

Translate this page into:
Identification of bioactive molecules from Triphala (Ayurvedic herbal formulation) as potential inhibitors of SARS-CoV-2 main protease (Mpro) through computational investigations
⁎Corresponding authors at: Department of Pharmaceutical Chemistry, Rasiklal M. Dhariwal Institute of Pharmaceutical Education & Research, Pune 411019, Maharashtra, India (M. Rudrapal); Department of Medical Laboratory Sciences, College of Applied Medical Sciences, Majmaah University, Al Majmaah 11952, Saudi Arabia (J. Khan). rsmrpal@gmail.com (Mithun Rudrapal), j.khan@mu.edu.sa (Johra Khan),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
Severe acute respiratory syndrome coronavirus disease (SARS-CoV-2) induced coronavirus disease 2019 (COVID-19) pandemic is the present worldwide health emergency. The global scientific community faces a significant challenge in developing targeted therapies to combat the SARS-CoV-2 infection. Computational approaches have been critical for identifying potential SARS-CoV-2 inhibitors in the face of limited resources and in this time of crisis. Main protease (Mpro) is an intriguing drug target because it processes the polyproteins required for SARS-CoV-2 replication. The application of Ayurvedic knowledge from traditional Indian systems of medicine may be a promising strategy to develop potential inhibitor for different target proteins of SARS-CoV-2. With this endeavor, we docked bioactive molecules from Triphala, an Ayurvedic formulation, against Mpro followed by molecular dynamics (MD) simulation (100 ns) to investigate their inhibitory potential against SARS-CoV-2. The top four best docked molecules (terflavin A, chebulagic acid, chebulinic acid, and corilagin) were selected for MD simulation study and the results obtained were compared to native ligand X77. From docking and MD simulation studies, the selected molecules showed promising binding affinity with the formation of stable complexes at the active binding pocket of Mpro and exhibited negative binding energy during MM-PBSA calculations, indication their strong binding affinity with the target protein. The identified bioactive molecules were further analyzed for drug-likeness by Lipinski’s filter, ADMET and toxicity studies. Computational (in silico) investigations identified terflavin A, chebulagic acid, chebulinic acid, and corilagin from Triphala formulation as promising inhibitors of SARS-CoV-2 Mpro, suggesting experimental (in vitro/in vivo) studies to further explore their inhibitory mechanisms.
Keywords
SARS-CoV-2
COVID-19
Triphala
Bioactive molecules
Molecular docking
Molecular dynamics simulation

1 Introduction
Coronavirus disease 2019 (COVID-19) is a highly infectious disease with a high rate of reproducibility. Severe acute respiratory syndrome coronavirus disease-2 (SARS-CoV-2) is the causative agent for COVID-19 (Rudrapal et al., 2020). This deadly disease has infected nearly every country on the planet, with 266,946,588 confirmed cases and 5,282,046 fatalities, according to the latest reports of World Health Organization (https://covid19.who). With the majority of the world under lockdown and the alarming threat of thousands of deaths every day, rapid drug discovery for the identification of anti-COVID drugs is an urgent need of the hour. Conventional drug discovery processes could take years. To aid in the rapid discovery of new drugs, computational approaches have been effective in identifying potential drugs/medications for COVID-19 therapy (Rudrapal et al., 2021a). The use of computational algorithms such as molecular docking, molecular dynamics (MD) simulation and density functional theory (DFT) method has made the search for drugs of natural or synthetic origin that target the viral system, particularly SARS-CoV-2 main protease more feasible while causing fewer side effects with improved therapeutic performance in human populations. In this context, repurposing the traditional herbal remedies such as Ayurvedic formulations can play a significant role in the development of effective therapeutic entities. Moreover, the uses of phytochemicals over synthetic drugs are advantageous since they are safer and a higher dose is well tolerated.
Ayurvedic formulation Triphala, which literary means Tri (three) and Phala (fruit) in Sanskrit, is a 'tridoshic rasayan' that balances and rejuvenates the “tridosha”, i.e., vata, pitta, and kapha, as well as promotes immunity and health. It is a polyherbal formulation made up of a proportional amount of dried fruit powder from three plants: Phyllanthus emblica (Indian gooseberry or Amalaki), Terminalia bellirica (Bibhitaki), and Terminalia chebula (Haritaki) (Peterson et al., 2017). In the clinical practice of Ayurveda, the formulation (Triphala) is consumed as a ‘health tonic’. It is also used to treat digestive problems such as gastric acidity, constipation and poor food assimilation. Additionally, several diseases such as fever, cough, jaundice, anemia, asthma, chronic ulcers, pyorrhea and leucorrhoea are also treated with Triphala. Additionally, obesity, inflammation, fatigue, cardiovascular disorders, liver dysfunction and ophthalmic problems are some other health ailments that Triphala is being recommended for. Moreover, it demonstrates cardiotonic properties by lowering serum cholesterol levels, reducing myocardial necrosis, enhancing blood circulation, and strengthening capillaries. Flavonoids, tannins, gallic acid, pectin, vitamin C, corilagin, and phyllaemblic compounds are present in in P. emblica fruits (Habib-ur-Rehman et al., 2007). Terminalia bellirica extract contains β-sitosterol, tannins and phenolic acids such as ellagic acid, gallic acid, galloyl glucose, ethyl gallate, and chebulaginic acid (Kumar and Khurana, 2018). The main bioactive active constituents present in T. chebula are chebulic, chebulinic acid, 2,4-chebulyl-D-glucopyranose, terflavin A, terchebin, ellagic acid, gallic acid, tannic acid, ethyl gallate, arjungenin, arjunglucoside I, punicalagin and luteolin.
Recently, computational techniques are being extensively used to investigate the inhibitory potential of phytocompounds against SARS-CoV-2 (Singh et al., 2021; Sharma et al., 2021; Bhardwaj et al., 2021). Numerous studies have been conducted to determine the efficacy of traditional medicine as anti-SARS-CoV-2 agents (Verma et al., 2020; Chakraborty et al., 2021; Dutta et al., 2021). Furthermore, effective design of SARS-CoV-2 inhibitors can be accomplished through the selection of druggable targets. Main protease (Mpro) or 3-chymotrypsin-like protease 3 (CLpro) cleaves non-structural proteins (Nsp) at eleven different sites and leads to the formation of Nsp4-Nsp16, which is essential for viral replication (Yang et al., 2005). The S protein and Mpro are important targets for designing SARS-CoV-2 inhibitors (Idroes et al., 2021). No adverse toxic effect is likely to be observed by inhibiting Mpro as there are no known similar enzymes in humans with similar cleavage specificity (Khater and Nassar, 2021). Papain-like protease (PLpro) is another enzyme similar to Mpro (Khan et al., 2021a); it generates Nsp1-Nsp3 by breaking at various sites, thereby generating essential proteins that suppress the immune system by speeding up the infection process in the host cell (Harcourt et al., 2004). One of the important targets is RNA-dependent RNA polymerase (RdRp) as the viral genome depends on it for its replication and transcription (Borgio et al., 2020; Rudrapal et al., 2021b). In literature (Ahmad et al., 2021), the possible potential of Indian medicinal plants and formulations against SARS-CoV-2/COVID-19 has been mentioned based upon the recommendation (https://www.ayush.gov.in/docs/ayush-Protocol-covid-19.pdf; https://www.ayush.gov.in/docs/ayurved-guidlines.pdf) of AYUSH (Ayurveda, Yoga, Naturopathy, Unani, Siddha, Sowa-Rigpa and Homoeopathy). The usefulness of Triphala formulation and other Ayurvedic herbal remedies as therapeutic supplements in COVID-19 has also been reported in some recent literature (Ozah, 2020). Khan et al. (2021b) reported the inhibitory potential of Triphala against spike glycoprotein of SARS-CoV-2 by molecular docking. In view of these information, in the urge to design new SARS-CoV-2 inhibitors by blockage of its main protease (Mpro), we performed structure-based molecular docking studies of bioactive molecules from Triphala formulation. The selected candidates were subjected to MD simulations for confirmation of the SARS-CoV-2 inhibitory potential of bioactive molecules. Further, in silico screening of the selected compounds was conducted with the application of Lipinski’s filters to determine their drug-likeness and ADMET properties.
2 Materials and methods
2.1 Protein preparation
The crystal structure of the Mpro of SARS-CoV-2 bound with non-covalent inhibitor X77 (N-(4-tert-butylphenyl)-N-[(1R)-2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl) ethyl]-1H-imidazole-4-carboxamide) at a resolution of 2.1 Å (PDB id: 6W63) (Rudrapal et al., 2021b) was retrieved from RCSB Protein data bank (https://www.rcsb.org/). The maestro's protein preparation wizard (version 2021-2 Schrodinger suite) was used to prepare proteins by removing all water within 5 Å distance from the ligand, filling amino acid side chains, adding missing hydrogen atoms, creating disulfide bonds, and optimizing hydrogen bonds. For restrained minimization, OPLS4 force field was used until the average RMSD (root-mean-square deviation) of the non-hydrogen atom converged to 0.30 Å, allowing heavy atoms to move sufficiently to release strained bonds, angles, and clashes (Roos et al., 2019; Kumar et al., 2021).
2.2 Ligand preparation
The Triphala formulation's bioactive molecules were chosen from the literature (Prasad and Srivastava, 2020), and their chemical structures were obtained from a popular online database, PubChem (https://pubchem.ncbi.nlm.nih.gov/). LigPrep was used to generate the ligands' three-dimensional (3D) coordinates (Chen and Foloppe, 2010). Ionization state (pH 7.4), tautomer, and chirality were predicted by Epik module. The OPLS4 force field was employed for energy minimization until an energetically stable confirmation was obtained (Roos et al., 2019).
2.3 Molecular docking study
The selected set of phytoconstituents was subjected for molecular docking against Mpro with the Extra Precision (XP) protocol of the glide docking module of Maestro (Friesner et al., 2006). The glide-receptor grid generation module was used with the default settings to identify the binding site, and the centroid of the bound ligand was taken into account for grid generation (Halgren et al., 2004). The single best pose for each molecule was saved and the compounds were then classified based on their docking scores, which represent free binding energies (Othman et al., 2021).
2.4 Molecular dynamics simulations
Following that, the top four compounds with the best binding free energies, namely terflavin A, corilagin, chebulagic acid, and chebulinic acid, were selected to further ensure the flexibility and stability at the binding site of Mpro. A 100 ns MD simulation was performed using GROnin-gen MAchine for Chemical Simulation (GROMACS) 2019.2 software (Abraham et al., 2015). The topologies of the protein and ligand were determined using the ‘pdb2gmx’ script and the GlycoBioChem PRODRG2 server (http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg), respectively. Energy minimization was accomplished using the GROMOS96 54a7 force field. The complex was solvated using the SCP water model, electrically neutralized by adding sodium ions and chloride ions using ‘gmxgenion’ script, equilibrated under an 0.3 ns isothermal-isochoric (NVT) and 0.3 ns isothermal-isobaric (NPT) ensembles and subjected to MD simulations (100 ns) (Ghosh et al., 2021). The pressure and temperature of the system were maintained at 1 atm and 300 K, respectively. The leap frog algorithm was used to integrate the motion equation in triplicate at a time step of 2 fs. Finally, the protein–ligand complexes were put to generate trajectories of RMSD, RMSF, Rg and H bond by GROMACS scripts. The trajectories were analyzed and visualized by VMD 1.9.3 (Humphrey et al., 1996).
2.5 MM-PBSA calculations
The binding free energies of protein–ligand complexes were quantified by the Molecular Mechanics Poisson Boltzmann Surface Area (MM-PBSA). This technique allows us to study the biomolecular interactions between the target and bioactive molecules. The g_mmpbsa script was utilized for MM-PBSA binding free energy calculations (Kumari et al., 2014). Van der Waals, polar solvation, and electrostatic contributions to energy were calculated. Solvent accessible surface area (SASA) was used to calculate non-polar energy contribution.
2.6 Drug-likeness, ADMET and toxicity prediction
The selected bioactive molecules were further checked for drug-likeness properties using Lipinski’s rule of five (RO5) (Hussain et al., 2021; Rudrapal and Sowmya, 2019). The ADMET properties including absorption, distribution, metabolism, excretion and toxicity of the selected were determined by the admetSAR server (Cheng et al., 2012; Ghost et al., 2021). Oral in silico toxicity prediction was performed using web-based server ProTox-II (Banerjee et al., 2018).
3 Results and discussion
3.1 Molecular docking
Triphala (Ayurvedic herbal formulation) is made up of dried fruits from three different herbal plants in equal parts (Jantrapirom et al., 2021; Tarasiuk et al., 2018). Following a thorough review of the literature on the three plants T. chebula, T. bellirica, and P. emblica, 19 bioactive molecules were chosen for molecular docking against the Mpro of SARS-CoV-2. The binding pocket around the inhibitor X77 of the target protein Mpro was used as a reference for determining the grid box. Docking analysis generated many configurations, which were scored to determine the most favorable binding modes. The bioactive molecules docked against Mpro of SARS-CoV-2 exhibited excellent binding, as evidenced by high docking scores that exceeded X77. The validation of the docking method was carried out by redocking of co-crystal ligand, X77 in the original ligand binding site of Mpro. The redocking was successful. The measured RMSD value of redocking was 0.4493 Å between the experimental docking pose of X77 and the natural binding mode of the native ligand. The superimposed conformation of the experimental ligand with the original co-crytal ligand, X77 is displayed in Fig. 2. The docking results and the binding free energies of 19 compounds are given in Table 1.
Compounds
XPG Score (kcal/mol)
Glide emodel (kcal/mol)
MMGBSA ΔG Bind (kcal/mol)
Terflavin A
−14.521
−123.949
−86.36
Corilagin
−12.845
−87.203
−68.70
Chebulagic acid
−11.359
−58.079
−55.83
Chebulinic acid
−11.217
−137.224
−64.05
Phyllaemblicin B
−10.237
−85.229
−52.22
Punicalagin
−9.388
−75.623
−70.60
Bellericoside
−9.290
−65.127
−54.02
Emblicanin B
−8.416
−65.211
−54.04
Quercetin
−7.641
−52.406
−43.32
Galloyl glucose
−7.625
−56.523
−43.99
Epicatechin
−7.548
−48.274
−46.39
Phyllaemblic acid
−7.510
−48.331
−37.68
Arjunglucoside I
−7.220
−53.970
−35.40
Luteolin
−7.186
−45.854
−41.32
Ellagic acid
−7.104
−46.060
−43.04
Terchebin
−6.850
−92.637
−61.45
Belleric acid
−6.595
−45.484
−46.38
Emblicanin A
−6.502
−109.354
−55.77
β-Sitosterol
−6.277
−46.520
−51.84
Apigenin
−6.072
−44.327
−40.62
Gallic acid
−5.930
−33.277
−28.29
Methyl gallate
−5.916
−37.714
−35.54
Arjungenin
−5.862
−47.716
−40.94
Ascorbic acid
−5.445
−27.964
−21.89
Syringic acid
−5.380
−34.749
−31.81
Ethyl gallate
−4.332
−38.651
−35.41
Phyllaemblicin B
−10.237
−85.229
−52.22
X77
−5.966
−5.966
−85.045
Terflavin A achieved the highest docking score, with a value of −14,521 kcal/mol. Corilagin, chebulagic acid, chebulinic acid and X77 had XP Gscore values of −12.845 kcal/mol, −11,359 kcal/mol, −11,217 kcal/mol, and −5,966 kcal/mol, respectively. Chemical structures of the four best-docked compounds and the native ligand X77 are depicted in Fig. 1. Chebulagic acid has a lower XP Gscore value than chebulinic acid, but the binding energy value of chebulinic acid is better (−64.05 kcal/mol). Furthermore, despite having a higher XP Gscore, the dG bind value of the X77 ligand is −85,045 kcal/mol, lower than corilagin, chebulagic acid, and chebulinic acid. The interactions with Mpro of the top four docked compounds terflavin A, chebulagic acid, chebulinic acid, corilagin, including the native ligand X77 are visualized in Fig. 3.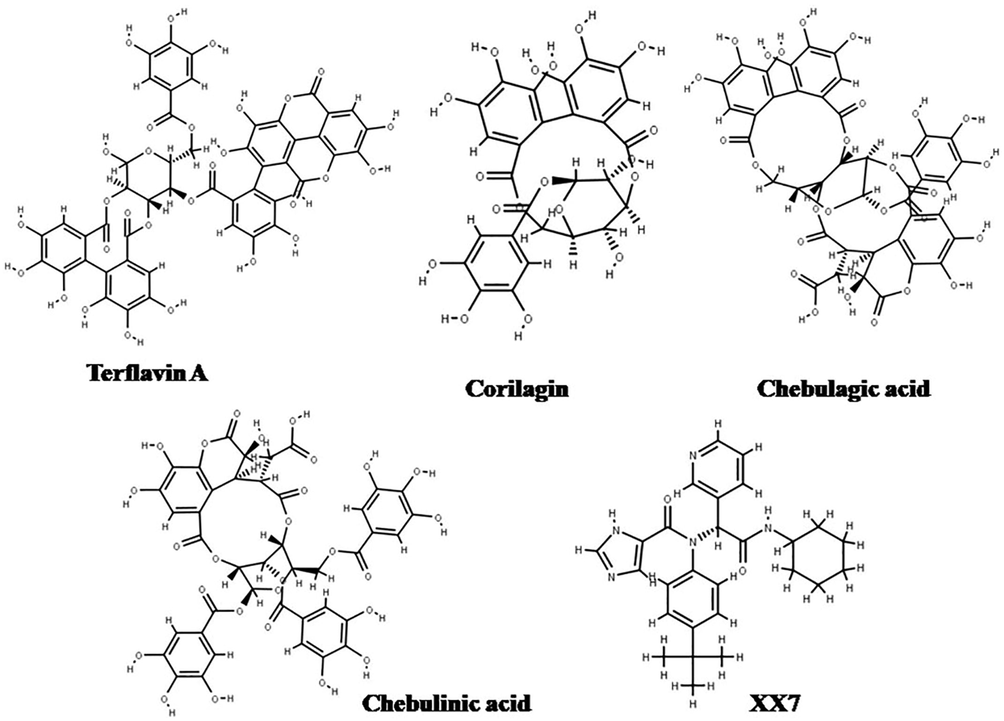
Chemical structures of best-docked compounds and native ligand X77.
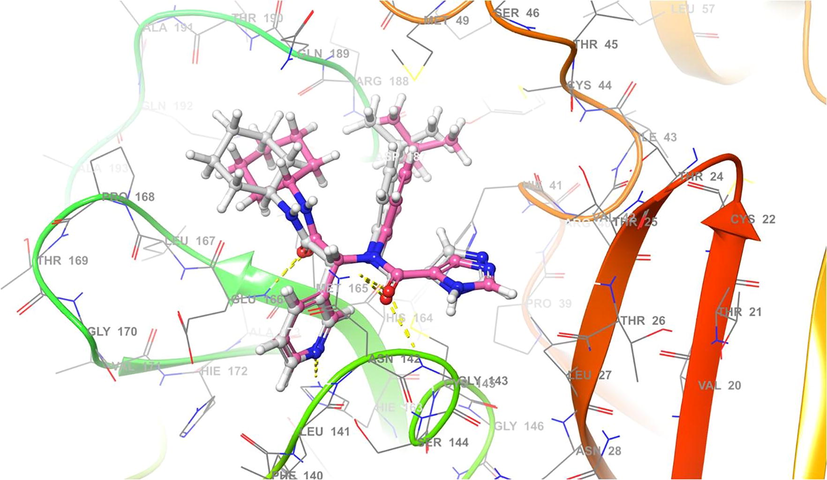
Redocking of X77 in the original binding site of Mpro. Superimposed conformation of the experimental ligand with the native ligand, X77. Gray indicates X77 docked and pink indicates X77cocrystal.
![Interaction between the ligands and main protease (Mpro) (PDB: 6W63). (a) Terflavin, (b) Chebulagic acid, (c) Chebulinic acid, (d) Corilagin, (e) X77 or N-(4-tert-butylphenyl)-N-[(1R)-2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl]-1H-imidazole-4-carboxamide.](/content/185/2022/34/3/img/10.1016_j.jksus.2022.101826-fig4.png)
Interaction between the ligands and main protease (Mpro) (PDB: 6W63). (a) Terflavin, (b) Chebulagic acid, (c) Chebulinic acid, (d) Corilagin, (e) X77 or N-(4-tert-butylphenyl)-N-[(1R)-2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl]-1H-imidazole-4-carboxamide.
Terflavin A, chebulagic acid, chebulinic acid, corilagin are types of hydrolysable tannins, which are typically found on Terminalia species leaves (L.). Terflavin A and corilagin are ellagitannin, while chebulagic acid and chebulinic acid are benzopyran tannin. Tannins can inhibit SARS-CoV-2 through binding with catalytic dyad residues of Mpro (Chang et al., 2019; Das et al., 2020). Tannic acid even inhibits both Mpro and TMPRSS2 protease, effectively suppressing SARS-CoV-2 (Wang et al., 2020). The catalytic residues of 3CLpro (or Mpro in the case of SARS-CoV-2) are Cys145 and His41, which are surrounded by other residues that confer substrate specificity (Rudrapal et al., 2021b; Singh et al., 2020).
Terflavin A interacts with Mpro on its active site by H-bonds at residues such as Asn142 (2.10 Å), Gly143 (2.13 Å), Ser144 (2.50 Å), Cys145 (2.25 Å), Glu166 (1.84 Å), Arg188 (2.13 Å), Gln189 (2.13 Å), and Thr190 (2.50 Å) and then H-carbon bonds at Met165 (1.66 Å) and Pro168 (1.74 Å) and pi-sulphur interactions at Cys44 (2.14 Å) and Met49 (2.43 Å) residues. Terflavin A exhibits H-bond with Cys145 (1.96 Å), but has no interaction with His41. Chebulagic acid forms H-bonds with residues Asn142 (1.67), Gly143 (2.18 Å), Glu166 (2.01 Å), Pro168 (2.97 Å), Arg188 (2.16 Å), and Gln189 (1.67 Å), H-carbon bonds in Met49 (1.90 Å) and Asp187 (1.61 Å) and pi-alkyl interactions in Met165 (1.72 Å). While the binding of free energy value is high, chebulagic acid does not interact with Cys145 or His41. Chebulinic acid forms H-bonds on the active site, namely residues Thr26 (1.89 Å), Phe140 (2.07 Å), Asn142 (2.18 Å), Cys145 (2.16 Å), Glu166 (2.26 Å), Gln189 (2.27 Å) and Thr190 (1.52 Å) as well as pi-alkyl, pi-pi stacked and pi-sigma interactions on Pro168 (2.04 Å), His41 (2.19 Å), and Gln189 (2.67 Å), respectively. Chebulinic acid, like Terflavin A, has only one H-bond with Cys145 (2.08 Å) and no interaction with His41. Corilagin forms H-bonds at residues Thr25 (1.87 Å), His41 (2.11 Å), Asn142 (1.03 Å), Cys145 (1.66 Å), and Glu166 (1.78 Å) then H-carbon bonds at Met165 (2.06 Å) and pi-alkyl and pi-sulphur interactions at residues Met49 (2.13 Å) and Cys145 (2.12 Å), respectively. As evidenced, H-bond interactions exist between corilagin and Cys145 (1.72 Å) and His41 (1.08 Å). As a native ligand, the X77 forms H-bonds at residues Asn142 (2.16 Å), Gly143 (1.89 Å), His163 (1.67 Å), and Glu166 (2.05 Å), then H-carbon bonds at Thr26 (2.10 Å) and Met165 (2.90) and pi-pi stacked and pi-sulphur interactions at residues His41 (2.17 Å), Met49 (2.04 Å), and Cys145 (1.14 Å), respectively. These findings support previous findings that the selected compounds bind to the catalytic residues of Mpro (Silva et al., 2021). The substrate-binding cleft contains a catalytic dyad composed of His41 and Cys145, which is located between domain I and II at the N-terminus, and a hydrogen-bonded water molecule bound to His41 acts as a third component of the catalytic triad (Tan et al., 2005).
3.2 Molecular dynamics simulation
MD simulations and other related techniques are on the verge to act as routine techniques for the discovery of drugs (De Vivo et al., 2016). In this study, the stability and dynamic behavior of the four ligand–protein (bound) complexes were verified by MD simulations for a 100 ns time. The RMSD, root mean square fluctuation (RMSF), radius of gyration (Rg) and solvent accessible surface area (SASA) graphs were plotted of the apoprotein, co-crystal ligand complex and four ligand–protein complexes (Junejo et al., 2021). The RMSD analysis provides information insight into the structural deviation and conformational stability and convergence during the time of the simulation. Thereby, in an equilibrated, stable system, the RMSD towards the end of MD simulations should be within 1–3 Å for a globular protein. The time-dependent RMSD graph was plotted for the backbone of the apoprotein, co-crystal ligand–protein complex and ligand (terflavin A, corilagin, chebulagic acid, chebulinic acid) protein (Mpro) complex. Fig. 4 (A) shows the RMSD values of apoprotein and co-crystal ligand–protein complex fluctuate in the beginning from 0.2 nm and at the end at around ∼0.3 nm, confirming the stability of the protein during 100 ns of simulation. At first, the RMSD values of the apoprotein and the protein–ligand complex are different, but after 50 nm, the RMSD values become stable and remain stable until 100 ns. Fig. 4 (B) shows the RMSD values of protein (Mpro) and ligand (terflavin A, corilagin, chebulagic acid, chebulinic acid) complex, indicated with colour code including Mpro-TerA (green), Mpro-Cor (pink), Mpro-Cgic (cyan), and Mpro-Cnic (indigo). The RMSD of Mpro-TerA fluctuates at the beginning until 70 ns (0.2 nm to ∼ 0.6 nm) and at 100 ns, it fluctuates at around ∼0.3 nm. The RMSD values of both the Protein-Corilagin (Mpro-Cor) and Protein-Chebulagic acid (Mpro-Cgic) complex are stable from the beginning (0.2 nm), and stay stable until 100 nm (0.25 nm), confirming the stability of both protein–ligand complexes during 100 ns of simulation. Whereas, Protein-Chebulinic acid complex fluctuats (between 0.2 nm and 0.3 nm) up to 70 ns, but after 70 nm, it becomes stable and stays stable (0.2 nm) until 100 nm. The overall RMSD results reveal that the binding of the ligands (corilagin, chebulagic acid, chebulinic acid) in the active site of Mpro is stable.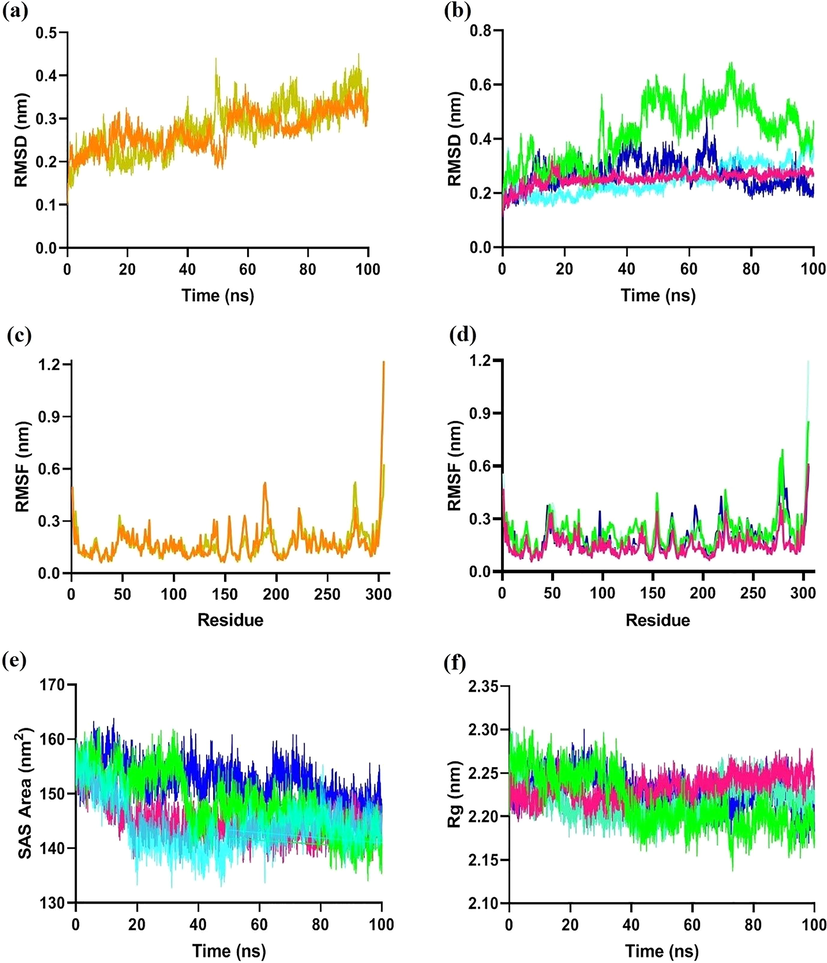
(a) Root-mean-square deviation (RMSD) analysis of apoprotein, and co-crystal ligand–protein complex; (b) Root-mean-square deviation (RMSD) analysis of ligand–protein complexes (Mpro-TerA, Mpro-Cor, Mpro-Cgic, and Mpro-Cnic); (c) Root-mean-square fluctuation (RMSF) analysis of apoprotein, and co-crystal ligand–protein complex; (d) Root-mean-square fluctuation (RMSF) analysis of ligand–protein complexes (Mpro-TerA, Mpro-Cor, Mpro-Cgic, and Mpro-Cnic); (e) SASA analysis and (f) Radius of gyration (Rg) of the apoprotein, co-crystal ligand–protein complex and protein–ligand complexes (Mpro-TerA, Mpro-Cor, Mpro -Cgic, and Mpro-Cnic).
The RMSF study gives data regarding the amino acid residues fluctuation from the reference structure. Fig. 4 (C) shows that the co-crystal ligand fluctuation is almost similar to apoprotein, whereas Fig. 4 (D) shows the fluctuation of the ligand's binding to active site residues Thr24, Thr25, His41, Cys44, Met49, Tyr54, Phe140, Asn142, Gly143, Cys145, His163, His164, Met165, Glu166, Leu167, Pro168, Asp187, Arg188, Gln189, and Thr190 in each of Mpro TerA, Mpro-Cor, Mpro-Cgic, and Mpro-Cnic indicates that there is nearly similar RMSF fluctuation compared to apoprotein and co-crystal ligand–protein residues. Thus, analysis of the RMSF suggested a stable protein–ligand complex since there are no significant changes at the active site's reference location after successful binding by the apoprotein.
Further, the ‘sasa' function was used to calculate the SASA to examine the accessibility of solvent molecules on the protein surface. The SASA value of apoprotein, co-crystal ligand–protein complex, and protein–ligand complexes Mpro-TerA, Mpro-Cor, Mpro-Cgic, and Mpro-Cnic were stable during 100 ns MD. Thus, it indicates that ligand–protein complexes have comparable stability to apoprotein and co-crystal ligand–protein complexes [Fig. 4 (E)].
During MD simulations, a study on the radius of gyration (Rg) indicates the protein compactness. Rg of the C α atoms of the Mpro SARS-CoV-2 protein was computed to ascertain conformational changes in it. The Rg graph was plotted for protein using 100 ns trajectories as represented in Fig. 4 (F). The Rg plot indicates that the Rg value apoprotein (mean: 2.20 nm) and protein–ligand complex (mean: 1.23 nm) are nearly identical. Finally, Rg results suggest that all secondary structure of protein complexes is closely packed during 100 ns MD simulation.
Previous literature has highlighted the potential impact of medicinal plants and their phytoconstituents against SARS-CoV-2 (Rudrapal et al., 2021b). Studies have reported that thioflavonoids and epicatechin-3-O-gallate interacted with (binding sites) Mpro and PLpro, and were considered as SARS-CoV-2 inhibitors (Gogoi et al., 2021; Ismail et al., 2021). Thus, bioactive molecules bind to or interact with the active residues which are present at the binding cavity of Mpro and PLpro.
3.3 MM-PBSA binding free energy (BFE)
Python script MmPbSaStat.py provided in the g_mmpbsa was utilized to calculate average BFE and its standard deviation error. The energy released during the interaction of protein–ligand is represented in the form of BFE. It is a cumulative sum of Van der Waals, polar salvation, electrostatic, and SASA energy. Precisely, the MM-PBSA method estimates the binding free energy of a ligand–protein complex as the difference between the free energy of the complex and the free energies of the unbound components (receptor and ligand). More negative the BFE, the more effectively the ligand binds to the protein (Table 2). The results indicate that all the forms of energy contributed fairly to the interactions between Mpro and the selected molecules. Mpro-TerA complex showed the least binding BFE (−233.782 kJ/mol) among the selected bioactive molecules followed by Mpro-Cgic (−221.916 kJ/mol). The binding free energy (BFE) of the X77 (−160.625 kJ/mol) is less negative compared to the selected bioactive molecules. A comparison of the BFE of all the complexes reveals that, except for corilagin, all of the other selected bioactive molecules outperform X77 in inhibiting the protein Mpro of SARS-CoV-2.
Complexes
ΔEbinding (kJ/mol)
SASA (kJ/mol)
ΔEpolar solvation (kJ/mol)
ΔEElectrostatic (kJ/mol)
ΔEVan der Waals (kJ/mol)
Terflavin A
−233.782 ± 19.600
−28.158 ± 1.819
201.507 ± 22.760
−41.154 ± 8.852
−365.977 ± 16.074
Corilagin
−134.539 ± 13.270
−18.924 ± 1.200
81.964 ± 16.865
−5.503 ± 7.878
−192.076 ± 12.655
Chebulagic acid
−221.916 ± 17.867
27.490 ± 1.279
171.064 ± 16.916
−34.604 ± 11.949
−330.887 ± 15.585
Chebulinic acid
−215.150 ± 20.128
29.860 ± 1.612
223.952 ± 19.289
−76.837 ± 13.932
332.405 ± 19.549
X77
−160.625 ± 15.840
−20.961 ± 1.249
123.945 ± 13.577
−29.776 ± 6.919
−233.832 ± 13.802
3.4 Drug-likeness, ADMET and toxicity properties
Lipinski's rule of five (Ro5) is a set of rules used to assess the drug-likeness of a chemical compound that has a pharmacological activity that could lead to it being an orally active drug for human use (Lipinski et al., 2001; Kalita et al., 2020). The Ro5 specifies that the molecular weight must be less than 500 Da, the number of H-bond donors must be less than 5, the number of H-bond acceptor must be less than 10, the xlogP must be less than 5, and the molar refractivity must be between 40 and 130 (Fatimawali et al., 2021; Kashyap et al., 2016; Sefren Geiner et al., 2020). Regarding this statement, Table 3 indicates that only compound X77 appears to meet the Ro5 criteria. However, strict adherence to Ro5 is not required for naturally occurring compounds or phytocomponents. Lipinski specifically states that the natural products and substrates of biological transporters are not covered by Ro5 (Lipinski et al., 2001; Rudrapal et al., 2017; Rudrapal et al., 2018). It is, therefore important to note here that the desired pharmacological efficacy of Ayurvedic/herbal formulations is possibility due to the multi-component nature (different components) of such formulations.
Compounds
Molecular Weight (Da)
H-bond donor
H-bond acceptor
LogP
Molar Refractivity
Violation
Terflavin A
1086
15
30
1.22
211.35
4
Chebulagic acid
954
10
27
−0.60
182.03
4
Chebulinic acid
956
12
27
0.90
185.99
4
Corilagin
634
8
18
1.18
125.43
3
X77
459
1
2
3.69
131.96
0
On its way to the target site, the drug candidate undergoes pharmacokinetic events such as absorption, distribution, metabolism, and excretion (ADME). ADME parameters, in conjunction with toxicity, are critical for the discovery and development of potential drug candidates (Guan et al., 2019). Not only should a high-quality drug candidate be efficacious against the therapeutic target, but it should also exhibit appropriate ADMET properties at the dosage for therapeutic purposes. Based on the results of the admetSAR and ProTox analyses (Table 4), all of the compounds were from any signs of toxicity such as hepatotoxic, carcinogenic, or mutagenic. The acute oral toxicity analysis reveals that all compounds are classified as class III (500 mg/kg LD50 5000 mg/kg), which means that they are not toxic, except for chebulagic acid, which is classified as category II (50 mg/kg LD50 500 mg/kg), indicating a moderate level of toxicity (Herkenne et al., 2008; Li et al., 2014). Typically, animal experiments are used to determine a compound's toxicity, carcinogenicity, and mutagenicity. However, this takes a long time and necessitates the sacrifice of animals. Thus, prediction of toxicity within in silico technique is a faster and less expensive alternative, as it is based on existing data on a compound's toxicity. As a result, it is used to develop models capable of predicting the toxicity, carcinogenicity, and mutagenicity of novel compounds.
Compounds
Human intestinal absorption
Human oral bioavailability
Hepatotoxicity (Prediction/Probability
Carcinogenicity (Prediction/Probability
Immunotoxicity (Prediction/Probability
Mutagenicity (Prediction/Probability
Acute Oral Toxicity (kg/mol)/Class
Predicted LD50 (mg/kg)
Terflavin A
0.8311 (+)
0.5571
Inactive/0.81
Inactive/0.79
Active/0.73
Inactive/0.64
2.212/III
5000
Chebulagic acid
0.8305 (+)
0.6000
Inactive/0.87
Inactive/0.79
Active/0.97
Inactive/0.57
2.987/III
420
Chebulinic acid
0.8094 (+)
0.6143
Inactive/0.87
Inactive/0.87
Active/0.91
Inactive/0.57
3.232/III
823
Corilagin
0.8598 (+)
0.5286
Inactive/0.85
Inactive/0.72
Active/0.72
Inactive/0.56
2.385/III
2260
X77
0.9422 (+)
0.5000
Inactive/0.71
Inactive/0.63
Active/0.64
Inactive/0.66
2.708/III
650
Bioavailability is a pharmacological term that refers to the level and amount to which a bioactive compound can be absorbed and made available (Kim et al., 2014). A compound's low oral bioavailability is a frequent issue during the drug design process. As a result, a drug may fail in clinical trials despite its high efficacy in vitro and in vivo experiments (Fukunishi et al., 2014). A compound is predicted to have low oral bioavailability if its bioavailability score is lower than 0.5. If the score is greater than 0.5, the compound is expected to have a high oral bioavailability (Rath et al., 2021). According to the findings, all compounds, except for X77, have a high oral bioavailability.
Toxic doses are frequently expressed as LD50 (mg/kg body weight). LD50 (median lethal dose) is a particular dose wherein 50% of the participants die after being exposed to a compound (Erhirhie et al., 2018). A compound is considered highly toxic if the LD50 value is higher than 300 mg/kg. If the LD50 is between 300 and 1,000 mg/kg, a compound is regarded as moderately toxic. If, on the other hand, the LD50 value of a compound is between 1,000 and 5,000 mg/kg, it is considered to be less or slightly toxic. Thus, chebulagic acid, chebulinic acid, and X77 are classified as moderately toxic, whereas corilagin and terflavin A are classified as less toxic.
4 Conclusions
To develop drugs from ancient Indian knowledge against SARS-CoV-2, we made an attempt to analyze the binding modes of the phytoconstituents of Triphala formulation by docking against the active site of Mpro. Terflavin A, corilagin, chebulagic acid and chebulinic acid demonstrated promising binding affinity with the target protein, Mpro. Molecular dynamics simulation was carried out for these selected bioactive molecules to further elucidate their binding affinity towards the Mpro and explored the obtained trajectories for RMSD, RMSF, Rg, and SASA. The trajectories were found to have increased flexibility and higher stability. Further, strong binding affinity between the bioactive molecules and Mpro were confirmed by low binding free energies, dynamic behaviors of complexes as well as residue contribution energies obtained by MM-PBSA. This study identifies four bioactive molecules from Triphala formulation as promising SARS-CoV-2 Mpro inhibitors; hence these bioactive molecules can be further examined using experimental studies for confirmation of their therapeutic efficacies.
Funding
The author would like to acknowledge the Deanship of Scientific Research, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia, for providing Grant through project number Covid19-2020-002-IRMC.
Conflicts of interest
The authors declare no conflicting interests.
7 Availability of data and material
Data and material will be made available after proper request to the corresponding author.
CRediT authorship contribution statement
Mithun Rudrapal: Software, Investigation. Ismail Celik: Software, Investigation. Johra Khan: . Mohammad Azam Ansari: Funding acquisition. Mohammad N. Alomary: Funding acquisition. Rohitash Yadav: Software, Investigation, Writing – original draft. Tripti Sharma: Software, Investigation, Writing – original draft. Trina Ekawati Tallei: Software, Investigation. Praveen Kumar Pasala: . Ranjan Kumar Sahoo: Funding acquisition. Shubham J. Khairnar: Funding acquisition. Atul R. Bendale: Funding acquisition. James H. Zothantluanga: Software, Investigation. Dipak Chetia: Software, Investigation. Sanjay G. Walode: Software, Investigation.
Acknowledgements
All MD simulations presented in the study were carried out with resources provided by TÜBTAK (Turkish Scientific and Technological Research Council), ULAKBIM (Turkish Academic Network and Information Centre), and TRUBA (High Performance and Grid Computing Centre). The author would like to acknowledge the Deanship of Scientific Research, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia, for providing Grant through project number Covid19-2020-002-IRMC.
References
- GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19-25.
- [CrossRef] [Google Scholar]
- Indian medicinal plants and formulations and their potential against COVID-19–preclinical and clinical research. Front. Pharmacol.. 2021;11:2470.
- [CrossRef] [Google Scholar]
- ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res.. 2018;46:W257-W263.
- [CrossRef] [Google Scholar]
- Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn.. 2021;39(10):3449-3458.
- [CrossRef] [Google Scholar]
- State-of-the-art tools unveil potent drug targets amongst clinically approved drugs to inhibit helicase in SARS-CoV-2. Arch. Med. Sci.. 2020;16:508-518.
- [CrossRef] [Google Scholar]
- Bromelain a potential bioactive compound: a comprehensive overview from a pharmacological perspective. Life. 2021;11:317.
- [CrossRef] [Google Scholar]
- A comprehensive review of the structure elucidation of tannins from Terminalia Linn. evidence-based complement. . Altern. Med.. 2019;2019:1-26.
- [CrossRef] [Google Scholar]
- Drug-like bioactive structures and conformational coverage with the LigPrep/ConfGen suite: comparison to programs MOE and catalyst. J. Chem. Inf. Model.. 2010;50:822-839.
- [CrossRef] [Google Scholar]
- Plants of the Genus Terminalia: an insight on its biological potentials, pre-clinical and clinical studies. Front. Pharmacol.. 2020;11
- [CrossRef] [Google Scholar]
- Role of molecular dynamics and related methods in drug discovery. J. Med. Chem.. 2016;59(9):4035-4061.
- [CrossRef] [Google Scholar]
- Appraisals of the Bangladeshi medicinal plant Calotropis gigantea used by folk medicine practitioners in the management of COVID-19: a biochemical and computational approach. Front. Mol. Biosci.. 2021;8
- [CrossRef] [Google Scholar]
- Advances in acute toxicity testing: strengths, weaknesses and regulatory acceptance. Interdiscip. Toxicol.. 2018;11:5-12.
- [CrossRef] [Google Scholar]
- Data on the docking of phytoconstituents of betel plant and matcha green tea on SARS-CoV-2. Data Br.. 2021;36:107049.
- [CrossRef] [Google Scholar]
- Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem.. 2006;49:6177-6196.
- [CrossRef] [Google Scholar]
- Prediction of synthetic accessibility based on commercially available compound databases. J. Chem. Inf. Model.. 2014;54(12):3259-3267.
- [CrossRef] [Google Scholar]
- Design, molecular docking, drug-likeness and molecular dynamics studies of 1,2,4-trioxane derivatives as novel P. falciparum Falcipain-2 (FP-2) inhibitors. Biotechnologia. 2021;102:257-275.
- [CrossRef] [Google Scholar]
- Identification of potential plant-based inhibitor against viral proteases of SARS-CoV-2 through molecular docking, MM-PBSA binding energy calculations and molecular dynamics simulation. Mol. Divers.. 2021;25(3):1963-1977.
- [CrossRef] [Google Scholar]
- ADMET-score – a comprehensive scoring function for evaluation of chemical drug-likeness. Medchemcomm. 2019;10:148-157.
- [CrossRef] [Google Scholar]
- Habib-ur-Rehman, Yasin, K.A., Choudhary, M.A., Khaliq, N., Atta-ur-Rahman, Choudhary, M.I., Malik, S., 2007. Studies on the chemical constituents of Phyllanthus emblica. Nat. Prod. Res. 21, 775–781. https://doi.org/10.1080/14786410601124664.
- Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem.. 2004;47:1750-1759.
- [CrossRef] [Google Scholar]
- Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol.. 2004;78:13600-13612.
- [CrossRef] [Google Scholar]
- In vivo methods for the assessment of topical drug bioavailability. Pharm. Res.. 2008;25:87.
- [CrossRef] [Google Scholar]
- Hussain, N., Kakoti, B.B., Rudrapal, M., Sarwa, K.K., Celik, I., Attah, E.I., Khairnar, S.J., Bhattacharya, S., Sahoo, R.K., Walode, S.G., 2021. Bioactive antidiabetic flavonoids from the stem bark of cordia dichotoma forst.: identification, docking and ADMET studies. Molbank 2021, M1234. https://doi.org/10.3390/M1234.
- The study of Calotropis Gigantea leaf metabolites from Ie Brouk geothermal area Lamteuba-Aceh Besar using molecular docking. IOP Conf. Ser. Earth Environ. Sci.. 2021;667:012072.
- [CrossRef] [Google Scholar]
- Targeting multiple conformations of SARS-CoV2 papain-like protease for drug repositioning: an in-silico study. Computers Biol. Med.. 2021;131:104295.
- [CrossRef] [Google Scholar]
- Pharmacological benefits of Triphala: a perspective for allergic rhinitis. Front. Pharmacol.. 2021;12
- [CrossRef] [Google Scholar]
- Antidiabetic bioactive compounds from Tetrastigma angustifolia (Roxb.) Deb and Oxalis debilis Kunth.: Validation of ethnomedicinal claim by in vitro and in silico studies. South African J. Bot.. 2021;132:164-175.
- [Google Scholar]
- Design, synthesis, antimalarial activity and docking study of 7-chloro-4- (2-(substituted benzylidene)hydrazineyl)quinolines. Med. Chem. (Los. Angeles).. 2020;16:928-937.
- [CrossRef] [Google Scholar]
- Synthesis, antimalarial activity evaluation and drug likeness study of some new quinoline-lawsone hybrids. Indian J. Pharm. Sci.. 2016;78
- [CrossRef] [Google Scholar]
- Evolution of RNA viruses from SARS to SARS-CoV-2 and diagnostic techniques for COVID-19: A review. Beni-Suef Univ. J. Basic Appl.. 2021;10:60.
- [Google Scholar]
- Discovery of potential inhibitors of the receptor-binding domain (RBD) of pandemic disease-causing SARS-CoV-2 spike glycoprotein from Triphala through molecular docking. Curr. Chinese Chem.. 2021;1:e220321192390
- [CrossRef] [Google Scholar]
- In silico molecular docking analysis for repurposing approved antiviral drugs against SARS-CoV-2 main protease. Biochem. Biophys. Reports. 2021;27:101032.
- [CrossRef] [Google Scholar]
- Critical evaluation of human oral bioavailability for pharmaceutical drugs by using various cheminformatics approaches. Pharm. Res.. 2014;31:1002-1014.
- [CrossRef] [Google Scholar]
- Phytochemistry and medicinal potential of the terminalia bellirica roxb. (bahera) Indian J. Nat. Prod. Resour.. 2018;9:97-107.
- [Google Scholar]
- Kumar, P.P., Shaik, R.A., Khan, J., Alaidarous, M.A., Rudrapal, M., Khairnar, S.J., Sahoo, R., Zothantluanga, J.H., Walode, S.G. (2021b). Cerebroprotective effect of aloe emodin: in silico and in vivo studies. Saudi J. Biol. Sci. doi: 0.1016/j.sjbs.2021.09.077.
- g_mmpbsa —A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model.. 2014;54:1951-1962.
- [CrossRef] [Google Scholar]
- In silico prediction of chemical acute oral toxicity using multi-classification methods. J. Chem. Inf. Model.. 2014;54:1061-1069.
- [CrossRef] [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169–409X(96), 00423–1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) Adv. Drug Deliv. Rev.. 2001;46:3-26.
- [CrossRef] [Google Scholar]
- Toward a treatment of antibacterial and antifungal infections: Design, synthesis and in vitro activity of novel arylhydrazothiazolylsulfonamides analogues and their insight of DFT, docking and molecular dynamic simulations. J. Mol. Struct.. 2021;130862
- [Google Scholar]
- Triphala: a useful therapeutic supplement during COVID-19 pandemic. J. Drug Deliv. Thera.. 2020;10(4):219-220.
- [CrossRef] [Google Scholar]
- Therapeutic uses of Triphala in ayurvedic medicine. J. Altern. Complement. Med.. 2017;23:607-614.
- [CrossRef] [Google Scholar]
- Oxidative stress and cancer: chemopreventive and therapeutic role of Triphala. Antioxidants. 2020;9:72.
- [CrossRef] [Google Scholar]
- Design, synthesis and molecular modeling studies of novel mesalamine linked coumarin for treatment of inflammatory bowel disease. Bioorg. Med. Chem. Lett.. 2021;41:128029.
- [CrossRef] [Google Scholar]
- OPLS3e: Extending force field coverage for drug-like small molecules. J. Chem. Theory Comput.. 2019;15:1863-1874.
- [CrossRef] [Google Scholar]
- Novel series of 1,2,4-trioxane derivatives as antimalarial agents. J. Enzyme Inhib. Med. Chem.. 2017;32:1159-1173.
- [CrossRef] [Google Scholar]
- Repurposing of phytomedicine-derived bioactive compounds with promising anti-SARS-CoV-2 potential: Molecular docking, MD simulation and drug-likeness/ADMET studies. Saudi J. Biol. Sci. 2021
- [CrossRef] [Google Scholar]
- In silico screening of phytopolyphenolics for the identification of bioactive compounds as novel protease inhibitors effective against SARS-CoV-2. J. Biomol. Struct. Dyn.. 2021;1–17
- [CrossRef] [Google Scholar]
- Coronavirus disease-2019 (COVID-19): an updated review. Drug Res.. 2020;70:389-400.
- [CrossRef] [Google Scholar]
- Design, synthesis, drug-likeness studies and bio-evaluation of some new chalconeimines. Pharm. Chem. J.. 2019;53:814-821.
- [CrossRef] [Google Scholar]
- Newer series of trioxane derivatives as potent antimalarial agents. Med. Chem. Res.. 2018;27:653-668.
- [CrossRef] [Google Scholar]
- The potential of leaf extract of Pangium edule Reinw as HIV-1 protease inhibitor: a computational biology approach. J. Appl. Pharm. Sci. 2020
- [CrossRef] [Google Scholar]
- An in-silico evaluation of different bioactive molecules of tea for their inhibition potency against non structural protein-15 of SARS-CoV-2. Food Chem.. 2021;346:128933.
- [CrossRef] [Google Scholar]
- Drug repurposing and computational modeling for discovery of inhibitors of the main protease (M pro) of SARS-CoV-2. RSC Adv.. 2021;11:23450-23458.
- [CrossRef] [Google Scholar]
- In-silico evaluation of bioactive compounds from tea as potential SARS-CoV-2 nonstructural protein 16 inhibitors. J. Tradit. Complement. Med. 2021
- [CrossRef] [Google Scholar]
- A comprehensive review on promising anti-viral therapeutic candidates identified against main protease from SARS-CoV-2 through various computational methods. J. Genet. Eng. Biotechnol.. 2020;18:69.
- [CrossRef] [Google Scholar]
- pH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol.. 2005;354:25-40.
- [CrossRef] [Google Scholar]
- Triphala: current applications and new perspectives on the treatment of functional gastrointestinal disorders. Chin. Med.. 2018;13:39.
- [CrossRef] [Google Scholar]
- Anti-SARS-CoV natural products with the potential to inhibit SARS-CoV-2 (COVID-19) Front. Pharmacol.. 2020;11
- [CrossRef] [Google Scholar]
- Tannic acid suppresses SARS-CoV-2 as a dual inhibitor of the viral main protease and the cellular TMPRSS2 protease. Am. J. Cancer Res.. 2020;10:4538-4546.
- [Google Scholar]
- Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol.. 2005;3:e324.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.101826.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




