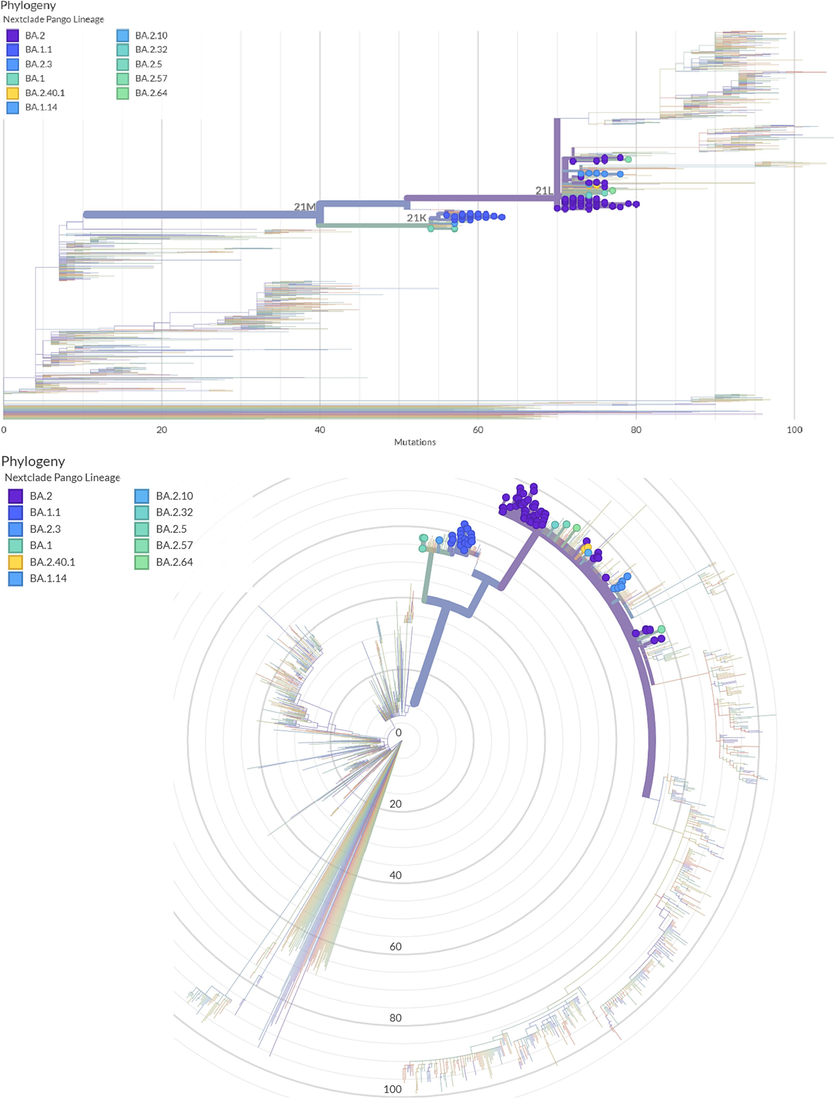
Maximum likelihood phylogenetic tree describing phylogenetic analysis for 94 SARS-COV2 sequences and the Wuhan-Hu-1 sequence. The rounded color strands around the tree indicate the PANGO lineage and the Nextstrain, representing study-generated sequences as compared with globally published SARS-COV2 sequences.
To elucidate the phylogenetic characteristics of the sequenced SARS-COV2 specimens, a Maximum Likelihood (ML) Phylogenetic tree was generated through Nextclade online tool ( https://clades.nextstrain.org/). Divergence from the Wuhan-1 reference strain, PANGO lineage, and Nextstain clade are indicated in the phylogenetic tree (Fig. 2). The highest prevalence of detected variants was from the major Omicron clade (21L); BA.2 (60 %) and other 21L occurrences were BA.2.3 (6 %), BA.2.40.1 (2 %), BA2.32 (1 %), BA.2.5 (1 %), BA.2.57 (1 %), and BA.2.64 (1 %). While the prevalence of 21K clade was: BA.1.1 (21 %), BA.1 (3 %), and BA.1.14 (1 %).
There were 284 single nucleotide alterations detected (Table S2) in which transitions constituted 76.4 % while transversions were 23.6 %. Transitions consisted of C > T (47.2 %), T > C (9.5 %), A > G (10.9 %), and G > A (8.8 %). Transversions consisted of G > T (7.4 %), and C > A (4.6 %) whereas other transversions with frequencies less than 4 % each. Those mutations lead to 148 amino acid substitutions distributed in the following genomic regions: 54 in ORF1a, 21 in ORF1b, 37 in Spike, 7 in ORF3a, 3 in ORF6, 5 in ORF7a, 1 in ORF8, 1 in ORF9b, 2 in Envelope, 5 in Membrane, and 10 in Nucleocapsid.
We also found that in addition to previously reported mutations of both 21K and 21L clades, 70 unique non-synonymous mutations were identified (Fig. 5). ORF1a constituted 39.1 %, while other genes are: ORF1b 15.2 %, S gene 5.4 %, ORF3a 3.3 %, ORF8 1.1 %, Membrane 2.2 %, Nucleocapsid 4.3 %, Envelope 1.1 %. Although the impact of these mutations is not yet known, they might be explored computationally as high-frequency mutations that can play a role in immune evasion.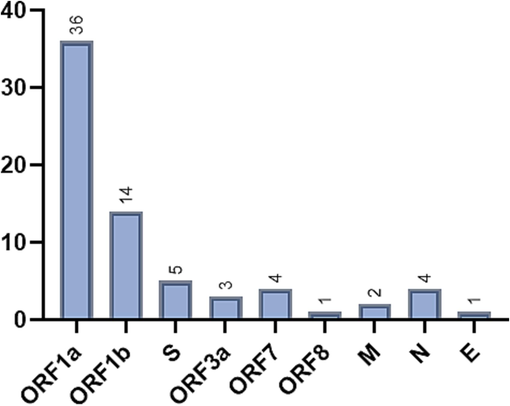
Unique non-synonymous mutations identified in 21K & 21L Omicron clades in Jeddah city.
3.1 3D modeling of Omicron Nucleocapsid protein
Predictive structural modeling of the Omicron-specific Nucleocapsid protein relied on incorporating high-frequency mutations in the sequencing output P13L (96.8 %), R203K (98.9 %), G204R (98.9 %), as well as S413R (70 %), and the deletions E31-, R32-, and S33- (100 %) were introduced to the SARS-COV2 Nucleocapsid protein amino acid sequence. The mutated sequence submitted to the i-TASSER server generated several structures, and the selected structure exhibited the best estimated TM-Score of 0.85 ± 0.08, C-Score of 0.98, and estimated RMSD of 4.8 ± 3.1 Å. The PyMol alignment showed that the two protein structures were not identical thus the Omicron alterations have impacted the folding of the Nucleocapsid protein mainly in the loop regions (Fig. 6) (see Fig. 7).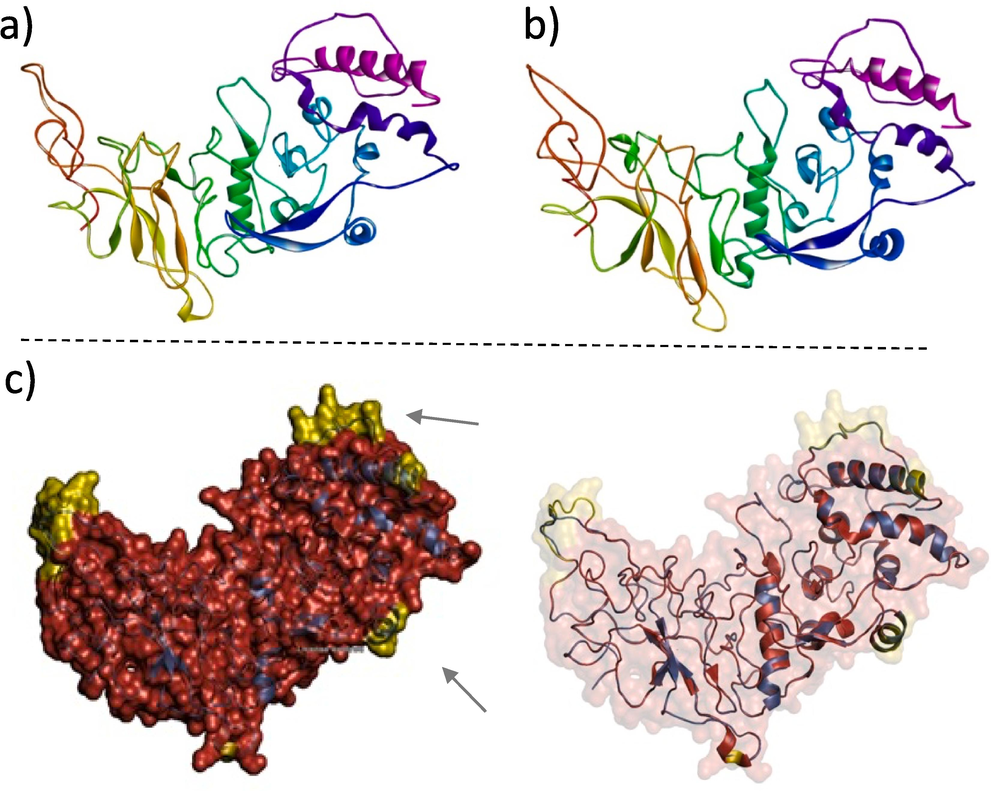
(A) EM structure of SARS-COV2 Nucleocapsid protein (PDB ID 8f5d). (B) Predicted structure of Omicron Nucleocapsid protein (modeled by i-TASSER server). (C) The PyMol alignment between the SARS-COV2 Nucleocapsid protein (red color) and the Omicron Nucleocapsid protein (modeled by I-TASSER server) (blue color) showed that the two structures were not identical. The non-aligned regions are in yellow and indicated by the arrows.
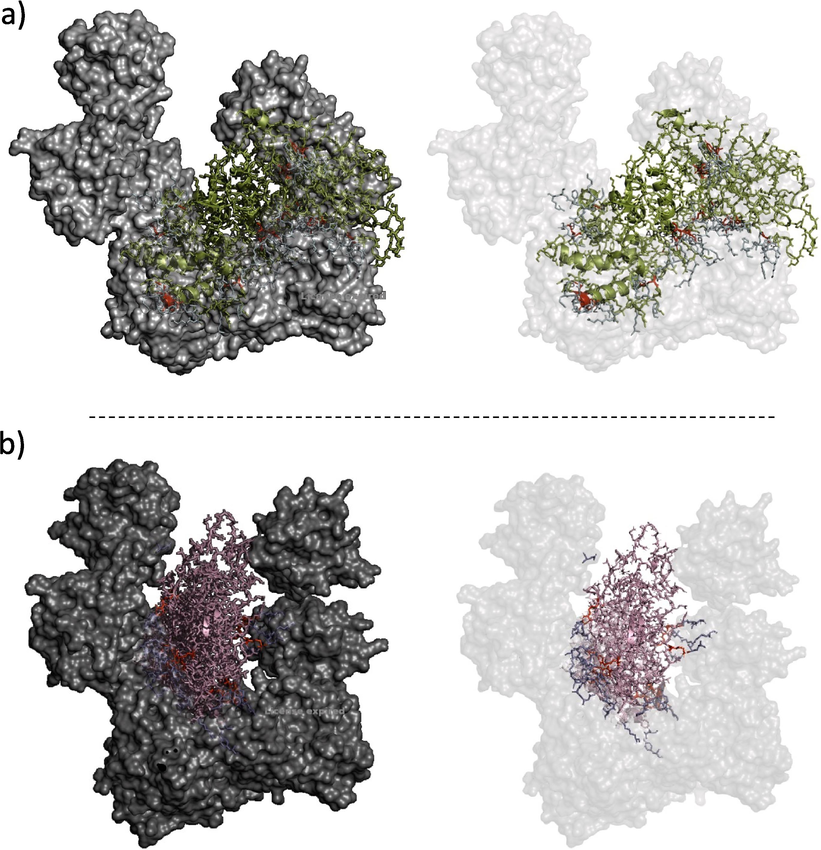
Molecular docking model of Nucleocapsid protein with human RIG-I protein as predicted by Patchdock.
3.2 Molecular docking study of interaction of SARS-COV2 nucleocapsid protein with human RIG-I protein
Patchdock was used to conduct comparative molecular docking to predict how both versions of the Nucleocapsid interacted with the human RIG-I protein that was experimentally demonstrated to interact with SARS-COV2 (Chen et al., 2020). The models exhibiting best-weightedhted scores were selected for further analysis (Table 1).
Protein
Cluster
Members
Representative
Weighted Score
SARS-COV2 Nucleocapsid
0
54
Center
−953.0
Lowest Energy
−1010.9
Omicron Nucleocapsid
0
46
Center
−875.5
Lowest Energy
−1054.1
To further analyze the docked structures, PyMol was used to visualize both interacting models. For the SARS-COV2 Nucleocapsid - RIG-I interaction a total of 15 RIG-I residues were shown to interact with SARS-COV2 Nucleocapsid with a total of 26 polar interactions. On the other hand, Omicron Nucleocapsid - RIG-I interaction showed a total of 8 RIG-I residues interacting with Omicron Nucleocapsid with a total of 10 polar interactions (Table 2).
SARS-COV2 N Protein
Omicron N Protein
RIG-I AA Residue
Number of Polar Interactions
RIG-I AA Residue
Number of Polar Interactions
Ser 26
1
Gln 300
1
Glu 136
4
Thr 260
2
Asp 63
1
Ala 261
1
Leu 64
2
Ser 191
1
Asp 81
1
Arg 192
1
Gln 70
1
Lys 230
2
Arg 191
1
Gly 233
1
Ser 193
1
Gly 235
1
Ser 194
2
Ser 413
3
Asp 415
3
Glu 366
1
Ser 416
3
Glu 367
1
Asp 415
1
Total Interactions
26
10
SARS-COV2 Nucleocapsid was found to interact with the two N-terminal domains (CARDS) (Ser 26, Asp 63, Leu 64, Gln 70, Asp 81, and Glu 136), the loop region (Arg 191, Ser 191, Ser 193, and Ser 194) and the central RNA helicase domain (Glu 366, Glu 367, Ser 413, Asp 415, and Ser 416). On the other hand, the Omicron Nucleocapsid protein was found to exhibit a lesser number of interacting RIG-I protein residues spread across the loop region (Ser 191, Arg 192, Lys 230, Gly 233, and Gly 235) between the N-terminal domains (CARDS) and the central RNA helicase domain (Thr 260, Ala 261, and Gln 300). Interestingly, the Omicron Nucleocapsid showed no interaction with the (CARDS).
4 Discussion
4.1 Mutational features of Omicron lineages
The 21K Omicron clade was designated a variant of concern since it has 62 defining non-synonymous mutations including 36 mutations of the Spike (S) gene. The 69/70 (Spike H69- and V70-) deletion in this variant causes S gene target failure in which the S-assay within the TaqPath test yields a negative result (A. Li et al., 2022) hence dubbed the “stealth” Omicron since it complicates surveillance. While the 3 amino-acid insertion in Spike of 'EPE' at position 214 was identified as an insertion hotspot in the N-terminal domain region of the Spike Protein (Gerdol et al., 2022). However, the insertion was detected in 15 out 25 Omicron 21K clade samples (BA.1 = 1 and BA.1.1 = 14).
The 21L Omicron clade shares 38 amino-acid mutations with 21K including 21 shared mutations in the S gene. While 21L Omicron lacks the defining 69/70 deletion found in 21K, it carries six additional S gene mutations, e.g., Spike: T19I, V213G, S371F, T376A, D405N, and R408S. Additionally, a 9- nucleotide deletion was reported at position 21633–21641 leading to three deletions and a substitution Spike: L24-, P25-, P26-, and A27S as described (Jung et al., 2022).
Higher affinity binding of the spike protein RBD with human ACE2 receptor has been linked to increased transmission (Zahradník et al., 2021). Considering the RBD region of the Spike (319–541), there are 14 mutations in the sequenced samples from Jeddah: Spike: G339D, S371F, S373P, S375F, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, and N501Y. Spike G339D, K440N, K493Q, and R498Q were predicted to be unfavorable for Omicron RBD binding with human ACE2 in comparison to the wild-type virus, while the A484E substitution was predicted to be favorable for the binding of Omicron RBD with human ACE2 by computational molecular dynamics simulations of ACE2 interactions with Omicron variant spike protein (Rath et al., 2022). According to Zahradnk et al. (2002), the substitutions Q498R and N501Y together have a greater affinity for the human ACE2 receptor. Additionally, a cluster of mutations at the furin cleavage site with the alterations H655Y, N679K, and P681H was linked to increased Transmission (Gong et al., 2021). R203K and G204R mutations in the Nucleocapsid (N) gene were also associated with greater virus loads. (Jung et al., 2022).
Several experimental studies have assessed the immune evasion characteristics of Omicron variants and concluded that three clusters of Spike substitutions are responsible of Omicron variants and concluded that three Spike substitutions cluster were linked with increased resistance to vaccine-induced humoral immunity and neutralizing antibodies: cluster one (S371L, S373P, and S375F), cluster two (N440K, and G446S), and cluster three (Q493, G496, Q498, and Y505H), in addition to previously characterized Spike amino acid substitutions K417N, S477N, T478K, E484A, and N501Y (Cao et al., 2022), (Cameroni et al., 2022). One of the frequently mutated genomic regions is ORF1a in which deletion of 3 amino acids at ORF1a L3674-, ORF1a S3675-, and ORF1a G3676- (corresponding to an NSP6 deletion 105–107) is speculated to facilitate escape from the innate immune response by impairing viral degradation capability of infected cells (Benvenuto et al., 2020). Subsequent investigations further demonstrated interaction/modulation of interferon signaling pathways with several SARS-COV2 proteins at multiple levels, thus SARS-CoV-2 escapes interferon suppression through interacting with pattern-recognition receptors and modulating antiviral pathways by, which in turn could lead to severe disease pathogenesis (Q. Liu et al., 2022a). Certainly, the most important characteristic of the Omicron variants is their immune evasion capability in both previously recovered COVID-19 patients as well as those vaccinated.
Further discussion of potential phenotypic consequences for high prevalence mutations (more than 65 %) in the study population is in Table 3.
Genomic Region
Amino Acid Substitution
Prevalence in Study Population
Impact of AA Substitution (Experimental and/or Computational)
ORF1a
135 S > R
72.30 %
Unknown
842 T > I
72.30 %
Unknown
1307 G > S
73.40 %
Unknown
3027 L > F
72.30 %
Unknown
3090 T > I
73.40 %
Unknown
3201 L > F
69.10 %
Unknown
3255 T > I
98.90 %
Potentially affecting NSP3/NSP5 processing as a result of its proximity to the cleavage site (Obermeyer et al., 2022)
3395P > H
97.80 %
Mutation in the coding region of protease, substitutions in this region were linked to impact transmission (Obermeyer et al., 2022)
ORF1b
314P > L
98.90 %
The P323L substitution in NSP12 corresponding to ORF1b: P314L imparts a growth advantage in combination with spike protein mutation D614G in comparison to wild type (Kannan et al., 2022)
1315 R > C
73.40 %
Unknown
1566 I > V
100 %
Unknown
S
142 G > D
72.30 %
positioned at the epitope attachment site on the N-Terminal Domain of spike protein, linked to viral adaptation with neutralizing antibodies (Shen et al., 2021)
213 V > G
70 %
led to five-fold reduction in the binding affinity of HLA-DRB1*03:01 thus contributing to immune evasion (Nersisyan et al., 2022)
339 G > D
98.90 %
Allows escape from neutralizing antibodies, affects T cell binding affinity with HLA molecules thus increasing infectivity and transmission of Omicron (Y. Li et al., 2022)
375 S > F
92.50 %
promotes decline in fusogenicity, reduction in ACE2 binding affinity, and efficacy of S cleavage (Kimura et al., 2022)
376 T > A
75.50 %
Reduced efficiency of S cleavage in BA.2 lineages in comparison to D614G (Pastorio et al., 2022)
405 D > N
77.60 %
In combination with Spike: R408S may decrease the neutralizing activity of S2A4 antibody and H014 antibody cocktail (Zhao et al., 2022)
408 R > S
77.60 %
In combination with Spike: D405N may decrease the neutralizing activity of S2A4 antibody and H014 antibody cocktail (Zhao et al., 2022)
417 K > N
88.30 %
Diminished Spike-ACE2 binding potency resulting from the lack of a salt bridge connecting K417 and D30 in ACE2, associated with the evasion of neutralizing antibodies from recuperating sera and vaccinations (Pondé, 2022)
440 N > K
70.20 %
linked to higher viral load and increased transmission (Tandel et al., 2021)
477 S > N
94.60 %
Strengthens Spike-ACE2 binding affinity (Singh et al., 2021)
478 T > K
94.60 %
Strengthens Spike-ACE2 by changing the binding free energy of RBD/ACE2 (Pondé, 2022)
484 E > A
94.60 %
It leads to decreased TMPRSS2 usage, failure of neutralization by recovered human sera, and evasion of various antibody cocktails in BA.1 and BA.2, especially when combined with H655Y. (Hu et al., 2022)
493 Q > R
94.60 %
Decreases efficacy of neutralizing antibody bamlanivimab (Guigon et al., 2022)
498 Q > R
95.70 %
Promotes favorable Spike-ACE2 interaction (da Costa et al., 2022)
501 N > Y
95.70 %
Increased fitness for replication in the upper airway due to higher affinity of spike protein with corresponding receptors (Y. Liu et al., 2022)
505 Y > H
94.60 %
Significant reduction in epitope recognition with no effect on Spike-ACE2 binding (Lin et al., 2022)
614 D > G
98.90 %
Improved viral fitness through increased replication in the upper and lower airway causing higher viral load and infectivity (Plante et al., 2021)
655H > Y
100 %
regulates the relative utilization by Omicron of the three entrance pathways (Cathepsin B/L-dependent, TMPRSS2, and metalloproteinase), and with no impact spike cleavage (Yamamoto et al., 2022)
679 N > K
100 %
Markedly more effective furin-directed cleavage of S protein at the S1/S2 position in comparison with wild type (Lubinski et al., 2022)
681P > H
100 %
Improves its cleavability by furin-like proteases, with no marked impact on membrane fusion or viral entrance (Lubinski et al., 2022)
764 N > K
96.80 %
Develops potential protease cleavage sites for serine protease SKI-1/S1P that is expressed in the upper respiratory tract but not inside the lungs (Maaroufi, 2022)
954 Q > H
100 %
In combination with N969K at the HR1 region of S cleavage site disrupts spike processing thus impairing infectivity (Pastorio et al., 2022)
969 N > K
100 %
In combination with Q954H at the HR1 region of S cleavage site disrupts spike processing thus impairing infectivity (Pastorio et al., 2022)
ORF3a
223 T > I
73.40 %
destabilizes the loop region of the protein structure at β7 - β8 pleated sheets junction (Bianchi et al., 2021)
E
9 T > I
100 %
Unknown
M
19 Q > E
91.50 %
postulated to play a part in nucleosome biogenesis, post-translational modifications, and viral assembly, and potentially contribute to immune evasion (Hossain et al., 2022)
63 A > T
98.90 %
ORF6
61 D > L
72.30 %
potential loss-of-function mutation due to its disruptive effect on ORF6 and reducing effective viral evasion from the innate immune response (Kehrer et al., 2022)
ORF9b
10P > S
96.80 %
Functions as an antagonist of Interferon (Hossain et al., 2022)
N
13P > L
96.80 %
Computationally showed diminished protein stability in comparison with wild type, and probable impact on RNA binding though it’s not fully understood (Oulas et al., 2021)
203 R > K
98.90 %
In combination with G204R it promotes Increased fitness for replication, thus enhancing infectivity and virulence. While the combination highly sensitive to neutralizing antibodies; the presence of Spike: N501Y/E484K promote immune escape (Wu et al., 2021)
204 G > R
98.90 %
In combination with R203K it promotes Increased fitness for replication, thus enhancing infectivity and virulence. While the combination highly sensitive to neutralizing antibodies; the presence of Spike: N501Y/E484K promote immune escape (Wu et al., 2021)
Based on GISAID reports, the Omicron (BA.1 & BA.1.1) were initially identified in Saudi Arabia in December 2021. We demonstrated the value of phylogenetic techniques in tracking the evolutionary changes of several lineages and sub-lineages of the Omicron variation in Jeddah, Saudi Arabia, through time in our genomic surveillance research. Our findings clearly demonstrated the change from 21K to 21L Omicron, with sub-lineages obviously co-circulating in the studied population. Whole genome sequencing for surveillance purposes can help identify mutations that may drive viral evolution and immune evasion. The binding affinity with the hACE2 receptor is still strong in spite of many alterations in the spike protein (Pascarella et al., 2022). Certainly, animal investigations of these lineages and sub-lineages are necessary to further evaluate this conjecture about phylogenetic and genomic analyses. Animal models have played a vital role in understanding the progression and variant diversity of COVID-19. Non-human primates, genetically modified mice, Syrian hamsters, and ferrets have been employed to study the virus's interaction with the host immune system, evaluate the efficacy of vaccines and therapeutics, and investigate the emergence and evolution of viral variants. Each model offers unique advantages and limitations, but collectively they have provided valuable insights into the pathogenesis of COVID-19. Animal models have allowed researchers to characterize the viral life cycle, identify key host factors involved in infection, investigate the immune response to SARS-CoV-2, evaluate the efficacy of vaccines and therapeutics, and study the emergence and evolution of viral variants. While animal models have been crucial for understanding COVID-19, it is important to recognize their limitations. No animal model perfectly replicates the human disease, and there can be significant differences in the immune response, disease progression, and susceptibility to different variants between humans and animals (Muñoz-Fontela et al., 2020).
As evident by the phylogenetic tree, over the course of our study, the Omicron lineage has evolved, and the successive variants and their sub-variants appear to become more transmissible and potentially immune-evasive. Therefore, continued monitoring programs are important for early warning of variants of concern that may have a deleterious impact on existing vaccines and population immunity. This is crucially significant in relation to Jeddah city, a major travel and religious tourism hub and a direct access point to Makkah. In addition to year-round visitors, during Hajj season almost 2 million people converge in Makkah from more than 185 countries in mass gatherings for four to five days for the annual pilgrimage which poses a critical challenge to preventing the spread and import/export of SARS-COV2 variants of concern (Badur et al., 2022).
4.2 Predicted impact of Omicron Nucleocapsid protein mutations on interaction with interferon signaling
The nucleocapsid (N) protein of SARS-CoV-2 is a versatile protein that plays a critical role in the viral life cycle. In addition to encapsidating the viral genome, the N protein interacts with various host cell proteins to manipulate the host immune response. One of the key ways in which the N protein evades the host immune response is by disrupting the interferon (IFN) signaling pathway (Bai et al., 2021).
IFN signaling is a crucial component of the innate immune response to viral infection. IFNs are a group of cytokines produced by host cells in response to viral infection. IFNs bind to IFN receptors on neighboring cells, triggering the expression of interferon-stimulated genes (ISGs). ISGs encode a variety of antiviral proteins, such as protein kinases, antiviral enzymes, and cell surface proteins that inhibit viral entry.
The N protein of SARS-CoV-2 can interfere with IFN signaling at multiple levels, including by disrupting the RIG-I pathway (Chen et al., 2020). RIG-I is a sensor protein that detects viral RNA and triggers the production of IFNs (Kawai and Akira, 2008). The N protein can bind to and inhibit RIG-I, preventing it from binding to viral RNA and becoming activated. The N protein can also interact with other proteins in the RIG-I pathway, such as MAVS and TBK1, to disrupt IFN signaling. One way in which the N protein suppresses RIG-I signaling is by binding to the DExD/H domain of RIG-I (Chen et al., 2020). The DExD/H domain is the ATPase domain of RIG-I, which is essential for RIG-I to bind to viral RNA and become activated. By binding to the DExD/H domain, the N protein prevents RIG-I from binding to viral RNA and becoming activated.
The N protein can also disrupt the interaction between RIG-I and MAVS (Chen et al., 2020). MAVS is an adaptor protein that is essential for RIG-I to signal to downstream kinases. By disrupting the interaction between RIG-I and MAVS, the N protein prevents RIG-I from signaling to downstream kinases and activating the IFN signaling cascade. In addition, the N protein can inhibit the phosphorylation of IRF3 by TBK1 (Chen et al., 2020). IRF3 is a transcription factor that is essential for IFN signaling. By inhibiting the phosphorylation of IRF3, the N protein prevents IRF3 from translocating to the nucleus and activating the transcription of IFN genes.
By disrupting the RIG-I pathway, the N protein of SARS-CoV-2 helps the virus evade the host immune response and replicate efficiently. The ability of the Nucleocapsid protein to suppress RIG-I-mediated interferon production is thought to be one of the key ways in which SARS-CoV-2 evades the host immune response. By suppressing IFN production, the Nucleocapsid protein allows the virus to replicate and spread more efficiently (Q. Liu et al., 2022b).
The human RIG-I protein is composed of three main regions: two caspase recruitment domains (CARDs) at the N-terminus, a central RNA helicase domain, and a C-terminal regulatory domain (CTD). The CARDs are protein domains that recruit and activate caspase proteases, which is an important step in the initiation of the innate immune response. The helicase domain unwinds double-stranded RNA (dsRNA), which is a critical step in the recognition of viral RNA by RIG-I. The CTD regulates the activity of RIG-I and contains phosphorylation sites that can be modified by kinases and phosphatases. This phosphorylation can either activate or repress RIG-I signaling (Kawai and Akira, 2008).
In this study, we sought to predict the impact of Omicron-specific mutations in the Nucleocapsid protein on its interaction with human RIG-I protein and to infer the potential impact on its ability to evade the immune response through suppression of IFN production.
Molecular docking showed that Omicron-specific Nucleocapsid protein exhibited reduced overall interaction with RIG-I with a total of 8 amino acid residues and 10 polar interactions in comparison to the SARS-COV2 Nucleocapsid protein which showed 15 interacting amino acid residues and a total of 26 polar interaction. Thus, it can be postulated from this model that Omicron-specific Nucleocapsid mutations P13L, R203K, G204R, as well as S413R, and the deletions E31-, R32-, and S33- have led to reduced impact on RIG-I interaction and subsequently INF production. However, this still has to be experimentally validated. While Omicron has exhibited overall increased immune evasion capability in comparison to previous SARS-COV2 variants; that can be attributed to the combination of the aforementioned mutations in the presence of Spike mutations N501Y/E484K that promote immune escape (Wu et al., 2021).
Computational methods have made significant progress in predicting protein–protein interactions (PPIs) and determining their binding energies, but they still face several limitations compared to experimental approaches. Experimental methods, such as yeast two-hybrid assays, co-immunoprecipitation, isothermal titration calorimetry, and X-ray crystallography, provide direct and accurate information about protein interactions and their binding energies. Computational methods, on the other hand, utilize various data sources and algorithms to predict PPIs and their binding energies. These methods include sequence-based methods, structure-based methods, and docking methods. Despite their advantages in terms of speed and cost-effectiveness, computational methods face limitations in accuracy, binding energy prediction, applicability, capturing dynamic interactions, and lack of experimental validation. Therefore, it is crucial to integrate computational methods with experimental approaches to gain a comprehensive understanding of protein interactions (Jessulat et al., 2011).
5 Conclusion
This genomic surveillance study reflects what happened in a limited window of time from February to April 2022 in Jeddah, Saudi Arabia. Through SARS-COV2 viral sequencing, this investigation has identified diverse lineages circulating in Jeddah between February 2022 and May 2022. The continued evolution and accumulation of non-synonymous mutations throughout the SARS-COV2 genome may play a role in increased disease severity, and immunological escape, thus posing a persistent public health challenge. These findings could serve as a foundation for future studies on the functional impact of mutations that have not been characterized and are crucial for the surveillance and development of booster vaccines. Furthermore, a predictive molecular docking study for the interaction of viral Nucleocapsid protein with human RIG-I protein indicated a shifting pattern of interaction that may be linked to enhanced immune evasion through suppression of interferon signaling pathways. Further validation of computational findings for predictive impact assessment of emerging mutations in successive variants is crucial to aid in understanding the impact on immune response and the need for updated vaccines and therapeutic agents.
Acknowledgements
This work was supported by the Deputyship for Research and Innovation, Ministry of Education, Saudi Arabia, through International Collaboration Grand Challenge Grant (Project #1095). The authors, therefore, extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number (1095).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Nextclade: clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021;6:3773.
- [CrossRef] [Google Scholar]
- MERS coronavirus outbreak: Implications for emerging viral infections. Diagn. Microbiol. Infect. Dis.. 2019;93:265-285.
- [CrossRef] [Google Scholar]
- In Vitro Evaluation of Leuconostoc mesenteroides Cell-Free-Supernatant GBUT-21 against SARS-CoV-2. Vaccines (Basel). 2022;10:1581.
- [CrossRef] [Google Scholar]
- Epidemiology, transmission dynamics and control of SARS: the 2002–2003 epidemic. Philos. Trans. R. Soc. Lond. B Biol. Sci.. 2004;359:1091-1105.
- [CrossRef] [Google Scholar]
- How will country-based mitigation measures influence the course of the COVID-19 epidemic? Lancet. 2020;395:931-934.
- [CrossRef] [Google Scholar]
- Meningococcal Disease and Immunization Activities in Hajj and Umrah Pilgrimage: a review. Infect. Dis. Ther.. 2022;11:1343-1369.
- [CrossRef] [Google Scholar]
- The SARS-CoV-2 Nucleocapsid Protein and Its Role in Viral Structure, Biological Functions, and a Potential Target for Drug or Vaccine Mitigation. Viruses. 2021;13:1115.
- [CrossRef] [Google Scholar]
- Evolutionary analysis of SARS-CoV-2: how mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J. Infect.. 2020;81:e24-e27.
- [CrossRef] [Google Scholar]
- Bhoyar, R.C., Jain, A., Sehgal, P., Divakar, M.K., Sharma, D., Imran, M., Jolly, B., Ranjan, G., Rophina, M., Sharma, S., Siwach, S., Pandhare, K., Sahoo, S., Sahoo, M., Nayak, A., Mohanty, J.N., Das, J., Bhandari, S., Mathur, S.K., Kumar, A., Sahlot, R., Rojarani, P., Lakshmi, J.V., Surekha, A., Sekhar, P.C., Mahajan, S., Masih, S., Singh, P., Kumar, V., Jose, B., Mahajan, V., Gupta, V., Gupta, R., Arumugam, P., Singh, A., Nandy, A., P. V., R., Jha, R.M., Kumari, A., Gandotra, S., Rao, V., Faruq, M., Kumar, S., Reshma G., B., Varma G., N., Roy, S.S., Sengupta, A., Chattopadhyay, S., Singhal, K., Pradhan, S., Jha, D., Naushin, S., Wadhwa, S., Tyagi, N., Poojary, M., Scaria, V., Sivasubbu, S., 2021. High throughput detection and genetic epidemiology of SARS-CoV-2 using COVIDSeq next-generation sequencing. PLoS One 16, e0247115. https://doi.org/10.1371/journal.pone.0247115.
- SARS-Cov-2 ORF3a: Mutability and function. Int. J. Biol. Macromol.. 2021;170:820-826.
- [CrossRef] [Google Scholar]
- Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature. 2022;602:664-670.
- [CrossRef] [Google Scholar]
- Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature. 2022;602:657-663.
- [CrossRef] [Google Scholar]
- SARS-CoV-2 Nucleocapsid Protein Interacts with RIG-I and Represses RIG-Mediated IFN-β Production. Viruses. 2020;13:47.
- [CrossRef] [Google Scholar]
- Assessment of mutations on RBD in the Spike protein of SARS-CoV-2 Alpha, Delta and Omicron Variants. Sci. Rep.. 2022;12:8540.
- [CrossRef] [Google Scholar]
- Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nat. Methods. 2023;20:512-522.
- [CrossRef] [Google Scholar]
- Emergence of a recurrent insertion in the N-terminal domain of the SARS-CoV-2 spike glycoprotein. Virus Res.. 2022;310:198674
- [CrossRef] [Google Scholar]
- Contribution of single mutations to selected SARS-CoV-2 emerging variants spike antigenicity. Virology. 2021;563:134-145.
- [CrossRef] [Google Scholar]
- Emergence of Q493R mutation in SARS-CoV-2 spike protein during bamlanivimab/etesevimab treatment and resistance to viral clearance. J. Infect.. 2022;84:248-288.
- [CrossRef] [Google Scholar]
- Unique mutations in SARS-CoV-2 Omicron subvariants’ non-spike proteins: Potential impacts on viral pathogenesis and host immune evasion. Microb. Pathog.. 2022;170:105699
- [CrossRef] [Google Scholar]
- Spike mutations contributing to the altered entry preference of SARS-CoV-2 omicron BA.1 and BA.2. Emerg Microbes Infect. 2022;11:2275-2287.
- [CrossRef] [Google Scholar]
- Jessulat, M., Pitre, S., Gui, Y., Hooshyar, M., Omidi, K., Samanfar, B., Tan, L.H., Alamgir, M., Green, J., Dehne, F., Golshani, A., 2011. Recent advances in protein–protein interaction prediction: experimental and computational methods. Expert Opin Drug Discov 6, 921–935. https://doi.org/10.1517/17460441.2011.603722.
- Omicron: What Makes the Latest SARS-CoV-2 Variant of Concern So Concerning? J. Virol.. 2022;96
- [CrossRef] [Google Scholar]
- Omicron SARS-CoV-2 variant: Unique features and their impact on pre-existing antibodies. J. Autoimmun.. 2022;126:102779
- [CrossRef] [Google Scholar]
- Toll-like Receptor and RIG-1-like Receptor Signaling. Ann. N. Y. Acad. Sci.. 2008;1143:1-20.
- [CrossRef] [Google Scholar]
- Kehrer, T., Cupic, A., Ye, C., Yildiz, S., Bouhhadou, M., Crossland, N.A., Barrall, E., Cohen, P., Tseng, A., Çağatay, T., Rathnasinghe, R., Flores, D., Jangra, S., Alam, F., Mena, N., Aslam, S., Saqi, A., Marin, A., Rutkowska, M., Ummadi, M.R., Pisanelli, G., Richardson, R.B., Veit, E.C., Fabius, J.M., Soucheray, M., Polacco, B.J., Evans, M.J., Swaney, D.L., Gonzalez-Reiche, A.S., Sordillo, E.M., van Bakel, H., Simon, V., Zuliani-Alvarez, L., Fontoura, B.M.A., Rosenberg, B.R., Krogan, N.J., Martinez-Sobrido, L., García-Sastre, A., Miorin, L., 2022. Impact of SARS-CoV-2 ORF6 and its variant polymorphisms on host responses and viral pathogenesis. bioRxiv 2022.10.18.512708. https://doi.org/10.1101/2022.10.18.512708.
- The SARS-CoV-2 Spike S375F Mutation Characterizes the Omicron BA.1 Variant. iScience. 2022;25:105720
- [CrossRef] [Google Scholar]
- Omicron and S-gene target failure cases in the highest COVID-19 case rate region in Canada—December 2021. J. Med. Virol.. 2022;94:1784-1786.
- [CrossRef] [Google Scholar]
- T-cell responses to SARS-CoV-2 Omicron spike epitopes with mutations after the third booster dose of an inactivated vaccine. J. Med. Virol.. 2022;94:3998-4004.
- [CrossRef] [Google Scholar]
- Characterization of SARS-CoV-2 Omicron spike RBD reveals significantly decreased stability, severe evasion of neutralizing-antibody recognition but unaffected engagement by decoy ACE2 modified for enhanced RBD binding. Signal Transduct. Target. Ther.. 2022;7:56.
- [CrossRef] [Google Scholar]
- Coronaviral Infection and Interferon Response: The Virus-Host Arms Race and COVID-19. Viruses. 2022;14:1349.
- [CrossRef] [Google Scholar]
- The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature. 2022;602:294-299.
- [CrossRef] [Google Scholar]
- Functional Evaluation of the P681H Mutation on the Proteolytic Activation of the SARS-CoV-2 Variant B.1.1.7 (alpha) Spike. iScience. 2022;25:103589
- [CrossRef] [Google Scholar]
- Maaroufi, H., 2022. The N764K and N856K mutations in SARS-CoV-2 Omicron BA.1 S protein generate potential cleavage sites for SKI-1/S1P protease. bioRxiv 2022.01.21.477298. https://doi.org/10.1101/2022.01.21.477298.
- Alterations in SARS-CoV-2 Omicron and Delta peptides presentation by HLA molecules. PeerJ. 2022;10:e13354.
- [Google Scholar]
- Role of Probiotics in the Management of COVID-19: A Computational Perspective. Nutrients. 2022;14:274.
- [CrossRef] [Google Scholar]
- Obermeyer, F., Jankowiak, M., Barkas, N., Schaffner, S.F., Pyle, J.D., Yurkovetskiy, L., Bosso, M., Park, D.J., Babadi, M., MacInnis, B.L., Luban, J., Sabeti, P.C., Lemieux, J.E., 2022. Analysis of 6.4 million SARS-CoV-2 genomes identifies mutations associated with fitness. Science (1979) 376, 1327–1332. https://doi.org/10.1126/science.abm1208.
- Generalized linear models provide a measure of virulence for specific mutations in SARS-CoV-2 strains. PLoS One. 2021;16:e0238665.
- [Google Scholar]
- Peculiar Variations of the Electrostatic Potential of Spike Protein N-terminal Domain Associated with the Emergence of Successive SARS-CoV-2 Omicron Lineages. J. Infect. 2022
- [CrossRef] [Google Scholar]
- Determinants of Spike infectivity, processing, and neutralization in SARS-CoV-2 Omicron subvariants BA.1 and BA.2. Cell Host Microbe. 2022;30:1255-1268.e5.
- [CrossRef] [Google Scholar]
- Spike mutation D614G alters SARS-CoV-2 fitness. Nature. 2021;592:116-121.
- [CrossRef] [Google Scholar]
- Physicochemical effect of the N501Y, E484K/Q, K417N/T, L452R and T478K mutations on the SARS-CoV-2 spike protein RBD and its influence on agent fitness and attributes developed by emerging variants of concern. Virology. 2022;572:44-54.
- [CrossRef] [Google Scholar]
- Scanning the RBD-ACE2 molecular interactions in the Omicron variant. Biochem. Biophys. Res. Commun.. 2022;592:18-23.
- [CrossRef] [Google Scholar]
- The Inhibition of SARS-CoV-2 and the Modulation of Inflammatory Responses by the Extract of Lactobacillus sakei Probio65. Vaccines (basel). 2022;10:2106.
- [CrossRef] [Google Scholar]
- Diagnostics for SARS-CoV-2 detection: A comprehensive review of the FDA-EUA COVID-19 testing landscape. Biosens. Bioelectron.. 2020;165:112454
- [CrossRef] [Google Scholar]
- PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res.. 2005;33:W363-W367.
- [CrossRef] [Google Scholar]
- Shen, L., Triche, T.J., Bard, J.D., Biegel, J.A., Judkins, A.R., Gai, X., 2021. Spike Protein NTD mutation G142D in SARS-CoV-2 Delta VOC lineages is associated with frequent back mutations, increased viral loads, and immune evasion. medRxiv 2021.09.12.21263475. https://doi.org/10.1101/2021.09.12.21263475.
- Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev. Med. Virol.. 2022;32
- [CrossRef] [Google Scholar]
- Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2. Sci. Rep.. 2021;11:4320.
- [CrossRef] [Google Scholar]
- Tandel, D., Gupta, D., Sah, V., Harinivas Harshan, K., 2021. N440K variant of SARS-CoV-2 has Higher Infectious Fitness. bioRxiv 2021.04.30.441434. https://doi.org/10.1101/2021.04.30.441434.
- The PyMOL Molecular Graphics System, Version 2.0, Schrödinger, LLC., n.d.
- Evidence for a mouse origin of the SARS-CoV-2 Omicron variant. J. Genet. Genomics. 2021;48:1111-1121.
- [CrossRef] [Google Scholar]
- Nucleocapsid mutations R203K/G204R increase the infectivity, fitness, and virulence of SARS-CoV-2. Cell Host Microbe. 2021;29:1788-1801.e6.
- [CrossRef] [Google Scholar]
- Yamamoto, M., Tomita, K., Hirayama, Y., Inoue, J., Kawaguchi, Y., Gohda, J., 2022. SARS-CoV-2 Omicron spike H655Y mutation is responsible for enhancement of the endosomal entry pathway and reduction of cell surface entry pathways. bioRxiv 2022.03.21.485084. https://doi.org/10.1101/2022.03.21.485084.
- I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res.. 2015;43:W174-W181.
- [CrossRef] [Google Scholar]
- SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol.. 2021;6:1188-1198.
- [CrossRef] [Google Scholar]
- Structural basis for mouse receptor recognition by SARS-CoV-2 omicron variant. In: Proceedings of the National Academy of Sciences 119. 2022.
- [CrossRef] [Google Scholar]
- Omicron SARS-CoV-2 mutations stabilize spike up-RBD conformation and lead to a non-RBM-binding monoclonal antibody escape. Nat. Commun.. 2022;13:4958.
- [CrossRef] [Google Scholar]
- A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med.. 2020;382:727-733.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2023.103081.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




