

Translate this page into:
Triple targeting of human IMPDH, SARS-CoV-2 RdRp, and Rhizopus oryzae RdRp: An in silico perspective
⁎Corresponding author at: Biophysics Department, Faculty of Sciences, Cairo University, Giza, Egypt. abdo@sci.cu.edu.eg (Abdo A. Elfiky) dr_abdo@cu.edu.eg (Abdo A. Elfiky)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Objectives: Mucormycosis has been reported associated with SARS-CoV-2 infections during the last year. The viral RNA-dependent RNA polymerase (RdRp) was proved to be a critical protein target in viral and fungal pathogens. The human inosine monophosphate dehydrogenase (IMPDH) is an evolved antiviral and antimicrobial therapeutic target. The aim is to triple-hit the viral and fungal RdRps and the human IMPDH. Methods: In the current study, molecular docking combined with molecular dynamics simulation (MDS) is utilized to test nucleotide inhibitors against the RdRps of SARS-CoV-2 and Rhizopus oryzae (the main causing agent of mucormycosis) RdRp. Additionally, the same inhibitors targeted the human Inosine monophosphate dehydrogenase (IMPDH). Results: The results reveal a comparable binding affinity of four nucleotide derivatives compared to remdesivir and sofosbuvir against both IMPDH and the RdRps of SARS-CoV-2 and Rhizopus oryzae. The binding affinities are calculated using different conformations of the RdRps after 100 ns MDS and trajectories clustering. Conclusions: The current study suggests the triple inhibition potential of four nucleotide inhibitors against SARS-CoV-2 & R. oryzae RdRps and the human IMPDH, while experimental validation is yet to be performed.
Keywords
Mucormycosis
Computational drug design
SARS-CoV-2
Nucleotide inhibitors
IMPDH
RdRp

1 Introduction
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and its associated pneumonia (COVID-19) caused health and economic burdens during the last two years. The currently developed pandemic of Mucormycosis in COVID-19 patients and COVID-19 recovered people is causing a debate about the therapeutics approved against COVID-19 (Werthman-Ehrenreich, 2021; Vaidyanathan, 2021). Several in silico studies are able to suggest possible medicines against COVID-19 even before the deposition of any solved structures for the viral proteins (Elfiky et al., 2021; Ibrahim et al., 2020; Sonousi et al., 2021). Some of the recommended drugs are now approved by the Food and Drugs Administration (FDA) and save lives (Sonousi et al., 2021; Beigel et al., 2020).
The viral RNA-dependent RNA polymerase (RdRp) is one of the most targeted viral proteins in many diseases caused by RNA viruses (Sonousi et al., 2021; Elfiky and Elshemey, 2018; Abdo, 2019; Elfiky, 2020). The active site aspartates of the RdRp is very conserved even in the fungal species of Mucorales (Elfiky, 2019a). On the other hand, the human Inosine monophosphate dehydrogenase (IMPDH) has been identified as a key enzyme in cell proliferation and differentiation regulation (Weber et al., 1996; Wu, 1994). IMPDH has recently emerged as an important therapeutic target in the expedition to discover drugs belonging to the immunosuppressive, antiviral, antimicrobial, and anticancer therapeutic era (Shu and Nair, 2008). IMPDH catalyzes the de novo synthesis of purine (guanine) nucleotides, the nicotinamide adenine dinucleotide (NAD)-dependent oxidation of inosine 5-monophosphate (IMP) to xanthosine 5-monophosphate (XMP). The first step of IMPDH reaction involves the attack of catalytic (Cys331) on substrate IMP followed by hydride transfer to NAD+, forming the covalent intermediate E-XMP*. E-XMP* is hydrolyzed during the second step, yielding the product XMP (Weber et al., 1976; Hedstrom and Wang, 1990).
Inhibition of IMPDH produces an overall reduction in guanine nucleotide pools, which subsequently interrupts DNA and RNA synthesis. Additionally, the inhibition of IMPDH is used as a strategy in immuno- suppressive therapy. The growth and differentiation of human lymphocytes are particularly dependent on the IMPDH- catalyzed de novo pathway for purine nucleotide synthesis (Allison et al., 1977). Inhibition of IMPDH leads to suppression of both T and B lymphocyte proliferation. For compounds targeting the IMP-binding site, development has focused on the nucleotide analog. The competitive IMPDH inhibitors for IMP, such as ribavirin (Striepen et al., 2004) and mizoribine (Chen et al., 2010), are nucleoside analogs that undergo phosphorylation to the active inhibitor (triphosphate) in vivo.
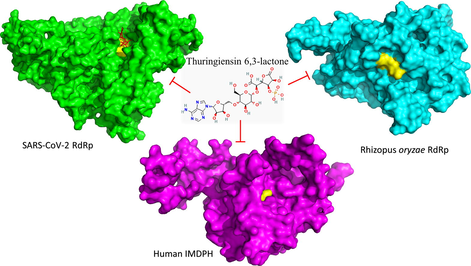
Thuringiensin (Thu), also known as β-exotoxin, is a thermostable secondary metabolite secreted by Bacillus thuringiensis. It has insecticidal activity against many insects, including species belonging to the orders Diptera, Coleoptera, Lepidoptera, Hymenoptera, Orthoptera, and Isoptera, and several nematode species. The insecticidal mechanism of Thu is not fully understood. However, it is known to be an ATP analog (Farkaš et al., 1969). The chemical formula of Thu is C22H32O19N5P, and it is composed of adenosine, glucose, phosphoric acid, and gluconic diacid. Thu inhibits RNA synthesis by competing with ATP on binding sites, affecting insect molting & pupation, and causing teratological effects at sublethal doses (Šebesta and Horska, 1970; Burgerjon et al., 1969; Ignoffo and Gregory, 1972). For a long time, the toxicity of Thu in animals has been a source of contention. Up to now, no conclusive evidence has been found that Thu is hazardous to humans. No significant differences in chromosome aberration rates were reported in rats fed with Thu compared with the controls in analyzing bone marrow mid cells, myeloma cells, and blood cells (Meretoja and Carlberg, 1977). Additionally, Thu was not observed in the liver, heart, kidney, spleen, and adrenal gland of mise after ingestion of 32P-labelled Thu (Liu et al., 2014).
In this study, our approach is to test four nucleotide inhibitors based on Thu against the RdRps of SARS-CoV-2 and Rhizopus oryzae in addition to the human IMPDH. Molecular docking combined with dynamics simulation emphasizes the binding effectiveness using computational tools that are yet to be validated experimentally.
2 Materials and methods
2.1 Structural retrieval and preparations
Thuringiensin (CID: 20056441) and its analogs were retrieved from the PubChem database in the SDF format then converted to PDB using Open Babel software (Kim et al., 2016; O'Boyle et al., 2011). These analogs include Thuringiensin 6,3-Lactone (CID: 102074352), 2-[(2R,3R,4R,5S,6R)-5-[[(2R,3S,4R,5R)-5-(6-Aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy]-3,4-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-3,5-dihydroxy-4-phosphonooxyhexanedioic acid (CID: 99213), and Adenosine 5′-(tetrahydrogentriphosphate), 5′-6-ester with D-glucose (CID: 194147). Sofosbuvir and Remdesivir were used as positive controls. AutoDock Tools 1.5.6 software was utilized to prepare the ligands for the docking (Morris et al., 2009). Kollman and Gasteiger charges were added to the ligands, and then the PDBQT files were generated for the docking experiments.
On the other hand, the 3D structures of the receptors were retrieved from the Protein Data Bank (https://www.rcsb.org//pdb) (Berman et al., 2003). The selected PDB files for the SARS-CoV-2 RdRp and the human IMPDH were 7BTF and 1NF7, respectively (Gao et al., 2020). No solved structure was deposited yet for the R. oryzae RdRp in the protein data bank, so we used the previously in silico generated all-atoms 3D-model by the Swiss Model webserver (Biasini et al., 2014; Elfiky, 2019a). This model was subjected to 100 ns MDS in a previous study and clustered through UCSF Chimera software (Pettersen et al., 2004). Seven different conformations of the R. oryzae model representing the most populated clusters are tested against the inhibitors. The structure 7BTF was subjected in a previous study to 100 ns MDS and then clustered into ten clusters. We used the representative structure from each cluster to test the binding of the nucleotide inhibitors.
2.2 Molecular docking
The receptors were stored in PDB format after removing the water molecules and ligands with PyMOL software (2.4.1). Then polar hydrogen atoms and Kollman charges were added to the receptors using Autodock Tools 1.5.6 then saved in PDBQT format for the docking experiments. Autodock Vina was used for the docking of the ligands to the receptor using flexible ligands and a flexible active site protocol (Trott and Olson, 2010). The grid box was centered at the active residues (D760 & D761 for SARS-CoV-2 RdRp and D193 & D194 for R. oryzae RdRp), defined by the native co-crystallized ligands, while box size was set to (40 × 40 × 40) Å3. On the other hand, the grid box size for the human IMPDH was set to be (40 × 60 × 64) Å3 and centered at (76.7, 83.8, 53.9). The active site for the human IMPDH was S68, N303, R322, S388, Y411, M414, and Q441. Other docking parameters were set to default while the exhaustiveness value was adjusted to 8 in all the docking trials. Discovery studio software is utilized to analyze the docking complexes (Jejurikar and Rohane, 2021).
Duncan's multiple range tests were performed for the calculated average binding affinities to demonstrate the differences using the IBM SPSS statistics 19 software.
3 Results and discussion
In the current study, four nucleotide inhibitors are tested against both host-cell IMPDH and pathogen's proteins (RNA-dependent RNA polymerase). These proteins proved their effectiveness as drug targets (Sonousi et al., 2021; Elfiky et al., 2020; Elfiky, 2019a; Trapero et al., 2018). Fig. 1 shows the 2D structures of the four nucleotide inhibitors which are Thuringiensin derivatives (CID: 99213, 194147, 20056441, and 102074352) and the positive control drugs (Sofosbuvir and Remdesivir). The FDA approves these drugs against RNA viruses such as Hepatitis C Virus, Ebola virus, and SARS-CoV-2 (Elfiky, 2017; Elfiky and Ismail, 2019; Elfiky, 2019b; Adem et al., 2021). The four Thuringiensin derivatives are based on the nucleotide adenosine (A), where the added moieties are present at the 5′ position of the ribose ring. On the other hand, the positive controls drugs (Remdesivir and Sofosbuvir) are adenosine triphosphate modified at position 1′ of the ribose (C≡N) and uridine derivative modified at position 1′ of the ribose (F, methyl), respectively.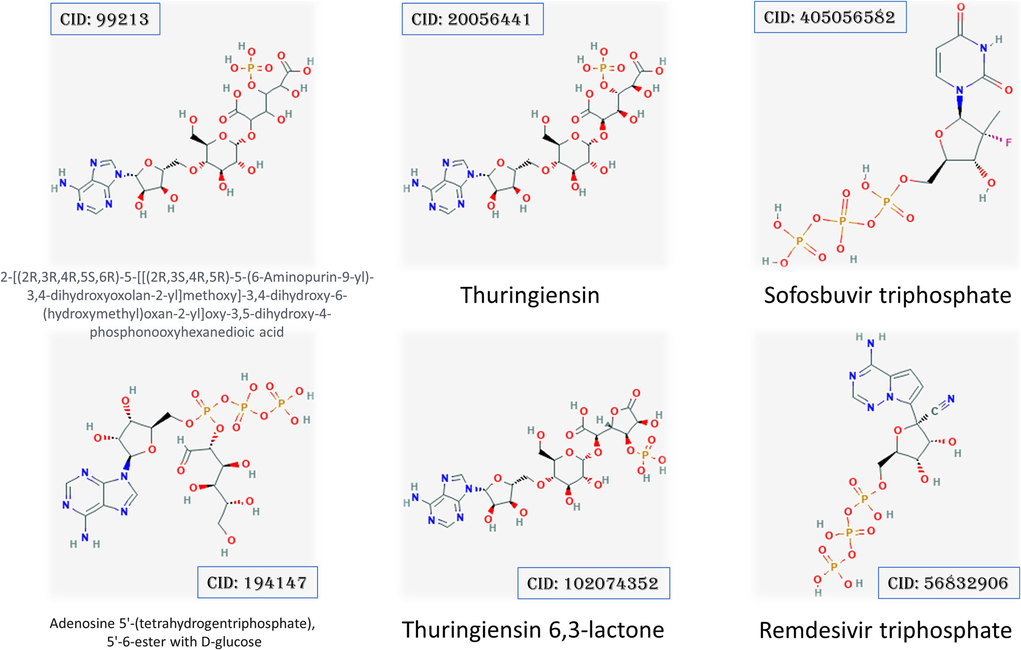
2D structures show the four Thuringiensin derivatives and the positive control drugs Sofosbuvir and Remdesivir with PubChem accession numbers.
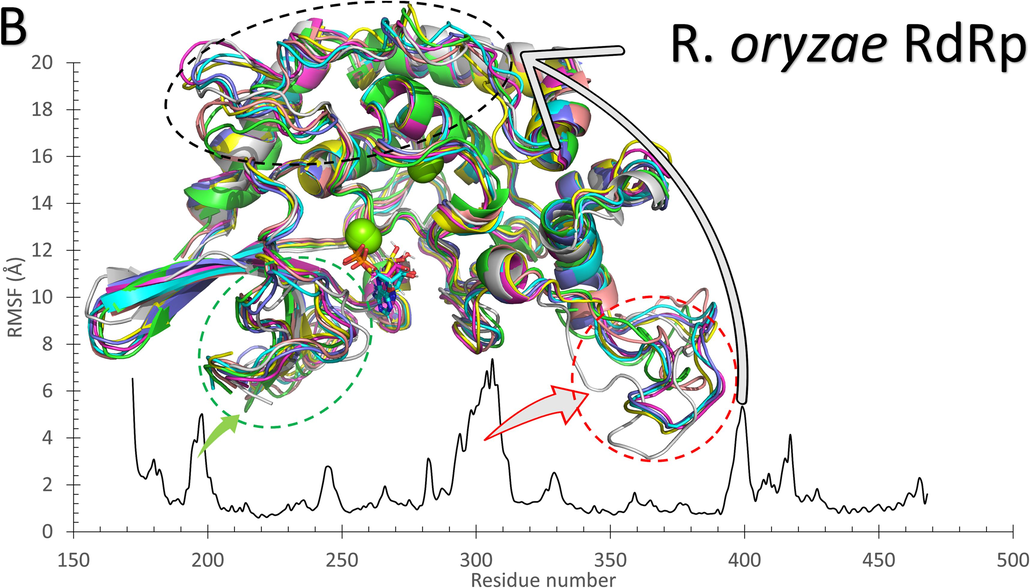
As reflected from Fig. 2, the systems of RdRp (SARS-CoV-2 (A) and R. oryzae (B)) are stable during the molecular dynamics simulation run with some disordered regions as depicted in the structure representatives (superimposed) and the per-residue root-mean-square fluctuation (RMSF) in Å. For example, SARS-CoV-2 RdRp has one high fluctuating region (residues 250–270) with an RMSF value >5 Å and apart from the active site (D760 and D761 shown in black sticks). On the other hand, R. oryzae has three high fluctuating regions, which are 190–205 (green arrow), 290–310 (red arrow), and 395–402 (black arrow). In addition, the root-mean-square deviations (RMSD) (Fig. 2C), the radius of gyration (RoG) (Fig. 2D), and surface accessible surface area (SASA) (Fig. 2E) versus the simulation time indicate systems stability during the 100 ns period.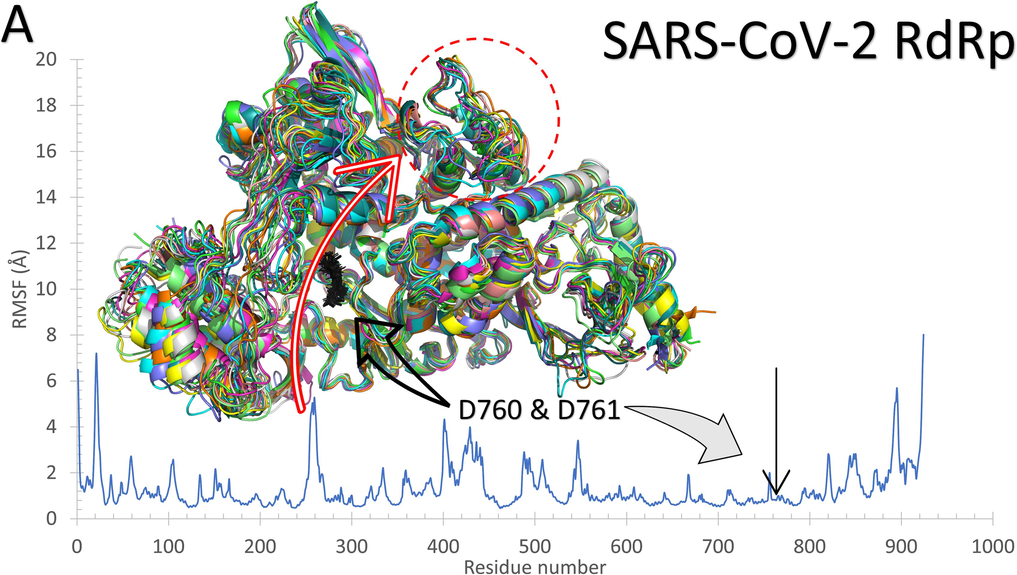
The structures representatives after the molecular dynamics simulations for the RdRp of SARS-CoV-2 (A) and R. oryzae (B). The per-residue root-mean-square fluctuations (in Å) are plotted for each protein along with the superimposed structures depicted in colored cartoons. The Root Mean Square Deviation (RMSD) in Å (C), Radius of Gyration (RoG) in Å (D), and Surface Accessible Surface Area (SASA) in Å2 (E) versus the simulation time (in ns) for the SARS-CoV-2 RdRp (blue) and Rhizopus oryzae RdRp (red).
The structures representatives after the molecular dynamics simulations for the RdRp of SARS-CoV-2 (A) and R. oryzae (B). The per-residue root-mean-square fluctuations (in Å) are plotted for each protein along with the superimposed structures depicted in colored cartoons. The Root Mean Square Deviation (RMSD) in Å (C), Radius of Gyration (RoG) in Å (D), and Surface Accessible Surface Area (SASA) in Å2 (E) versus the simulation time (in ns) for the SARS-CoV-2 RdRp (blue) and Rhizopus oryzae RdRp (red).
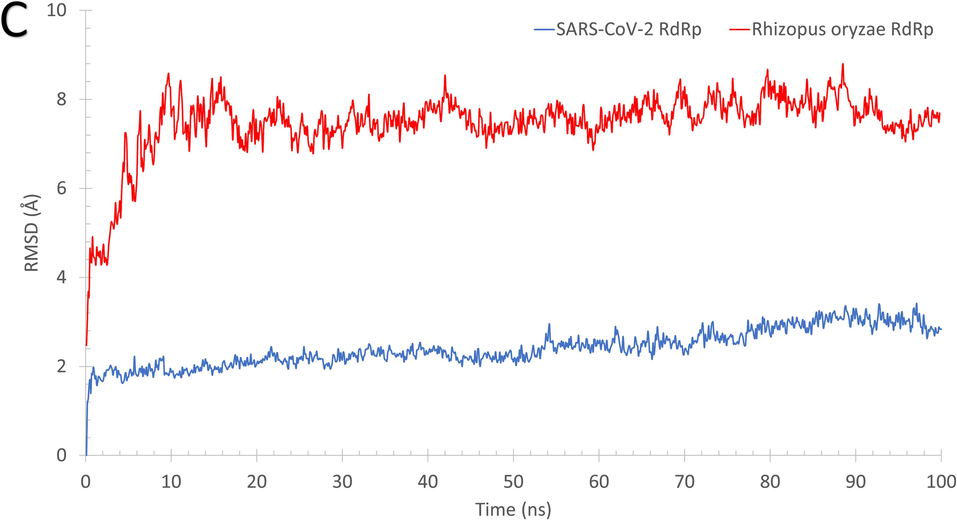
The structures representatives after the molecular dynamics simulations for the RdRp of SARS-CoV-2 (A) and R. oryzae (B). The per-residue root-mean-square fluctuations (in Å) are plotted for each protein along with the superimposed structures depicted in colored cartoons. The Root Mean Square Deviation (RMSD) in Å (C), Radius of Gyration (RoG) in Å (D), and Surface Accessible Surface Area (SASA) in Å2 (E) versus the simulation time (in ns) for the SARS-CoV-2 RdRp (blue) and Rhizopus oryzae RdRp (red).
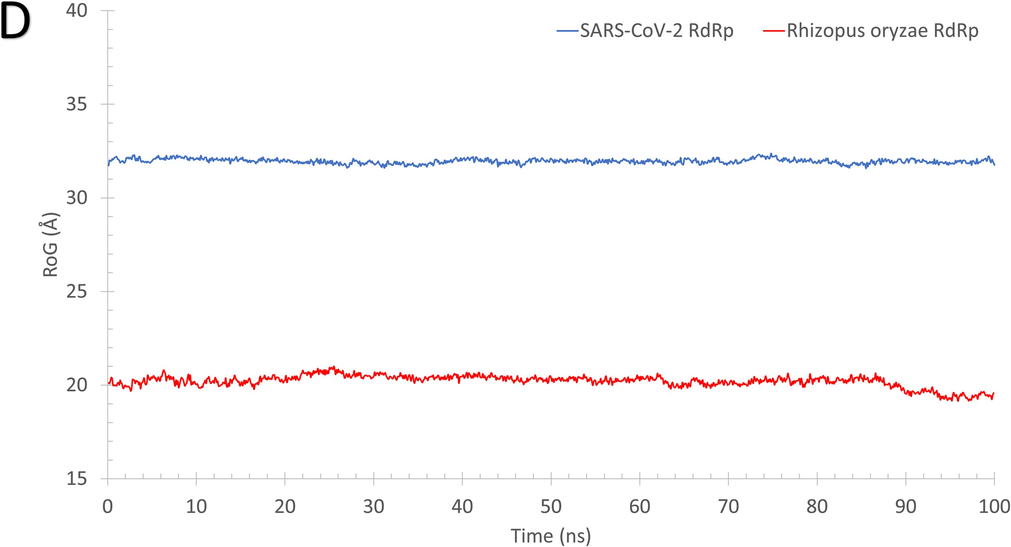
The structures representatives after the molecular dynamics simulations for the RdRp of SARS-CoV-2 (A) and R. oryzae (B). The per-residue root-mean-square fluctuations (in Å) are plotted for each protein along with the superimposed structures depicted in colored cartoons. The Root Mean Square Deviation (RMSD) in Å (C), Radius of Gyration (RoG) in Å (D), and Surface Accessible Surface Area (SASA) in Å2 (E) versus the simulation time (in ns) for the SARS-CoV-2 RdRp (blue) and Rhizopus oryzae RdRp (red).
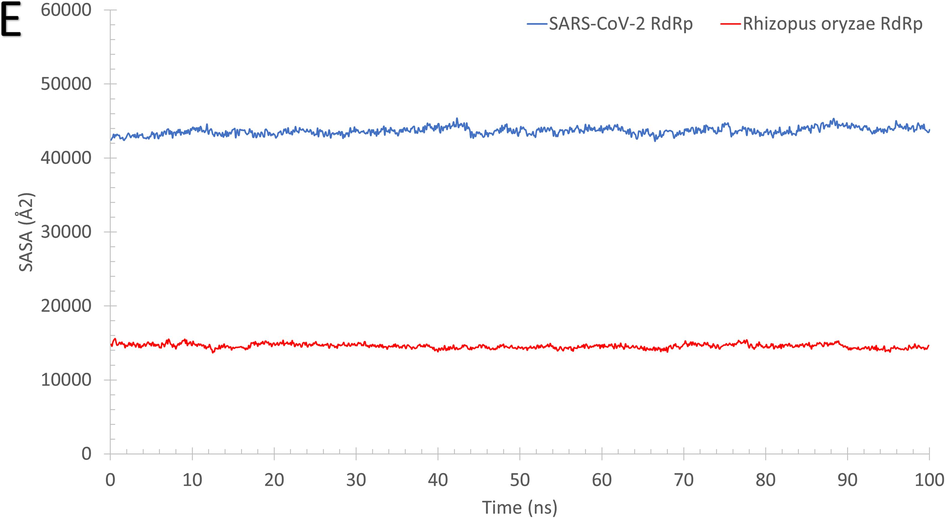
The structures representatives after the molecular dynamics simulations for the RdRp of SARS-CoV-2 (A) and R. oryzae (B). The per-residue root-mean-square fluctuations (in Å) are plotted for each protein along with the superimposed structures depicted in colored cartoons. The Root Mean Square Deviation (RMSD) in Å (C), Radius of Gyration (RoG) in Å (D), and Surface Accessible Surface Area (SASA) in Å2 (E) versus the simulation time (in ns) for the SARS-CoV-2 RdRp (blue) and Rhizopus oryzae RdRp (red).
3.1 Binding affinities of the nucleotide inhibitors against the pathogenic proteins
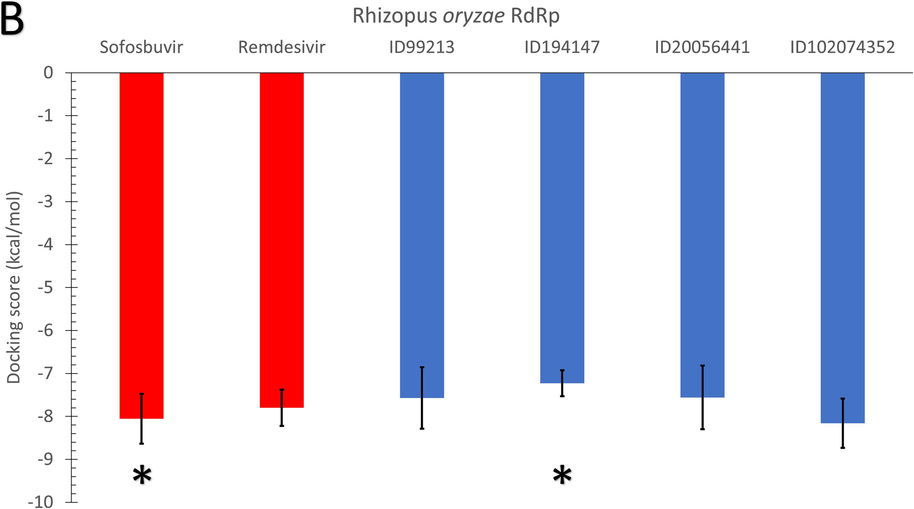
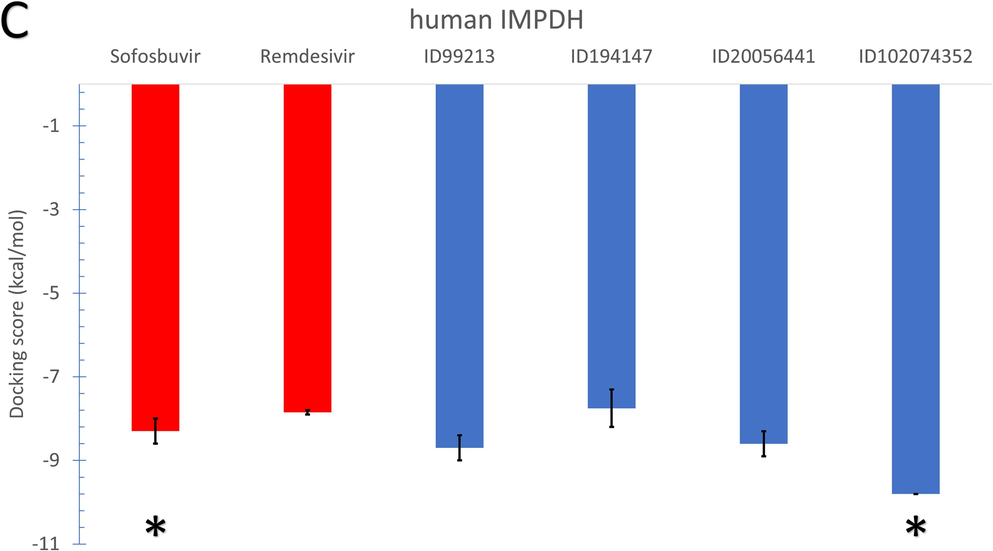
As shown in Fig. 3, the average binding affinities of the four nucleotide inhibitors (blue columns) (CID: 99213, 194147, 20056441, and 102074352) against SARS-CoV-2 RdRp (A), Rhizopus oryzae RdRp (B), and the human IMPDH (C) are in the same range of the positive control drugs. In Fig. 3, the error bars represent the standard deviations, while red columns represent the positive control drugs (Sofosbuvir and Remdesivir). The docking trials are performed on different conformations of the RdRp proteins generated from the 100 ns MDS runs, while Each conformation resembles a cluster from the trajectories. The MDS's role was to equilibrate the protein systems and ensure different possible conformations during the simulation period. According to the Duncan multiple range tests, asterisks in Fig. 3 indicate significantly different mean values (p-value <0.05).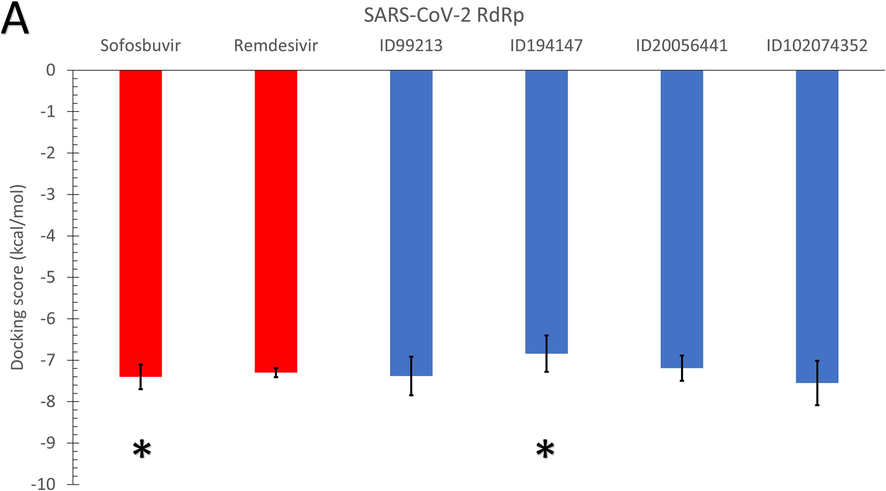
The average binding affinities (in kcal/mol) for the four adenosine derivatives (blue columns) and the positive controls (red columns) against (A) SARS-CoV-2 RdRp, (B) Rhizopus oryzae RdRp, and (C) the human IMPDH. Error bars resemble the standard deviation (SD) of the mean. After MDS cluster analysis, different conformations of the proteins are used in the docking experiments.
The average binding affinities (in kcal/mol) for the four adenosine derivatives (blue columns) and the positive controls (red columns) against (A) SARS-CoV-2 RdRp, (B) Rhizopus oryzae RdRp, and (C) the human IMPDH. Error bars resemble the standard deviation (SD) of the mean. After MDS cluster analysis, different conformations of the proteins are used in the docking experiments.
The average binding affinities (in kcal/mol) for the four adenosine derivatives (blue columns) and the positive controls (red columns) against (A) SARS-CoV-2 RdRp, (B) Rhizopus oryzae RdRp, and (C) the human IMPDH. Error bars resemble the standard deviation (SD) of the mean. After MDS cluster analysis, different conformations of the proteins are used in the docking experiments.
The average binding affinity for the four nucleotide inhibitors lies between −6.84 kcal/mol (194147 versus SARS-CoV-2 RdRp) and −9.8 kcal/mol (102074352 versus human IMPDH). For the SARS-CoV-2 RdRp, the average binding affinity for the four nucleotide inhibitors is −7.24 kcal/mol, while the average binding affinities of sofosbuvir and remdesivir against SARS-CoV-2 RdRp are −7.4 and −7.3 kcal/mol, respectively. Additionally, the average binding affinity of the four nucleotide inhibitors against R. oryzae RdRp is −7.63 kcal/mol, while it is −8.06 and −7.8 kcal/mol for the sofosbuvir and remdesivir, respectively. These values indicate that the Thuringiensin derivatives have comparable average binding affinities to that of Sofosbuvir and Remdesivir against the viral and fungal RdRps. We used Duncan's multiple range tests to better understand the difference between the average binding energies (p-value <0.05). The compound 194147 has significantly higher average binding energies (lower binding affinity) than Sofosbuvir in the SARS-CoV-2 and R. oryzae RdRps.
On the other hand, the average binding affinity of the four nucleotide inhibitors against the human IMPDH is −8.71 kcal/mol, which is lower (better) than the average binding affinity of the positive controls (-8.3 and −7.85 kcal/mol for sofosbuvir and remdesivir, respectively). The best compound from the Thuringiensin derivatives in binding all the proteins is the Thuringiensin 6,3-lactone (CID: 102074352). This compound has a significantly different average value compared to Sofosbuvir in binding the human IMPDH.
In Table 1, according to Duncan's multiple range tests, significantly different means are given different letters for each parameter. For example, Remdesivir can bind the three proteins with no significant difference in mean values. At the same time, Sofosbuvir and 194147 are significantly able to attach to the human IMDPH compared to SARS-CoV-2 RdRp. On the other hand, the compounds 99213, 20056441, and 102074352 are significantly better bound to the human IMDPH than other proteins.
SARS-CoV-2 RdRp
Rhyzopus oryzae RdRp
Human IMPDH
Sofosbuvir
−7.400a ± 0.30
−8.057a,b ± 0.58
−8.300b ± 0.30
Remdesvir
−7.300a ± 0.11
−7.800a ± 0.42
−7.850a ± 0.05
CID:99213
−7.380a ± 0.46
−7.571a ± 0.72
−8.700b ± 0.30
CID:194147
−6.840a ± 0.44
−7.229a,b ± 0.30
−7.750b ± 0.45
CID:20056441
−7.190a ± 0.31
−7.557a ± 0.74
−8.600b ± 0.30
CID:102074352
−7.550a ± 0.54
−8.157a ± 0.58
−9.800b ± 0.00
In order to check the binding mode of each ligand to the three proteins, the interaction map of the complexes is mined by the Discovery studio software. Based on the analysis, the compounds' binding modes against SARS-CoV-2 RdRp, R. oryzae RdRp, and human IMPDH are explored and tabulated. Table 2 lists the interactions established for each ligand (Thuringiensin derivatives and the positive control drugs) after docking to the active site of the proteins. The main type of interaction established upon docking is the formation of Hydrogen bonds (H-bonds). An average of 13 H-bonds is formed between the positive control drugs and the protein. For the Thuringiensin derivatives, the average number of H-bonds formed is 14, while few hydrophobic contacts, salt bridges (red), and halogen bonds (green) are reported in Table 2. The residues shown in bold are the active site residues in each protein. For example, the most-reported residues from SARS-CoV-2 RdRp that take part in the interactions with the nucleotide inhibitors are D760(12), D761(9), D618(9), R555(8), E811(6), S814(6), and Y619(5), ranked by the number of reported interaction established upon docking. For the R. oryzae RdRp, the most reported residues to interact with the fungal RdRp are E27(15), D56(12), R14(10), D193(8), G142(5), S146(5), D194(5). On the other hand, the residues from the human IMPDH that reported to interact with the nucleotide inhibitors are D364(14), D274(12), S276(8), H93(6), G326(6), D256(5), C331(5), and T333(5).
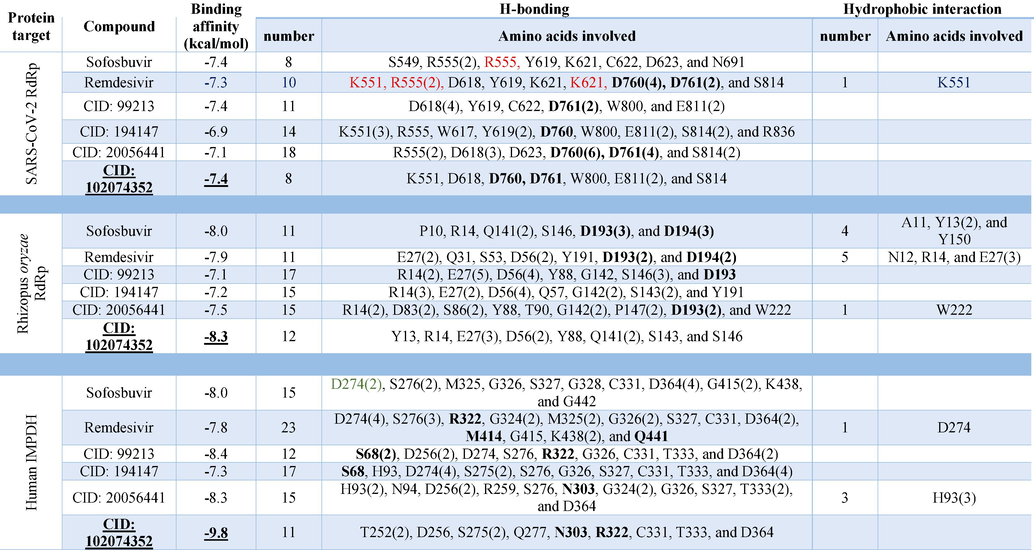
Fig. 4 shows the interactions that established between Thuringiensin 6,3-lactone (CID: 102074352), and the SARS-CoV-2 RdRp (top left), R. oryzae RdRp (top write), and the human IMPDH (down). This compound shows the best average binding affinities against the three proteins (significantly differ than sofosbuvir in IMPDH, p-value <0.05) compared to other nucleotide inhibitors. Fig. 4 is represented by Discovery studio software, where the ligands are shown in the sticks, and the interacting residues are depicted in lines. The only type of interaction is the formation of H-bonds (dashed lines). Thuringiensin 6,3-lactone formed 8 H-bonds to SARS-CoV-2 RdRp with residues K551, D618, D760, D761, W800, E811(2), and S814. For R. oryzae RdRp Y13, R14, E27(3), D56(2), Y88, Q141(2), S143, and S146 formed 12 H-bonds with Thuringiensin 6,3-lactone. At the same time, residues T252(2), D256, S275(2), Q277, N303, R322, C331, T333, and D364 of the human IMPDH formed 11 H-bonds with Thuringiensin 6,3-lactone.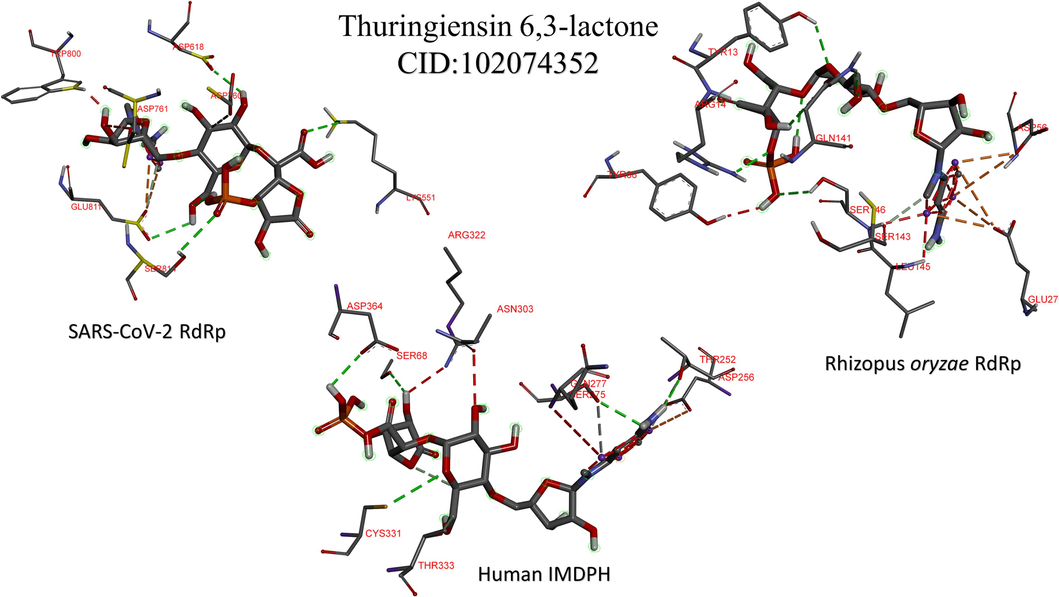
The interaction patterns for the best adenosine derivative (Thuringiensin 6,3-lactone) after docking into the active site of SARS-CoV-2 RdRp (top left), Rhizopus oryzae RdRp (top right), and the human IMPDH (bottom). The residues from the proteins that form contacts to the ligands are depicted in lines, while the ligands are in the sticks. H-bonds are shown in dashed lines.
The current work reveals the favorable binding affinity of four adenosine derivatives against the SARS-CoV-2 RdRp, Rhizopus oryzae RdRp, and the human IMPDH. Thuringiensin 6,3-lactone (CID: 102074352) has the best binding affinity to the three proteins from which it is significantly different compared to sofosbuvir in binding IMPDH, p-value <0.05. Hence can be a potential inhibitor against COVID-19/Mucormycosis coinfection, while experimental validation is yet to be performed.
4 Conclusion
Remdesivir proves its effectiveness against COVID-19 (reducing the hospitalization time). Four Thuringiensin derivatives (adenosine based) are tested against SARS-CoV-2 RdRp, Rhizopus oryzae RdRp, and the human IMPDH using in silico techniques. The results reveal the binding potential of the Thuringiensin derivative (Thuringiensin 6,3-lactone) against the viral, fungal, and human proteins. These adenosine derivatives can be potential inhibitors of the dual SARS-CoV-2 (COVID-19) R. oryzae (Mucormycosis) coinfection. Further, experimental validations are suggested for the best compound (Thuringiensin 6,3-lactone) as future work.
Funding
This paper is based upon work supported by This paper is based upon work supported by The Science and Technology Development Fund (STDF), grant no. 44575.
Authors' contribution
A.H. owned the idea, wrote the methods, and revised the manuscript. A.E. & A.E. drafted the manuscript, make calculations, and generated figures and tables. All the authors wrote and approved the final draft of the manuscript.
Acknowledgment
Bibliotheca Alexandrina is used to performing the MDS calculations. This paper is based upon work supported by The Science and Technology Development Fund (STDF), grant no. 44575.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 2.4.1 PV. The PyMOL Molecular Graphics System, Version 2.4.1 Schrödinger, LLC.
- Novel guanosine derivatives as anti-HCV NS5b polymerase: A QSAR and molecular docking study. Med. Chem.. 2019;15:130-137.
- [Google Scholar]
- Caffeic acid derivatives (CAFDs) as inhibitors of SARS-CoV-2: CAFDs-based functional foods as a potential alternative approach to combat COVID-19. Phytomedicine. 2021;85:153310
- [Google Scholar]
- The role of de novo purine synthesis in lymphocyte transformation. Ciba Found. Symp. 1977:207-224.
- [Google Scholar]
- SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res.. 2014;42:W252-W258.
- [Google Scholar]
- Teratology of the Colorado potato beetle, Leptinotarsa decemlineata, as provoked by larval administration of the thermostable toxin of Bacillus thuringiensis. J. Invertebr. Pathol.. 1969;14:274-278.
- [Google Scholar]
- Triazole-linked inhibitors of inosine monophosphate dehydrogenase from human and Mycobacterium tuberculosis. J. Med. Chem.. 2010;53:4768-4778.
- [Google Scholar]
- Zika virus: novel guanosine derivatives revealed strong binding and possible inhibition of the polymerase. Futur. Virol.. 2017;12:721-728.
- [Google Scholar]
- The antiviral Sofosbuvir against mucormycosis: an in silico perspective. Futur. Virol.. 2019;14:739-744.
- [Google Scholar]
- Novel guanosine derivatives as anti-HCV NS5b polymerase: A QSAR and molecular docking study. Med. Chem.. 2019;15:130-137.
- [Google Scholar]
- Novel guanosine derivatives against Zika virus polymerase in silico. J. Med. Virol.. 2020;92:11-16.
- [Google Scholar]
- Molecular dynamics simulation revealed binding of nucleotide inhibitors to ZIKV polymerase over 444 nanoseconds. J. Med. Virol.. 2018;90:13-18.
- [Google Scholar]
- The anti-HCV, Sofosbuvir, versus the anti-EBOV Remdesivir against SARS-CoV-2 RNA dependent RNA polymerase in silico. Mol. Divers. 2020
- [Google Scholar]
- Molecular dynamics and docking reveal the potency of novel GTP derivatives against RNA dependent RNA polymerase of genotype 4a HCV. Life Sci.. 2019;238:116958
- [Google Scholar]
- The structure of exotoxin of Bacillus thuringiensis var. gelechiae. Preliminary communication. Collect. Czech. Chem. Commun.. 1969;34:1118-1120.
- [Google Scholar]
- Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779-782.
- [Google Scholar]
- Mycophenolic acid and thiazole adenine dinucleotide inhibition of Tritrichomonas foetus inosine 5'-monophosphate dehydrogenase: implications on enzyme mechanism. Biochemistry. 1990;29:849-854.
- [Google Scholar]
- COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect.. 2020;80:554-562.
- [Google Scholar]
- Effects of Bacillus thuringiensis β-exotoxin on larval maturation, adult longevity, fecundity, and egg viability in several species of Lepidoptera. Environ. Entomol.. 1972;1:269-272.
- [Google Scholar]
- Thuringiensin: A thermostable secondary metabolite from bacillus thuringiensis with insecticidal activity against a wide range of insects. Toxins. 2014;6:2229-2238.
- [Google Scholar]
- The effect of Bacillus thuringiensis and of cell-free supernatants of some other bacteria on the mitotic activity of human lymphocytes. FEMS Microbiol. Lett.. 1977;2:109-111.
- [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30:2785-2791.
- [Google Scholar]
- UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem.. 2004;25:1605-1612.
- [Google Scholar]
- Mechanism of inhibition of DNA-dependent RNA polymerase by exotoxin of Bacillus thuringiensis. Biochimica et Biophysica Acta (BBA)-Nucleic Acids and Protein. Synthesis. 1970;209:357-367.
- [Google Scholar]
- Inosine monophosphate dehydrogenase (IMPDH) as a target in drug discovery. Med. Res. Rev.. 2008;28:219-232.
- [Google Scholar]
- Novel adenosine derivatives against SARS-CoV-2 RNA-dependent RNA polymerase: an in silico perspective. Pharmacol. Rep.. 2021;73:1754-1764.
- [Google Scholar]
- Gene transfer in the evolution of parasite nucleotide biosynthesis. PNAS. 2004;101:3154-3159.
- [Google Scholar]
- Fragment-Based Approach to Targeting Inosine-5'-monophosphate Dehydrogenase (IMPDH) from Mycobacterium tuberculosis. J. Med. Chem.. 2018;61:2806-2822.
- [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [Google Scholar]
- Coronavirus variants are spreading in India—what scientists know so far. Nature. 2021;332
- [Google Scholar]
- Key enzymes of IMP metabolism: transformation and proliferation-linked alterations in gene expression. Adv. Enzyme Regul.. 1976;14:3-24.
- [Google Scholar]
- Mucormycosis with orbital compartment syndrome in a patient with COVID-19. Am. J. Emerg. Med.. 2021;42:264. e265
- [Google Scholar]
- Mycophenolate mofetil: molecular mechanisms of action. Perspect. Drug Discovery Des.. 1994;2:185-204.
- [Google Scholar]




