

Translate this page into:
Unsymmetrical aromatic disulfides as SARS-CoV-2 Mpro inhibitors: Molecular docking, molecular dynamics, and ADME scoring investigations
⁎Corresponding authors at: Laboratory of Molecular Chemistry and Environment, University of Biskra, BP145, 07000 Biskra, Algeria (S. Belaidi). samirchtita@gmail.com (Samir Chtita), prof.belaidi@gmail.com (Salah Belaidi), mmogren@ksu.edu.sa (Muneerah Mogren AlMogren)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
COVID-19 pandemic caused by very severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) agent is an ongoing major global health concern. The disease has caused more than 452 million affected cases and more than 6 million death worldwide. Hence, there is an urgency to search for possible medications and drug treatments. There are no approved drugs available to treat COVID-19 yet, although several vaccine candidates are already available and some of them are listed for emergency use by the world health organization (WHO). Identifying a potential drug candidate may make a significant contribution to control the expansion of COVID-19. The in vitro biological activity of asymmetric disulfides against coronavirus through the inhibition of SARS-CoV-2 main protease (Mpro) protein was reported. Due to the lack of convincing evidence those asymmetric disulfides have favorable pharmacological properties for the clinical treatment of Coronavirus, in silico evaluation should be performed to assess the potential of these compounds to inhibit the SARS-CoV-2 Mpro.
In this context, we report herein the molecular docking for a series of 40 unsymmetrical aromatic disulfides as SARS-CoV-2 Mpro inhibitor. The optimal binding features of disulfides within the binding pocket of SARS-CoV-2 endoribonuclease protein (Protein Data Bank [PDB]: 6LU7) was described. Studied compounds were ranked for potential effectiveness, and those have shown high molecular docking scores were proposed as novel drug candidates against SARS-CoV-2. Moreover, the outcomes of drug similarity and ADME (Absorption, Distribution, Metabolism, and Excretion) analyses have may have the effectiveness of acting as medicines, and would be of interest as promising starting point for designing compounds against SARS-CoV-2. Finally, the stability of these three compounds in the complex with Mpro was validated through molecular dynamics (MD) simulation, in which they displayed stable trajectory and molecular properties with a consistent interaction profile.
Keywords
COVID-19
SARS-CoV-2
Disulfides
Molecular docking
Molecular dynamics
Main protease

- COVID-19
-
coronavirus disease 2019
- SARS-CoV-2
-
severe acute respiratory syndrome coronavirus-2
- WHO
-
World Health Organization
- Mpro
-
Main protease
- PDB
-
Protein Data Bank
- SMILES
-
simplified molecular input line entry system
- CYP
-
cytochrome P450
- MD
-
molecular dynamics
- RMSD
-
root-mean-square deviation
- RMSF
-
root-mean-square fluctuation
- Rg
-
radius of gyration
- SASA
-
solvent-accessible surface area
- HBD
-
hydrogen bond donor
- HBA
-
hydrogen bond acceptor
- RB
-
rotatable bond count
- PSA
-
polar surface area
Abbreviations
1 Introduction
The ongoing COVID-19 outbreak caused by novel coronavirus, which emerged in Wuhan by the end of December 2019, is still posing a serious problem to global health. At the time of writing this paper, the disease has caused more than 452 million affected cases and more than 6 million deaths worldwide (http://covid19.who.in). This crisis has pushed government and scientist throughout the world to intensify their efforts to develop treatments and to prevent the rapid spreading of this disease. Unfortunately, there is no available drug treatment against COVID-19 approved yet. Luckily, several vaccines candidates are now available, and WHO authorize some of them for emergency use.
SARS-CoV-2 agent is closely related to that of SARS-CoV, and this allows previously defined protein structures to be used to quickly build a drug discovery model for the novel coronavirus (Hui et al., 2020). The main protease (Mpro), which have a significant importance in the replication process of the virus, has been considered as a potential target in the development of novel SARS inhibitors (Anand et al., 2003; Holmes, 2003; Leng and Bentwich, 2003; Marra et al., 2003; Poutanen et al., 2003; Snijder et al., 2003; Thiel et al., 2003; Rota et al., 2003). Despite, MERS-CoV, SARS-CoV and SARS-CoV-2 agent belong to beta-coronaviruses category; they are in fact slightly distinguished from each other. Recent studies have shown that SARS-CoV-2 shares approximately 80% nucleotide identity and 89.10% nucleotide similarity with SARS-CoV. Therefore, the main protease of SARS-CoV, 3CLpro, has been the target of several in silico investigations to develop novel inhibitors candidates (Tahir UlQamar et al., 2020; Aouidate et al., 2020; Fouedjou et al., 2021). The 3CLpro has a high sequence identity rate between nCoV and nCoV-2; hence, their 3CLpro are likely homologous and have identical structures and functions. Moreover, SARS-CoV and SARS-CoV-2 agents infect cells in the same way and use the same protein machinery to multiply inside the host cell.
Asymmetric disulfides have become powerful resources in combinatorial chemistry and pharmaceutical research (Chtita et al.; Ameta et al.), due to their various biological activities. In fact, they exhibit anticancer activity (Branowska et al.), antibacterial activity (Kumar et al.), as well as antiviral activity by blocking the interaction between arenavirus Z protein and myeloid cell leukemia protein (Garcia et al.). These compounds are known also for their herbicidal activity (Wang et al.).
Keeping in view these features mentioned above, we propose herein to study the behavior of SARS-CoV 3CLpro inhibitors towards SARS-CoV-2 Mpro. In fact, we have performed an in silico study using docking, Dynamic, and ADME of 40 aromatic disulfides known as inhibitors of SARS-CoV 3CLpro (Wang et al., 2017; Chtita et al., 2021). The study was realized in respect of the active site of the recently crystallized SARS-CoV-2 Mpro (Protein Data Bank [PDB]: 6LU7) (Liu et al., 2020).
2 Material and methods
(S3 of the Supplementary Material).
3 Results and discussion
In this investigation, a set of 40 unsymmetrical aromatic disulfides was studied in silico to highlight their possible binding energy and interaction modes with the active site of SARS-CoV-2 Mpro, using the AutoDock Vina software. The estimated binding energy of the binding site of the 6LU7 enzyme structure is summarized in Table 1 for all the studied compounds. According to the outcomes obtained from docking screening, six compounds (6, 7, 8, 9, 10, and 11) with the strongest binding energies were chosen to depict the binding mode of the disulfide inhibitors.
N°
Binding energies
N°
Binding energies
N°
Binding energies
N°
Binding energies
8
−7.9
15
−6.4
12
−6.0
3
−5.6
6
−7.2
27
−6.4
25
−6.0
31
−5.6
11
−7.2
14
−6.3
13
−5.9
34
−5.5
9
−7.1
28
−6.3
35
−5.9
38
−5.5
10
−7.1
29
−6.2
36
−5.9
40
−5.5
7
−7.0
30
−6.2
5
−5.8
39
−5.4
4
−6.7
32
−6.2
17
−5.8
1
−5.3
16
−6.6
18
−6.1
20
−5.8
21
−5.3
19
−6.5
37
−6.1
24
−5.8
22
−5.2
26
−6.5
2
−6.0
33
−5.7
23
−5.1
Before docking molecules 6, 7, 8, 9, 10, 11 into the Mpro receptor pocket, we first validated the 3D model of the Mpro protein. For this, we compared the types of residues with which the N3 inhibitor interacted in the 6LU7 complex obtained from the protein core library (https://www.rcsb.org/structure/6LU7) (Fig. S2-A) with the residues with which the rigid structure of the N3 inhibitor interacted after it was prepared in PDBQT format (Fig. S2-B) following the same protocol of validation reported in a study performed by Daoui et al.
From the two-dimensional visualizations shown in Fig. S2-A and S2-B, we can notice that the prepared N3 ligand interacts with the same residue that the crystallized N3 ligand interacted with. Therefore, we can consider that the model of the Mpro protein complexed with the N3 inhibitor is valid and that molecular docking on the Mpro protein can be performed in this work.
The interactions between the inhibitor and surrounding residues of SARS-CoV-2 Mpro are illustrated in the 2D and 3D schematics, which they were obtained by importing docking results into the Discovery Studio Visualizer. Fig. 1 and Table 2 show the amino acids that participate in the pattern of interactions between the ligand and enzyme with a significant partial energy contribution to total energy of interaction. Most of these interactions include Van der Waals interactions, hydrophobic contacts (Pi-Pistacked, Pi-Pi T-shaped, amide-Pistacked, alkyl Pi-Sigma, and Pi-alkyl), hydrogen bonds (hydrogen bond, carbon, and Pi-donor), electrostatic, carbonyl, and specific atom-aromatic ring (Pi-cation, Pi-donor, Pi-halogen, and Pi-carbon) and provide insight into understanding molecular recognition (Du et al., 2016). Fig. 1 depict the docking interactions of the most active molecules (6, 7, 8, 9, 10, and 11) based on docking studies. Fig. 2 show Docked conformation poses of ligands 6, 7, 8, 9, 10 and 11 inside the Mpro receptor pocket and the superimposed view of these ligands.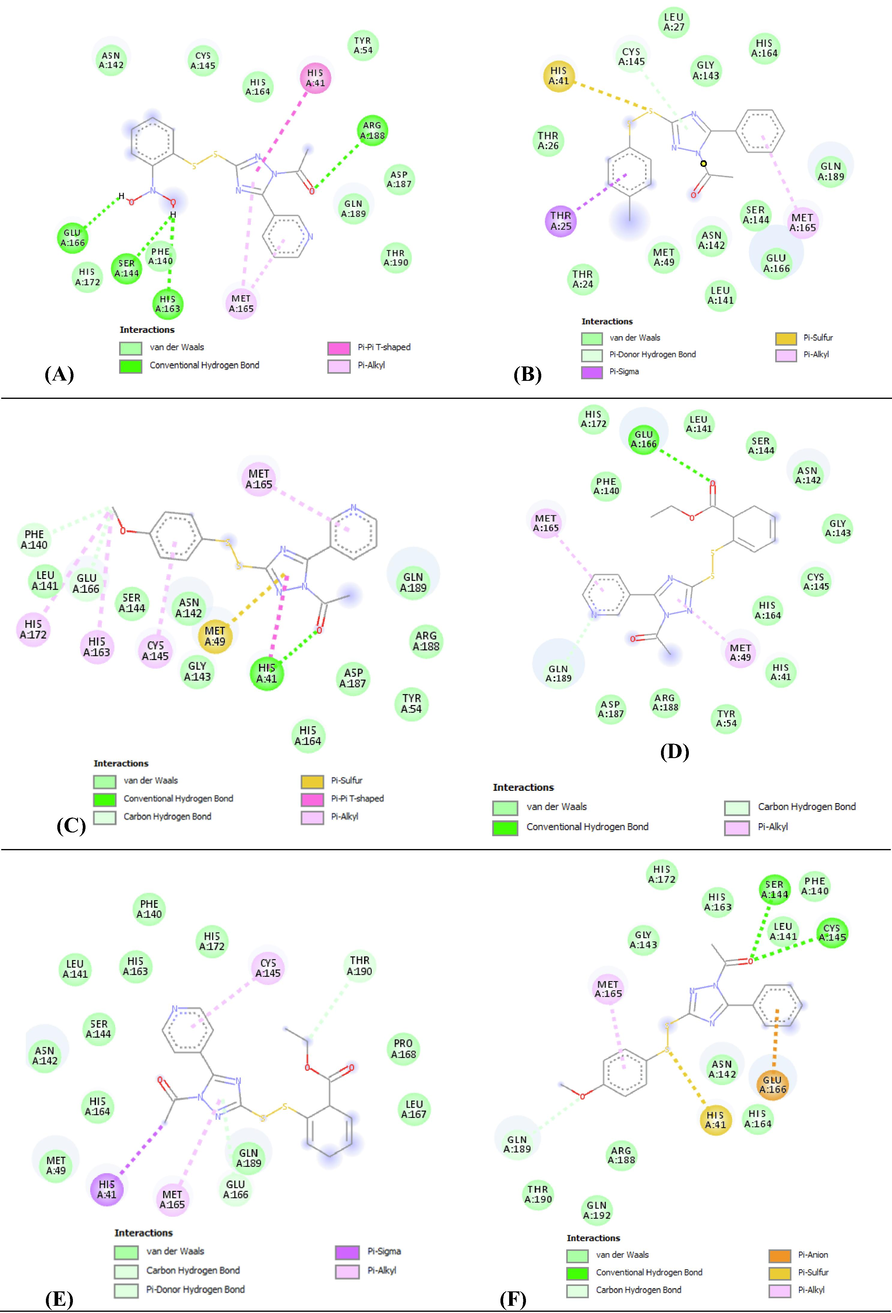
Different interactions and key residues for the inhibitor binding between 6LU7 and compounds (A) 8, (B) 6, (C) 11, (D) 9, (E) 10 and (F) 7.
Residues
Distance
Interaction type
Residues
Distance
Interaction type
6
CYS145
3.60
Pi-Donor
9
GLU166
3.11
Hydrogen Bond
THR25
3.66
Pi-Sigma
GLN189
3.55
Carbon Hydrogen Bond
HIS41
4.61
Pi-Sulfur
MET49
4.84
Pi-Alkyl
MET165
5.26
Pi-Alkyl
MET165
4.59
Pi-Alkyl
7
SER144
3.03
Hydrogen Bond
10
THR190
3.47
Carbon Hydrogen Bond
CYS145
3.76
Hydrogen Bond
GLU166
4.19
Pi-Donor
GLN189
3.39
Carbon Hydrogen Bond
HIS41
3.72
Pi-Sigma
GLU166
4.12
Pi-Anion
MET165
5.23
Pi-Alkyl
HIS41
4.36
Pi-Sulfur
CYS145
5.25
Pi-Alkyl
MET165
4.45
Pi-Alkyl
11
HIS41
3.15
Hydrogen Bond
8
Glu166
2.19
Hydrogen Bond
PHE140
3.27
Carbon Hydrogen Bond
Ser144
2.48
Hydrogen Bond
GLU166
3.55
Carbon Hydrogen Bond
His163
2.64
Hydrogen Bond
MET49
4.31
Pi-Sulfur
Arg188
3.11
Hydrogen Bond
HIS41
4.21
Pi-Pi T-Shaped
Leu141
2.81
Acceptor-Acceptor
CYS145
5.10
Pi-Alkyl
His41
5.63
Pi-Pi T-Shaped
MET165
4.35
Pi-Alkyl
Met165
5.12
Pi-Alkyl
HIS163
4.74
Pi-Alkyl
Met165
5.07
Pi-Alkyl
HIS172
4.81
Pi-Alkyl
(A) RMSD of the backbone of SARS-CoV-2 Mpro with compounds 6, 7, and 11 over 100 ns MD simulation. (B) RMSF of Cα atoms of SARS-CoV-2 Mpro with compounds 6, 7, and 11.
Moreover, the results show that compound8is the most promising ligand, which bound with SARS-CoV-2 Mpro via multiple hydrogen bonding and hydrophobic contacts. The binding site is mostly located in a hydrophobic cleft lined by the following amino acids: His41, Tyr54, Phe140 Leu141, Asn142, Ser144, Cys145, His163, His164, Met165, Glu166, His172, Asp187, Arg188, Gln189, and Thr190. There are four hydrogen bond interactions with four different amino acids, Ser144, His163, Glu166, and Arg188, at distances of 2.48, 2.64, 2.19, and 3.11 Å, respectively; one acceptor–acceptor interaction with Leu141 at a distance of 2.81 Å; and two hydrophobic Pi-alkyl interactions with Met165 at 5.07 and 5.12 Å; further enquiry indicated the presence of an Pi-Pi T-shaped interaction with His41 at a distance of 5.63 Å (Fig. 3A). The high affinity of compound 8 is also associated by Van der Waals forces created on the backbone of the amide substituents with Tyr54, Phe140, Asn142, Cys145, His164, His172, Asp187, Gln189, and Thr190, which helped create a strong cohesive environment, thereby stabilizing the complex formed.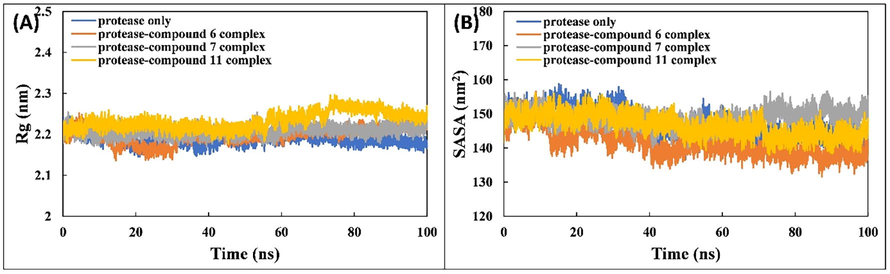
(A) Rg of the backbone of SARS-CoV-2 Mpro and associated complexes with compounds 6, 7, and 11 over 100 ns MD simulation. (B) SASA of SARS-CoV-2 Mpro and associated complexes with compounds 6, 7, and 11 over 100 ns MD simulation.
Binding ability of compound 6 is also very favorable. A hydrogen bond formed with Cys145 of the 6LU7 at a distance of 3.60 Å. The docking score for the best interaction pose is −7.20 kcal/mol.
The conformational energy of the ligand is optimized by the presence of three hydrophobic interactions (Pi-alkyl interaction with Met165 at a distance of 5.25 Å, Pi-Sigma with Thr25 at a distance of 3.66 Å, and Pi-sulfur with His41 at a distance of 4.61 Å) that largely involve charge transfers to help intercalate the ligand in the binding site of the 6LU7. The high affinity of compound 6 is associated with Van der Waals forces between the backbone of the amide substituents with Thr24, Thr26, Leu27, Met49, Leu141, Asn142, Gly143, Ser144, His164, Glu166, and Gln189, which undoubtedly created a strong cohesive environment to stabilize the complex formed (Fig. 1B).
Compound 11 also has a favorable binding affinity and is associated with three H-bonds formed with the amino acids of 6LU7 (one Pi-donor hydrogen bond with His41 at a distance of 3.15 Å and two C–H interactions with Phe140 and Glu166 at distances of 3.27 and 3.55 Å, respectively). The docking score for the best interaction is −7.20 kcal/mol. The conformational energy of the ligand was minimized by the presence of five hydrophobic interactions (one Pi-sulfur with Met49 at 4.31 Å and four Pi-alkyl interactions with Cys145, His163, Met165, and His172 at distances of 5.10, 4.74, 4.35, and 4.81 Å, respectively), which largely involve charge transfer to intercalate the ligand in the binding site of 6LU7; further enquiry indicated the presence of an Pi-Pi T-shaped interaction with His41 at a distance of 4.21 Å. The high affinity of compound 11 is associated with Van der Waals forces created on the backbone of the amide substituents with Tyr54, Leu141, Asn142, Gly143, Ser144, His164, Asp187, Arg188, and Gln189 (Fig. 1C).
The binding ability of compound 9 has an interaction score of −7.10 kcal/mol. Two hydrogen binds formed with two amino acids (one Pi-donor hydrogen bond with Glu166 at a distance of 3.11 Å and one C–H interaction with Gln189 at a distance of 3.55 Å). The conformational energy of the ligand is minimized by the presence of two Pi-alkyl interactions with Met49 and Met165 at distances of 4.84 and 4.59 Å, respectively. As with the other compounds, Van der Waals forces are present on the backbone of the amide substituents with His41, Tyr45, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His164, His172, Asp187, and Arg188 (Fig. 1D).
The docking score for the best interaction of compound 10 is −7.10 kcal/mol, with two hydrogen bonds formed with two amino acids (one Pi-donor hydrogen bond with Glu166 at a distance of 4.19 Å and one C–H interaction with Thr190 at a distance of 3.47 Å). The energy of the ligand is minimized by the abundance of two Pi-alkyl interactions with Cys145 and Met165 at distances of 5.25 and 5.23 Å, respectively, and one Pi-Sigma interaction with His41 at a distance of 3.72 Å.
The high affinity of compound 10 is linked with the presence of Van der Waals interactions with Met49, Phe140, Leu141, Asn142, Ser144, His163, His164, His172, Leu167, Pro168, and Gln189 (Fig. 1E), which undoubtedly creates a strong cohesive environment.
Compound 7 firmly binds at the target site of 6LU7 with three hydrogen bonds composed with three amino acids (two Pi-donor hydrogen bonds with Ser144 and Cys145 at distances of 3.03 and 3.76 Å, respectively, and one C–H interaction with Gln189 at a distance of 3.39 Å). The stability of this compound in the binding site is attributed to the three hydrophobic interactions (one Pi-alkyl with Met165 at a distance of 4.45 Å, one Pi-anion with Glu166 at a distance of 4.12 Å, and one Pi-sulfur with His41 at a distance of 4.36 Å). The high affinity of compound 7 is also associated with the presence of Van der Waals forces with Phe140, Leu141, Asn142, Gly143, His163, His164, His172, Arg188, Thr190, and Gln192 (Fig. 1F). The docking score for the best interaction pose is −7.00 kcal/mol.
Furthermore, the high binding energy of ligand 8, which bears the common (1-(3-(phenyldisulfaneyl)-5-(pyridin-n-yl)-1H-1,2,4-triazol-1-yl)ethan-1-one; where n = 3,4,5) structure with ligands 9, 10 and 11 can be explained by the number of hydrogen bonds formed between amino acids and radicals inserted in the group of 1(3-(phenyldisulfaneyl). We notice that the binding energy of ligand 8 (Table 2, Fig. 3A) decreases drastically when the number of hydrogen bonds between the (R-phenyl) disulfanyl group and the predicted active sites of Mpro decrease. The nitrogen group of the ligand 8 forms three regular hydrogen bonds, while the ethyl acetate and methanol groups of the ligand 9 (Fig. 1D), 10 (Fig. 3E) and 11 (Fig. 1C) form a single hydrogen bond. Therefore, the effect of substitution of radical nitrogen in the structure of the ligand 8 by each of the ethyl acetate and methanol radicals in the structure of ligands 9, 10, and 11, respectively, could be a decisive reason to explain the observed divergence between the binding energies of ligands’ structures.
On the other hand, the substitution of the (pyridin-n-yl) group in the structure of ligands 8, 9, 10 and 11 by the (phenyl) group in ligands 6 (Fig. 1B) and 7 (Fig. 1F) did not produce any hydrogen bonding interaction with any amino acid and also the binding energies of each ligand 6 and 7 did not improve, while the binding energy of these ligands decreased from −7.2 Kcal/mol (Table 3) to −7.0 Kcal/mol (Table 3). Thus, the substitution of pyridin-n-yl with phenyl was not favorable to improve the binding energy. MW, molecular weight; HBD, hydrogen bond donor; HBA, hydrogen bond acceptor. RB, rotatable bond count; PSA, polar surface area.
Lipinski
Veber
Log S
MW
Log P
HBA
HBD
Violations
PSA
N. RB
Violations
Value
Class
6
341.45
3.43
5
3
0
98.38
5
0
−4.91
Moderately
7
357.45
3.45
4
0
0
107.61
6
0
−4.68
Moderately
8
373.41
2.36
6
0
0
157.09
6
1
−3.99
Soluble
9
400.47
3.37
6
0
0
137.57
8
0
−4.23
Moderately
10
400.47
3.37
6
0
0
137.57
8
0
−4.23
Moderately
11
358.44
3.02
5
0
0
120.50
6
0
−4.01
Moderately
3.1 Biological interpretation
The inhibitory biological activity of unsymmetrical aromatic disulfides against the SARS-CoV Mpro has been previously evaluated in vitro (Wang et al., 2017). Due to the close similarity between the SARS-CoV and SARS-CoV-2 viruses, we have opted for investigating in silico the behavior of disulfides regarding SARS-CoV-2 Mpro. Choosing this enzyme as a target gives important leads in antiviral treatment because it participates in the proteolytic processing of replicase polyproteins; therefore, it plays a major role in viral gene expression and replication. Thus, the inhibition of this enzyme could hamper the replication of the viral genome and then the multiplication of SARS-CoV-2. In this study, we suggest that molecules that could inhibit 3CLproof SARS-CoV may result in the same interest to inhibit ofSARS-CoV-2 Mpro, because of their high sequence similarity. This hypothesis was investigated using MD analysis.
Based on the MD results, the two critical amino acids in the active site of the target are Cys145 and His41. The inhibition of SARS-CoV 3CLpro has been reported to mainly occur via an irreversible covalent bond with Cys145 and, in other several cases, by reversible interaction with Cys145 and His41, which form a catalytic dyad, where Cys145 acts a nucleophile (Ramajayam et al., 2010; Shao et al., 2008; Jacobs et al., 2013; Thanigaimalai et al., 2013a; Liu et al., 2014; Thanigaimalai et al., 2013b) and His41 acts as a base that polarizes and deprotonates the nucleophile (Cys145) to increase its reactivity; this dyad is then attacked by the inhibitor to form an intermediate complex before releasing the enzyme. In our study, compounds 6, 7, 10, and 11 display interactions with Cys145 and His41, suggesting that these molecules could have an inhibitory activity against SARS-CoV-2 Mpro. For compounds 8 and 9, even if they form a stable complex, they will have no pharmacological effect because there is no stabilization of Cys145 or His41. SARS-CoV-2 Mpro is a cysteine protease, in which the unsymmetrical aromatic disulfides may play the role of an inhibitor, which competes with the substrate (competitive inhibitor). We believe that compounds 6, 7, 10, and 11 could be potential inhibitors for this enzyme and, consequently, may act as drug candidates against COVID-19. To confirm this assumption, an in vitro study against SARS-CoV-2 Mpro is recommended.
3.2 Pharmacokinetics and drug-likeness prediction
Pharmacokinetics and drug-likeness prediction for the six compounds were performed using SwissADME (Daina et al., 2017) to evaluate individual ADME behaviors of target compounds.
Despite the fact that chemical compounds have activity and selectivity, they may not be viable candidates for drug development. There are a number of rules and models to follow in the classification of such compound as potential drug candidate. The most widely accepted one is Lipinski’s rule of five (Lipinski et al., 1997). If two parameters are out of range, “poor absorption or permeability is possible”.
The compound may be absorbed in gastrointestinal tract if any one of the parameters is not met. Hence, Lipinski’s rule suggests that active molecules with good bioavailability and oral absorption have molecular weights <500, Log P < 5, less than five hydrogen bond donor (HBD) groups, and less than 10 hydrogen bond acceptor (HBA) groups (Duchowicz and Castro, 2009). A candidate that violates more than one of these criteria is less likely to be developed as a prospective oral drug.
The studied compounds are confronted to the Lipinski criteria, the results show that only four compounds listed in Table 4 meet perfectly these criteria. Thus, there would be no problem with oral bioavailability for all proposed compounds. ΔEvdW, Van der Waals energy; ΔEele, electrostatic energy; ΔEPSE, polar solvation energy. ΔESASA, solvent-accessible surface area energy; ΔEBE, binding energy.
Ligands
Type of energy
Compound 6
Compound 7
Compound 11
ΔEvdW
−147.514 ± 1.392
−136.670 ± 1.930
−183.500 ± 0.981
ΔEele
−58.916 ± 1.199
−29.588 ± 0.956
−36.933 ± 0.666
ΔEPSE
153.653 ± 1.979
100.179 ± 1.726
146.483 ± 1.085
ΔESSASA
−16.717 ± 0.120
−15.168 ± 0.151
−16.820 ± 0.096
ΔEBE
−69.489 ± 1.388
−81.212 ± 1.267
−90.842 ± 1.079
The Veber rule defines drug-likeness constraints as rotatable bond (RB) count ≤10 and polar surface area (PSA) ≤140 (Veber et al., 2002). PSA is strongly related to the absorption properties of active compounds. The obtained PSA for compound 8 is greater than 140, suggesting that this compound has strong polarity and is not readily absorbed by membranes. In addition, the obtained PSAs for compounds 6, 7, 9, 10, and 11 are all less than 140; which indicates that these active compounds have good oral absorption; namely good solubility and better penetration through protein membranes. The number of RBs for the six tested compounds are less than 10; as the number of RBs increases, the molecules become more flexible and more adaptable for efficient interaction with a particular binding pocket. The screening of potential candidates using Veber’s rules, allowed us to select only five compounds (6, 7, 9, 10, and 11) that meet the criteria of drug-likeness assessment, whereas, compound 8 is rejected for reason of violation of one Veber criterion (Table 3).
In drug discovery schemes, oral administration and high solubility will favor complete absorption, while poor solubility limits the absorption of the drug in the gastrointestinal tract. Data listed in Table 4 suggests that compounds 6, 7, 9, 10, and 11 have good to moderate solubility, the Log S value is between −6 and −4. Thus, the afore mentioned compounds could exhibit good oral adsorption. The lower value of Log S obtained for compound 6 indicates that is hydrophilic and is more soluble (−4 < LogS < −2), and that absorption takes place in the gastrointestinal tract. The surface area and lipophilicity of this organ influences the rate of absorption of compounds. Compounds 6, 7, and 11 display high predicted gastrointestinal absorption (Table S3).
Five isoenzymes of the CYP enzyme system are mainly involved in the metabolism of most drugs. All six compounds are predicted to be inhibitors of CYP2C19 and CYP1C2. This suggested that they may be metabolized in the liver. Moreover, they are predicted not to be inhibitors of isoenzyme CYP2D6. This is very useful, because these compounds are expected to not have CYP metabolism interactions with other drugs and would not present an increased hepatotoxicity risk.
Drug oral bioavailability is the fractional extent of the drug dosage that finally reaches the therapeutic active site; an acceptable probability score of 55% indicates that the drug candidate has met the rule of five. All candidate compounds have shown a score of 55%, indicating good bioavailability.
For the parameters of the distribution, the blood–brain barrier (BBB) protects the brain from harmful substances. The capacity of a medicine to cross into the brain is a critical criterion to consider when lowering side effects and toxicities or improving the efficacy of treatments having pharmacological action in the brain. It is believed that no compounds pass through the BBB; also, the results show that there is no P-gp substrate for all the screened compounds.
When a drug is bioavailable and administered at the proper dose rate, it reaches a steady-state level. After evaluating characteristics of compound permeability through various barriers, we predicted the excretion parameters total renal clearance (log mL/min/kg); OCT2 substrate.
The OCT2 is a protein transporter that has a vital contribution to renal absorption, disposition and clearance of drug compounds. Evaluation of the transfer of a candidate compound by OCT2 provides useful information as regards not only its clearance but also its potential contraindications. In silico analysis revealed that all compounds except 6 and 7 were not substrates of human OCT2, which is important for the excretion of cationic molecules. So, compounds 6 and 7 are predicted to undergo a renal uptake process and stay longer in the body unlike other compounds that will not have a problem with renal absorption.
The faster the excretion process, the greater the compound's CLTOT value, From Table S4 it can be seen that the log CLTOT value of the test compounds ranges from −0.151 to 0.045 ml/min/kg and from these values can be predicted that all compounds tested are relatively rapidly excreted from the body.
3.3 Molecular dynamics simulations
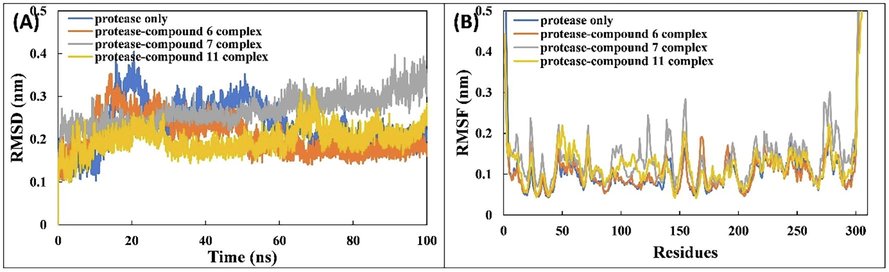
The MD simulation of Mpro and associated complexes was performed for 100 ns. The RMSDs of the proteins and associated complexes were calculated with respect to their initially prepared structures (Fig. 2A). The RMSD values of each system reached equilibrium after 40 ns and then leveled out. The average RMSD of Mpro alone is 0.232 ± 0.052 nm. The RMSDs of complexes of compounds 6, 7, and 11 are 0.206 ± 0.043, 0.265 ± 0.034, and 0.197 ± 0.028 nm, respectively. The RMSD curves of Mpro alone and all associated complexes were similar with only minor deviations. The RMSD of the entire system is less than 0.3 nm, indicting the good stability of the complexes in the aqueous environment.
The average RMSF values of backbones Cα atoms of Mpro alone and associated complexes are calculated to assess dynamic behavior of amino acids of the SARS-CoV-2 Mpro. The analysis of RMSF gives useful insights regarding the structural fluctuations of different regions of the protein. Greater fluctuations in residues signify a less stable nature of the protein. The RMSF of Mpro alone and associated complexes is shown in Fig. 5B and remained below 0.2 nm for the majority of the amino acids of Mpro, indicating the stability of the ligands with the test protein. However, several fluctuations are observed at the N- and C-termini that may be due to the hanging position of these residues. Several fluctuations are also found in the certain regions of Mpro in the presence of ligands, which may be due to the dynamic behavior of ligands within the bonding region. The average RMSF of individual atoms of ligands were also calculated to check the dynamics of ligand and showed some variation, denoting the dynamical shift from their initial position.
The Rg of Mpro in the absence and presence of ligands was also calculated (Fig. 3A) as this indicates the stability of protein during MD simulation. In general, a compact protein shows less variation in Rg values compared with those of an expanded protein. The average Rg values of Mpro alone and in complex with compounds 6, 7, or 11 were 2.188 ± 0.013, 2.198 ± 0.018, 2.207 ± 0.012, and 2.233 ± 0.021 nm, respectively. These values signify the stability of the ligands with Mpro. The stability was further verified by calculating the changes in SASA. The SASA of Mpro and associated complexes is shown in Fig. 3B. The SASA of all complexes are similar to Mpro alone, further validating the stability of the ligands with the protein.
The interactions of ligands with Mpro were studied by calculating the hydrogen bond profiles. The number of hydrogen bonds with respect to time is shown in Fig. 4A. Hydrogen bonds are present between the ligands and Mpro over the entire simulation period. The residues of Mpro that are involved in hydrogen bond formation with compound 6areHis41, Asn142, Gly143, Ser144, Cys145, His163, and Gln189. Similarly, Thr24, Thr25, Thr26, His41, Ser46, Met49, Leu50, Asn142, Gly143, Ser144, Cys145, His163, Glu166, and Gln189 are involved in hydrogen bond formation with compound 7, and Ser144, His163, Arg188, Gln189, and Gln192 formed hydrogen bonds with compound 11; the lower number of hydrogen bonds formed with compound 11 may be due to the interaction of water molecules at the binding sites.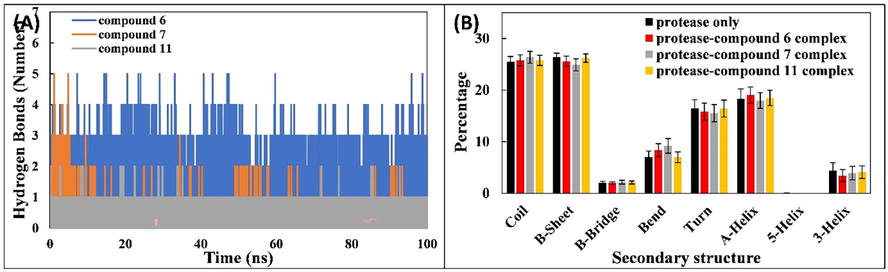
(A) Number of hydrogen bonds between SARS-CoV-2 Mpro and ligands (compounds 6, 7, and 11). (B) Percentage of secondary structure in Mpro and when in complex with ligands (compounds 6, 7, and 11).
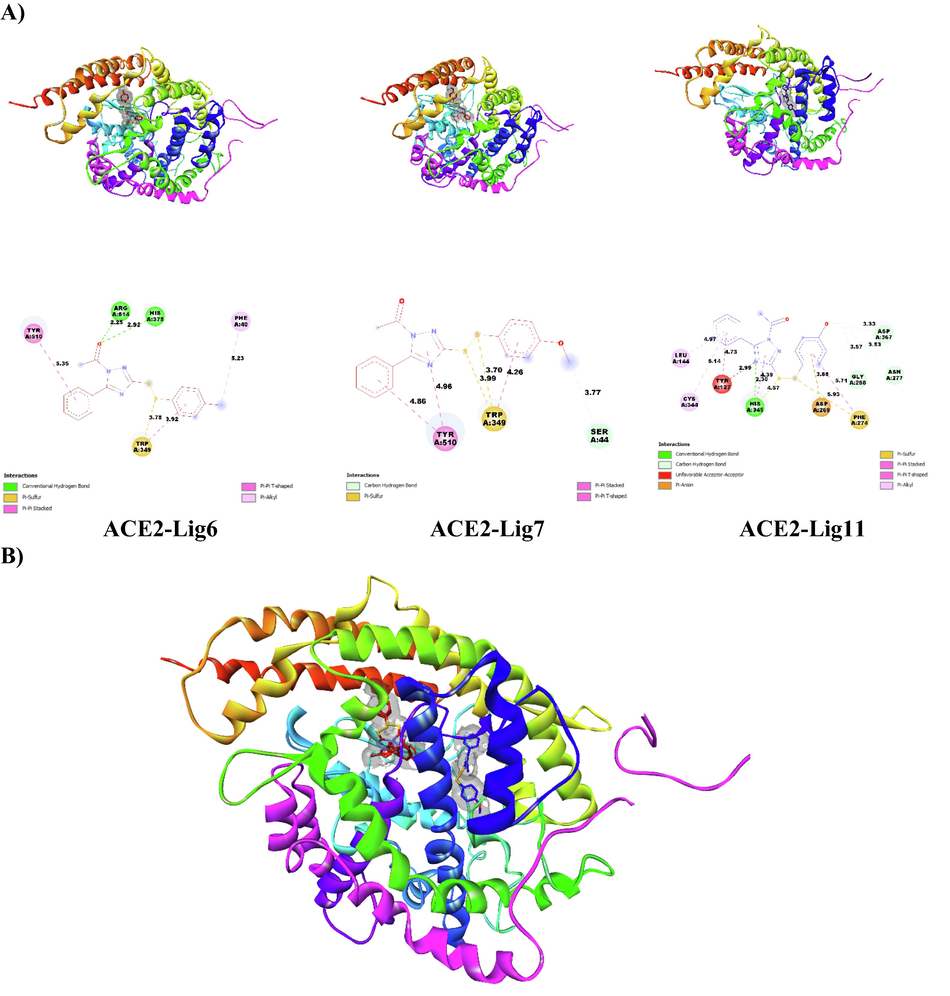
(A) Docked conformation poses of ligands 6, 7 and 11 inside the ACE2 receptor pocket and their two-dimensional visualization; (B) superimposed view of ligands 6, 7 and 11 inside the ACE2 receptor pocket.
The effect of binding of compounds 6, 7, and 11 on the secondary structure of Mpro was also assessed. The percentages of secondary structures in Mpro and associated complexes with compound 6, 7, and 11 are shown in Fig. 4B. The percentages of coil, β-sheet, β-bridge, bend, turn, and α-helix were 25.44%, 26.35%, 2.02%, 7.05%, 16.44%, and 18.30%, respectively. The interaction of ligands with Mpro resulted in insignificant changes in the secondary structures of the protein. This data further validates the structural stability of Mpro in complex with compound 6, 7, or 11.
The binding energies and forces involved in the complexation of compounds 6, 7, and 11 with Mpro were studied using the MM-PBSA analysis. The binding energy was calculated for 100 snapshots extracted from the trajectory at equal intervals of the entire MD simulation.
The different energies observed in the principal interaction of the ligands with Mpro are presented in Table 4. In typical drug-protein interactions, the major forces observed are hydrogen bonds, electrostatic forces, hydrophobic interactions and Van der Waals forces, which positively or negatively contribute to the total binding energy (Qais et al., 2021). The energetic contribution of the Van der Waals forces is the major contributor and most favorable in the overall binding. Electrostatic energy and SASA energy also positively contributed to the binding of all ligands with Mpro. In contrary, altered solvation energy binding of all ligands to Mpro.
The total average binding energy for binding of compounds 6, 7, and 11 with Mprowas −69.489 ± 1.388, −81.212 ± 1.267, and −90.842 ± 1.079 kJ/mol.
3.4 Ligand-receptor interactions with human ACE2 receptor
To further support the results of molecular docking and molecular dynamics as well as ADMET properties related to the potential candidate molecules 6, 7 and 11 to inhibit SARS CoV-2 Mpro, we evaluate the ability of these molecules to interact with the active sites of the human ACE2 receptor (PDB 1R4L) (https://www.rcsb.org/structure/1R4L). This is to predict the most important interactions likely to occur in the inhibition of human ACE2 activity by the disulfide derivatives. We perform this procedure in order to predict the modes of interaction of molecules 6, 7 and 11 with human ACE2 receptors responsible for viral binding to host cells (Mohapatra et al.; Abdalla et al.) and to identify active reference sites likely to be targeted by disulfide derivatives to disrupt the interaction of SARS-CoV-2 virus with ACE2 receptors and thus inhibit entry of this virus. For this purpose, we prepared the ACE2 protein and its ligands 6, 7 and 11 with the same tools and methodology used to prepare the main protease of SARS-CoV-2 in this work. Coordinates (x = 40.605, y = 2.495, 22.919) and size (80x80x80) containing the entire ACE2 protein structure were identified as the insertion site for the 3 ligands. The summary of the obtained results by molecular docking is presented in Table 5 and the different conformations poses of the ligands in the ACE2 receptor pocket are presented in Fig. 5.
Ligands
Receptor
Binding Affinity (kcal/mol)
Electrostatic Interaction
Hydrogen-Binding
InteractionHydrophobic Interaction
6
ACE2
−8.3
Trp349
Arg514, His378
Tyr510, Trp349, Phe40
7
−8.0
Trp349
Ser44
Tyr510
11
−8.5
Asp269, Phe274
His345, Gly268, Asp367, Aqn277, Tyr127
Leu144, Gly344, Tyr127, His345, Phe274
Through the results presented in Table 5, we notice that the binding energies of ligands 6, 7 and 11 with the predicted reference sites are high and close to each other.
Therefore, it is difficult to prefer one molecule among others, and this may be an appropriate reason to suggest the three-screened molecules as potential inhibitors of ACE2 as well as Mpro towards removing Coronavirus in the future.
Finally, we suggest the disulfides 6, 7 and 11 as SARS CoV-2 Mpro inhibitors because of their good interaction ability with tested protein. The compounds have also excellent bioavailability and do not pose toxicity concerns. The complexes of these compounds are also stable when simulated in aqueous medium. Therefore, based on these observations, the selected three compounds are potential candidates of SARS CoV-2 Mpro inhibitors.
4 Conclusion
In this work, molecular docking was performed on 40 unsymmetrical aromatic disulfides known as anti-SARS-CoV agents. Many of these compounds have shown remarkable binding interactions with SARS-CoV-2 Mpro. The outcomes have shown that the selected three disulfides (6, 7, and 11) exhibit potent ability to fight SARS-CoV-2in-silico. Also, they have shown good stability in MD simulation with SARS-CoV-2 Mpro in aqueous conditions. In addition, many of the investigated disulfides have shown good oral bioavailability and high gastrointestinal absorption. Finally, further studies should be performed in vitro and in vivo to assert if these compounds could be drug candidates to treat SARS-CoV-2. This work could constitute an in-silico approach toward the identification of novel inhibitors of anti-SARS-CoV-2.
Funding
We would like to express our gratefulness to the “Agence Universitaire de la Francophonie (AUF)” for funding this research project (reference: AUF-DRM6588. Title: Composé dérivés de l’eugénol: hémisynthèse ciblée via la relation structure activité et investigation de l’activité anti-SARS-CoV-2).
Conflict of interest
The authors declare no conflict of interest, financial or otherwise.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763-1767.
- [Google Scholar]
- Identification of a novel dual-target scaffold for 3CLpro and RdRp proteins of SARS-CoV-2 using 3D-similarity search, molecular docking, molecular dynamics and ADMET evaluation. J. Biomol. Struct. Dyn. 2020
- [CrossRef] [Google Scholar]
- QSAR study of unsymmetrical aromatic Disulfides as potent avian SARS-CoV main protease inhibitors using quantum chemical descriptors and statistical methods. Chemomet. Intell. Lab. Syst.. 2021;210(15):104266
- [CrossRef] [Google Scholar]
- Discovery of potent SARS-CoV-2 inhibitors from approved antiviral drugs via docking screening. Comb. Chem. High Throughput Screening. 2021;24(3):441-454.
- [CrossRef] [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7(1):42717.
- [Google Scholar]
- Insights into protein–ligand interactions: mechanisms, models, and methods. Int. J. Mol. J. Sci.. 2016;17(2):144.
- [Google Scholar]
- QSPR studies on aqueous solubilities of drug-like compounds. Int. J. Mol. Sci.. 2009;10(6):2558-2577.
- [Google Scholar]
- Cameroonian Medicinal plants as Potential candidates of SARS-CoV-2 Inhibitors. J. Biomol. Struct. Dyn. 2021
- [Google Scholar]
- The continuing 2019-nCoV epidemic threat of novel coronaviruses to globalhealth-the latest 2019-nCoV outbreak in Wuhan, China. Int. J. Infect. Dis.. 2020;91:264-266.
- [Google Scholar]
- Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndromecoronavirus (SARS-CoV) 3CL protease. J. Med. Chem.. 2013;56(2):534-546.
- [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 1997;23(1–3):3-26.
- [Google Scholar]
- Synthesis, modification and docking studies of 5-sulfonyl isatin derivatives as SARS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem.. 2014;22(1):292-302.
- [Google Scholar]
- Liu, X., Zhang, B., Jin, Z., Yang, H., and Rao, Z. The Crystal Structure of COVID-19 Main Protease In Complex With an Inhibitor N3 Complex (PDB ID:6lu7), 2020.
- The genome sequence of the SARS-associated coronavirus. Science. 2003;300(5624):1399-1404.
- [Google Scholar]
- Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med.. 2003;348(20):1995-2005.
- [Google Scholar]
- Glyburide inhibits non-enzymatic glycation of HSA: an approach for the management of AGEs associated diabetic complications. Int. J. Biol. Macromol.. 2021;169:143-152.
- [Google Scholar]
- Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorg. Med. Chem.. 2010;18(22):7849-7854.
- [Google Scholar]
- Rota, P.A., Oberste, M.S., Monroe, S.S., Nix, W.A., Campagnoli, R., Icenogle, J.P., Peñaranda, S., Bankamp, B., Maher, K., Chen, M., Tong, S., Tamin, A., Lowe, L., Frace, M., DeRisi, J.L., Chen, Q., Wang, D., Erdman, D.D., and Peret, T.C.T., Burn1, C., Ksiazek, T.G., Rollin, P.E., Sanchez, A., Liffick, S., Holloway, B., Limor, J., McCaustland, K., Olsen-Rasmussen, M., Fouchier, R., Günther, S., Osterhaus, A., Droste4, C., Pallansch, M.A., Anderson, L.J., Bellini, W.J., 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science, 300, 1394–1399.
- Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol.. 2003;331(5):991-1004.
- [Google Scholar]
- Design, synthesis, and evaluation of trifluoromethyl ketones as inhibitors of SARS-CoV 3CL protease. Bioorg. Med. Chem.. 2008;16(8):4652-4660.
- [Google Scholar]
- Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: design, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem.. 2013;68:372-384.
- [Google Scholar]
- Eur. J. Med. Chem.. 2013;65:436-447.
- Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol.. 2003;84(9):2305-2315.
- [Google Scholar]
- Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal.. 2020;10(4):313-319.
- [CrossRef] [Google Scholar]
- Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem.. 2002;45(12):2615-2623.
- [Google Scholar]
- Discovery of unsymmetrical aromatic disulfides as novel inhibitors of SARS-CoV main protease: chemical synthesis, biological evaluation, molecular docking and 3D-QSAR study. Eur. J. Med. Chem.. 2017;137:450-461.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102226.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




