

Translate this page into:
Synthesis, physicochemical, thermal, XDR/HSA-interactions of Trans-(1E,2E)-Benzil-O,O-dimethylsulfonyl dioxime: Cis-trans isomerization, DFT and TD-DFT investigation
⁎Corresponding authors. nalzaqri@ksu.edu.sa (Nabil Al-Zaqri), warad@najah.edu (Ismail Warad)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
In this study, a novel trans-(1E,2E)-benzil-O,O-dimethylsulfonyl dioxime (BDMDO) monomer was prepared via a one-step dehydrochlorination reaction. BDMDO was characterized by 1H and 13C NMR, UV–Vis, and FT-IR spectroscopies, CHN elemental analysis, mass spectrometry, energy-dispersive X-ray spectroscopy, and TGA. According to the X-ray diffraction data, the BDMDO crystal structure was solved as a trans-isomer dioxime. The lattice structure was stabilized by two types of H-bonds and a sufficient number of interesting non-covalent supramolecular interactions such as the C—H⋯H—C bonds. Molecular electrostatic potential measurements and Hirschfeld surface analysis were performed to understand these interaction modes. The cis–trans isomerization in BDMDO as well as the structural, vibrational, NMR, highest occupied molecular orbital/lowest unoccupied molecular orbital bandgap, density of states, and electronic properties were computed by the density functional theory and subsequently compared with available experimental 1H and 13C NMR, UV–Vis, and FT-IR spectral data. The thermogravimetric/derivative thermogravimetric behavior of BDMDO was determined experimentally under an open-room atmosphere.
Keywords
Dioxime
XRD
HSA
MEP
NMR

- DFT
-
Density functional theory
- DOS
-
Density of states
- EDX
-
Energy-dispersive X-ray
- GIAO
-
Gauge-including atomic orbital
- HSA
-
Hirschfeld surface analysis
- MEP
-
Molecular electrostatic potential
- MS
-
Mass spectrometry
- TS
-
Transition state
- XRD
-
X-ray diffraction
Abbreviations
1 Introduction
Oximes (>C=N-O–H) and their derivatives such as > C=N-O-SO2-R are useful building blocks in the preparation of organics with SNO polar atoms (Türkkan et al., 2012). Generally, oximes are prepared by the reaction of ketones or aldehydes with hydroxylamine. They are considered as key synthetic precursors because they can be easily converted to carbonyl, amino, nitro, and cyano groups (Özyürek et al., 2014). This makes oximes essential in laboratories, industries, and in our daily life. For example, in Japan, perillaldehyde-oxime or perilla sugar is used as a synthetic sweetener as it is two thousand times sweeter than sucrose (Adebayo et al., 2017). Oximes are also used in medicine to treat organophosphate poisoning as antibiotics such as mesylate, gemi-floxacin obidoxime chloride, and pralidoxime chloride (Türkkan et al., 2012) and as pesticides and fungicides such as diazinon and malathion (Adebayo et al., 2017). In addition to their antifungal, antioxidant, antiviral, and antibacterial properties, oximes reportedly exhibit tumor growth inhibition activities (Adebayo et al., 2017; Türkkan et al., 2012; Lorke et al., 2008; Pohanish, 2017; Aakeröy et al., 2013; Talley et al., 2000). Furthermore, owing to their antioxidant activities, oximes have numerous industrial applications, e.g., in plastics, textiles, detergents, radical scavengers, and paint materials(Talley et al., 2000; Türkkan et al., 2012; Adebayo et al., 2017; Lorke et al., 2008; Pohanish, 2017). The chelating properties of oximes expand their applications and have attracted attention relating to their use as polar polydentate ligands. In this role, oximes have been used to evaluate metal ions in environmental samples, e.g., uranium in seawater, nickel and cobalt in tap water and those present in plant foodstuffs and meat (Görgülü and Dede, 2019; Zengin et al., 2019; Motaleb and Selim, 2019; Warad et al., 2018a). Moreover, metal complexes using oximes as ligands have been used as catalysts because of their mode of coordination and structural stability (Gup et al., 2017; Stoumpos et al., 2010; Westcott et al., 2016; Bulatov et al., 2014). Information pertaining to the energetic, structural, molecular, vibrational, magnetic, and electronic behavior of these compounds has been obtained by the density functional theory (DFT), which has facilitated investigation into the design, synthesis, and characterization of novel materials based on oximes (Aouad et al., 2018).
To the best of our knowledge, neither the experimental spectra of trans-(1E,2E)-benzil-O,O-dimethylsulfonyl dioxime (BDMDO) nor DFT-quantum chemical calculations have been reported to date. In this study, we have reported the synthesis and characterization of the trans-dioxime, DMDO. The DFT/experimental structural and spectral parameters were matched in this study. Hirschfeld surface analysis (HSA) and molecular electrostatic potential (MEP) computational analysis were employed to understand the X-ray diffraction (XRD) packing connections. The cis–trans isomerization process was computed successfully by DFT. The thermal behavior of the desired dioxime was determined experimentally using thermogravimetry/derivative thermogravimetry (TG/DTG).
2 Experimental section
2.1 Instruments, software, and calculations
The chemicals used in this study were purchased from Sigma-Aldrich Chemicals and used as received. The XRD structural analysis was performed on a Rigaku RAXIS-RAPID 1000 diffractometer fitted with a charge-coupled device. The UV–Vis analysis was conducted on a Thermo Scientific Genesys 10 s spectrophotometer. Mass spectrometry (MS) measurements were conducted on a Varian MAT 711 spectrometer. The CHN elemental analysis was conducted using Perkin-Elmer Series I1 CHNSlO Analyzer 2400 and IR analysis was performed using a Nicolet Nexus spectrometer. The 1H and 13C NMR spectra were recorded on a JEOL ECS 400 or LAMBDA 400 spectrometer. DFT calculations at the B3LYP level of theory with a 6–311 G(p) basis set were performed using the Gaussian09 software (Wolff et al., 2012). The XRD structure was solved and refined using the SHELXT program)Sheldrick, 1997) and HSA was carried out using the CRYSTAL EXPLORER 3.1 software (Frisch et al., 2009).
2.2 Synthesis
A portion of methanesulfonyl chloride (0.8 g, 7.2 mmol) was dissolved in a mixture of 8 mL of pyridine and 40 mL of tetrahydrofuran (THF). The solution was cooled to −5 °C, and treated dropwise with 0.84 g (3.5 mmol) of diphenylglyoxime in THF for 1 h. The mixture was continuously stirred for 6 h at room temperature, and subsequently treated with 40 mL of dichloromethane and 200 mL of water in a 500 mL separating funnel. The dichloromethane layer was separated, followed by evaporation to obtain the desired product as white crystals in 80% yield (Warad et al., 2010). 1H NMR (CDCl3, ppm): δ 3.5 (s, 6H), 7.2–8.0 (3 m, 10H-ph), 13C NMR δ 28.0 (2CH3), 120.0–130.0 (12C-ph), 134.5 (2C=N) (See Table 1).
Chemical formula
C16H16N2O6S2
CCDC
794,517
Mr
396.43
Crystal system, space group
Triclinic, P
Temp. (K)
293
a, b, c (Å)
5.392 (2), 9.300 (4), 9.420 (4)
α, β, γ (°)
93.933 (9), 90.198 (11), 106.375 (9)
Z
4
V (Å3)
452.0 (3)
µ (mm−1)
0.33
Radiation type
Mo Kα
Diffractometer
Rigaku RAXIS-RAPID
Crystal size (mm)
0.50 × 0.20 × 0.20
No. of measured, independent, and observed [I > 2σ(I)] reflections
2059, 2059, 1741
Rint
0.052
θmax (°)
58.8
(sin θ/λ)max (Å−1)
0.649
R[F2 > 2σ(F2)], wR(F2), S
0.047, 0.157, 1.05
No. of reflections, parameters
2059, 119
Δρmax, Δρmin (e Å−3)
0.33, −0.43
3 Results and discussion
3.1 Synthesis, CHN elemental analysis, and energy-dispersive X-ray (EDX) analyses
BDMDO was synthesized by the dehydrogenation of 1 equiv. of (1E,2E)-benzil dioxime and 2 equiv. of methanesulfonyl chloride in dry THF using pyridine as a soft base. The elemental analysis indicated the theoretical values for the formation of a 1:2 mononuclear dioxime, as shown in Scheme 1.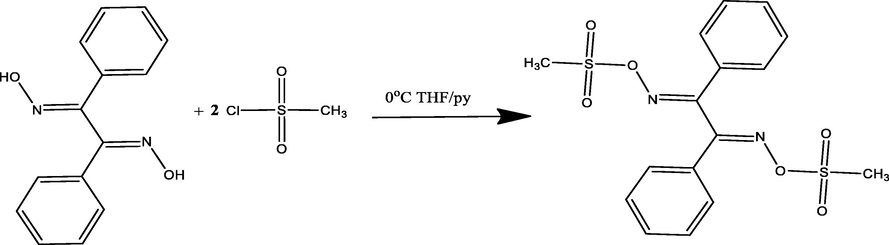
Synthesis of BDMDO.
The EDX analysis results reflected the stereochemistry of the components that combined to form DMDO. The EDX analysis results of the BDMDO structure indicated the presence of four types of atoms, including S, O, N, and C atoms, in its backbone, as shown in Fig. 1a.
(a) EDX and (b) MS of DMDO.
The MS results of BDMDO (m/z = 396.1 (observed), 396.52 (theoretical), Fig. 1b), exhibited sufficient correlation with C16H16N2O6S2: Anal. Calcd C, 48.47; H, 4.07; N, 7.07, found: C, 48.36; H, 4.08; N, 7.01%.
3.2 X-ray single crystal diffraction and DFT calculations
The ORTEP and DFT-optimized structures of BDMDO are illustrated in Fig. 2a and b, respectively. The bond length and angle values obtained by DFT and XRD are summarized in Table 2. BDMDO was crystallized in the lattice with triclinic and P space groups without any solvent molecules in the crystal lattice. Only the anti-isomer was detected by XRD, where the N–SO2CH3 functional groups were pointed far away from each other because of steric hindrance. No sterically disfavored syn isomer was detected by XRD, which is consistent with the previous report of the cis oxime being the less stable isomer [24]. The C⚌N—O—S torsion angle of 177.8° indicated a preference for close planarity; the N⚌C⋯C⚌N dihedral angle was found to be 180°, which is assumed to be a stable trans-conformation isomer of BDMDO. The S-O, O—N, and C⚌N bond lengths of 1.619, 1.438, and 1.281 Å, respectively, and the O—N⚌C, S—O—N, and S—O—C angles of 109.32°, 110.26°, and 102.25°, respectively, were consistent with literature data (Plater et al., 2019). In the XRD packing system, the BDMDO molecules were linked by 2H-bond types, such as CPh-H⋯O=SO2- and CMeH⋯O=SO2- with 2.611 and 2.702 Å bond lengths, respectively, as shown in Fig. 2c, and interesting supramolecular non-covalent interactions such as C—H⋯H—C bonds with a bond length of 2.378 Å, as shown in Fig. 2d.
(a) ORTEP and (b) optimized B3LYP/6-311G(d) structures of BDMDO, and (c) H-bonds and (d) C—H⋯H—C bond interactions.
Bond no.
Bond (Å)
Exp. XRD
DFT/B3LYP
Angles no.
Angles (°)
Exp. XRD
DFT/B3LYP
1
S(1)
O(1)
1.618(2)
1.6907
1
O(1)
S(1)
O(2)
101.8(1)
108.44
2
S(1)
O(2)
1.412(2)
1.4589
2
O(1)
S(1)
O(3)
110.0(1)
109.68
3
S(1)
O(3)
1.413(2)
1.4564
3
O(1)
S(1)
C(8)
102.3(1)
94.24
4
S(1)
C(8)
1.743(2)
1.7969
4
O(2)
S(1)
O(3)
120.1(1)
121.15
5
O(1)
N(1)
1.438(3)
1.4266
5
O(2)
S(1)
C(8)
111.0(1)
110.04
6
N(1)
C(7)
1.281(3)
1.2857
6
O(3)
S(1)
C(8)
109.9(1)
109.94
7
C(1)
C(2)
1.383(4)
1.395
7
S(1)
O(1)
N(1)
110.3(1)
110.25
8
C(1)
C(6)
1.372(3)
1.3782
8
O(1)
N(1)
C(7)
109.3(2)
110.96
9
C(2)
C(3)
1.352(4)
1.3556
9
C(2)
C(1)
C(6)
119.9(2)
119.28
10
C(3)
C(4)
1.353(4)
1.3544
10
C(6)
C(7)
C(7)
120.8(2)
121.22
There was an excellent agreement between the DFT/XRD bond lengths and angles, as shown in Fig. 3a and c, respectively. The graphical correlation (R2) of the bonds is 0.9798 (Fig. 3b), while that of the angles is 0.9801 (Fig. 2d). Thus, a significant correlation was established between the XRD/DFT structural parameters.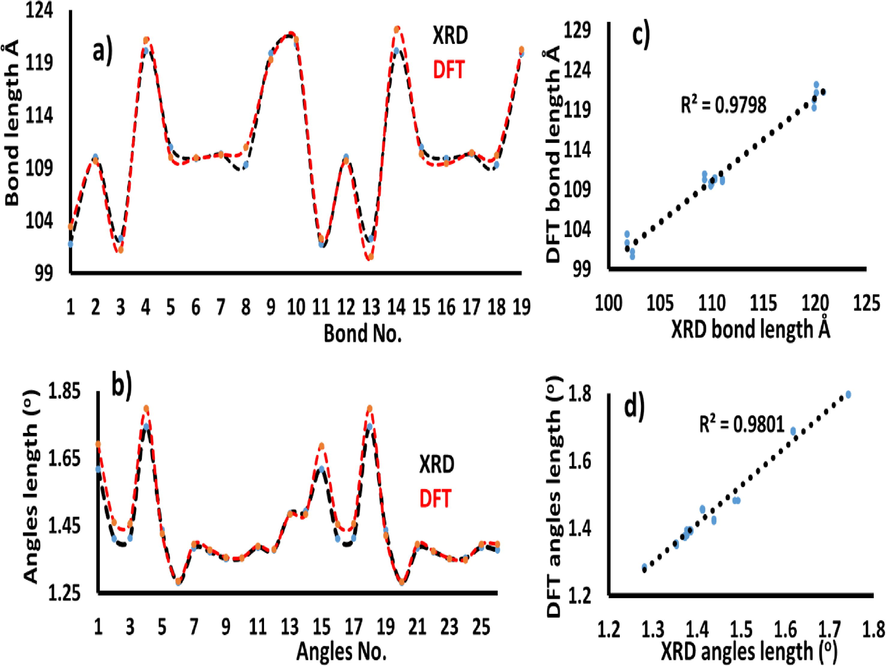
(a) XRD/DFT bond length relation and its graphical correlation; (b), (c) DFT/XRD angle relation and its graphical correlation (d).
3.3 Trans–cis isomerization in DMDO
Based on the analysis of the XRD patterns recorded in this study, the trans-isomer was preferentially isolated over the cis-isomer in accordance with steric effect rules. As other researchers have been able to detect cis isomers of oximes by XRD, we believe that it is possible for the dioxime to be isomerized from trans to cis under mild conditions.
Therefore, in the gaseous state and by ignoring any molecular interaction, the trans–cis isomerization in BDMDO was simulated using DFT/B3LYP/6-311G(d) via a 180° flip rotation around the highlighted Csp2–Csp2 bond, as shown in Scheme 2. The transition state (TS) structure of BDMDO was investigated using the QTS2 model. The stereo-chemical variation between the trans and cis isomers was simulated by performing a 180° flip rotation around the highlighted N⚌C*⋯C*⚌N sp2–sp2 bond; a dramatic change occurred in the dihedral angle of trans N⚌C⋯C⚌N from 180° to 0° as in the cis-isomer; meanwhile, the DFT/QTS2 system TS dihedral angle was found to be ∼ 90°. The global-minimum energy of the trans-isomer was determined to be − 1976.31730871 a.u. with Etrans = 0.0 kJ (zero reference energy) as it was more stable compared to the cis-isomer. Meanwhile, the global-minimum energy of the cis-isomer was determined to be − 1976.31495695 a.u. with Ecis = 6.10 kJ. The energy of the TS was found to be − 1976.31307314 a.u. with ETS = 11.12 kJ. The TS structure was recorded in between the trans–cis structures with a N⚌C⋯C⚌N dihedral angle of ∼ 90°. The isomer energy profile was consistent with the XRD result since it displayed the trans-isomer as the more stable isomer (less sterically hindered) as compared to the cis-isomer (more steric hindrance). The trans–cis isomerization energy in the gaseous state was very low, which is not surprising because a simple 180° single bond Csp2–Csp2 flip rotation was performed, as shown in Fig. 4.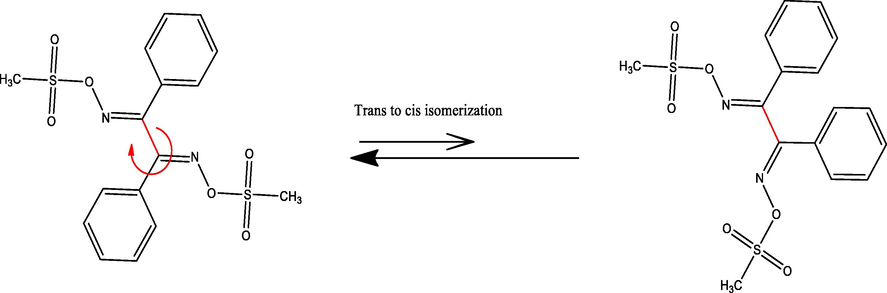
Trans–cis isomerization studied by DFT.
![Energy and structure profiles of trans–[TSQTS2]–cis isomerization.](/content/185/2021/33/2/img/10.1016_j.jksus.2020.101298-fig6.png)
Energy and structure profiles of trans–[TSQTS2]–cis isomerization.
3.4 Hirschfield and fingerprint (FP) surface computation
The calculations for elucidating HSA and FP(Titi et al., 2020; Barakat et al., 2018; Warad et al., 2018b; Aouad et al., 2019) of the molecular surface interactions in BDMDO were performed using the original crystallographic information (Fig. 5). The HSA result reflected the formation of a cross-like structure with four red points around the O atoms, which built the O⋯H bonds. No N…H bonds were detected, which supports the XRD packing result. The percentage ratio of atom…atom FP contacts decrease in the order of H⋯H (32.0%) > H⋯O (22%) > H⋯C (6%) > H⋯N and H⋯S (<1%), as shown in Fig. 5d.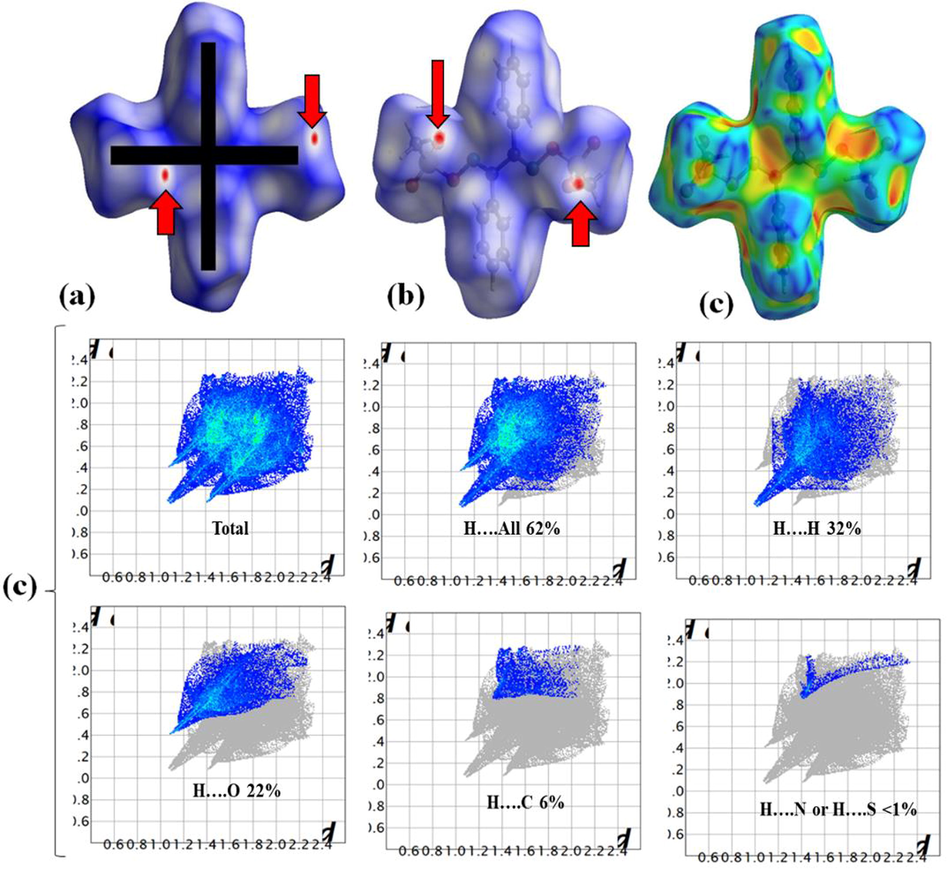
(a) dnorm, (b) shape index, (c) curvedness structures, and (d) inside/outside 2D-FP plot of BDMDO.
3.5 NMR investigation
The σ bonds for the C and H atoms in BDMDO are theoretically supported by the gauge-including atomic orbital (GIAO) approach, and the experiments were performed with CDCl3 as the solvent. The 1H NMR spectrum showed a signal at 3.5 ppm, corresponding to the CH3 group of methylsulfonyl, and three peaks, which belonged to the aromatic hydrogen between 7.2 and 8.0 ppm, as illustrated in Fig. 6a. The GIAO calculated σH reveals an excellent correlation with the corresponding experimental result, as shown in Fig. 6b (σH of CH3 is ∼ 3.45 and σH of H-Ph is ∼ 7–8 ppm).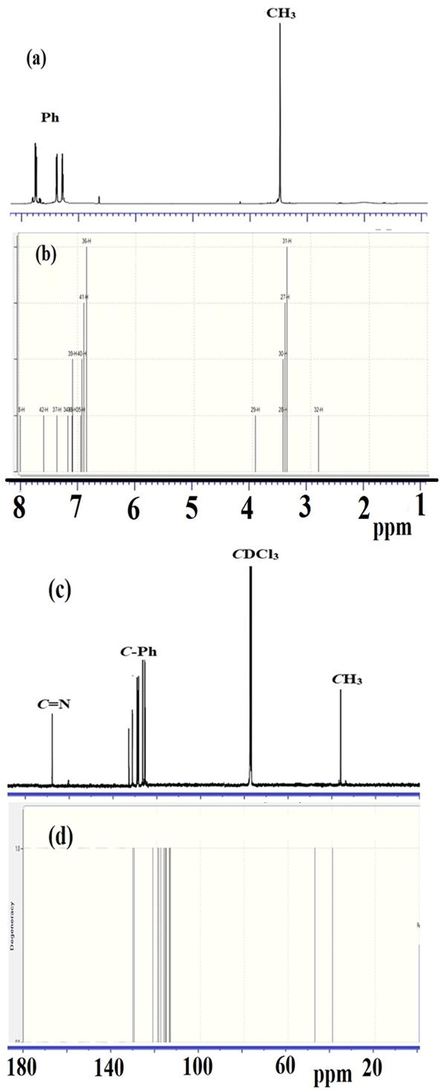
1H NMR: (a) experimental and (b) GIAO, and 13C NMR: (c) experimental and (d) GIOA spectra of BDMDO.
The experimental 13C NMR spectrum showed one signal for three carbons in the aliphatic region corresponding to the CH3 group at 28 ppm, six signals corresponding to 12C-Ph at 120–130 ppm, and one signal corresponding to two carbons of C⚌N at 165 ppm, as shown in Fig. 6c. The GIAO calculated carbon chemical shifts revealed a good correlation with the experimental result except for the C⚌N carbon, which was observed at 134.5 ppm, as shown in Fig. 6d.
3.6 Experimental/DFT–IR investigation
The experimental IR spectrum of BDMDO exhibited many bands corresponding to CPh-H, CMe-H, C⚌N, S⚌O, N—O, S—O, and S—C stretching vibrations. The stretching vibrations of the main functional groups are indicated as follows: the broadest band in the BDMDO spectra was assigned to ʋC=N at 1602 cm−1; aromatics exhibited multiple bands in the 3110–3020 cm−1 region due to CPh-H. For the methoxy group, CMe-H appeared in the 2880–2800 cm−1 range, and the sulfonyl group S⚌O bands appeared in the 1380–1260 cm−1 range. Other functional groups were assigned to their expected positions, as shown in Fig. 7a. The IR scaled following the DFT/B3LYP/6-311G(p) method was found to be close to the experimental result (Fig. 7b). The DFT frequencies were slightly higher than those obtained from the experimental IR analysis because the B3LYP-IR was computed in the gaseous state, while the experimental IR analysis was performed in the solid-state. In general, an excellent correlation was observed between the experimental and DFT/IR analyses, as evidenced by the correlation coefficient of 0.9984 (see Fig. 7c).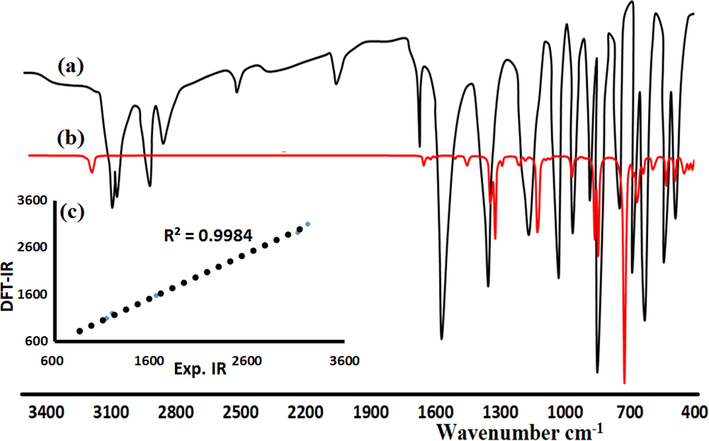
Infrared spectra of BDMDO: (a) Experimental, (b) B3LYP/IR, and (c) Exp./DFT relation.
3.7 UV–Visible and TD-DFT/B3LYP comparison
The UV–Visible spectral measurements and time-dependent DFT (TD-DFT) calculations using B3LYP exchange correlation functional with 6-311G(p) basis set were performed on BDMDO in methanol and combined as shown in Fig. 8. The observed and DFT-predicted wavelengths were in the UV region. In the experimental UV–Vis spectrum, only a π → π* broad electron band with λmax = 225 nm was recorded and there was no other peak in the visible area. The TD-DFT spectrum was consistent with this observation but with a very small red-shift (λmax = 228 nm with Δλ = 3 nm) (Fig. 8b). The TD-DFT peak corresponds mainly to the electron transitions from HOMO to LUMO.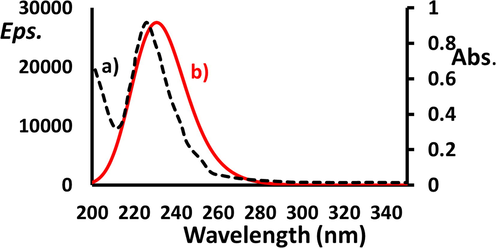
UV–Visible and TD-DFT spectra of BDMDO in MeOH.
3.8 MEP, HOMO/LUMO, and density of states
The MEP is one of the best theoretical methods for predicting the reactive centers in molecules. It was plotted for BDMDO as shown in Fig. 9a. The electron-rich (negative charge) area is indicated with red color around the O atoms, while the positive charge (electron-poor) area is highlighted with blue color around the methyl protons. The red color of the O region is very suitable for electrophilic attack by the blue-colored electrophilic protons to form H⋯O bonds. The green predominant regions correspond to a potential halfway (red and blue region) in between and do not induce sufficient binding. Such results match well with the XRD and HSA results. The HOMO/LUMO molecular orbital shapes and energy levels were computed using DFTB3LYP/6-311G(d), as illustrated in Fig. 9b. The computed fragment molecular orbital provided sufficient information about the HOMO/LUMO energy gap, ΔELUMO–HOMO, and the electronic properties of the HOMO and LUMO (Bulatov et al., 2014; Aouad et al., 2018; Wolff et al., 2012; Sheldrick, 1997; Frisch et al., 2009; Warad et al., 2010; Plater et al., 2019). The chemical reactivity of the systems corresponds to the ΔELUMO–HOMO value and is associated with the chemical stability of the molecules. Since both HOMO and LUMO have negative energy values, BDMDO should have been stabilized; moreover, the ΔELUMO–HOMO was found to be 0.118 a.u. with 3.211 eV, which is a very small energy gap that facelifted the electron in accordance with the π-electrons of the 2C = N conjugation structural system. The small energy gap increased the chemical reactivity, softness, and polarizability of the desired compound [24]. The value of ΔEgap = 3.22 eV obtained by the DOS calculation (Fig. 9d) is consistent with the energy level calculated by the HOMO/LUMO method.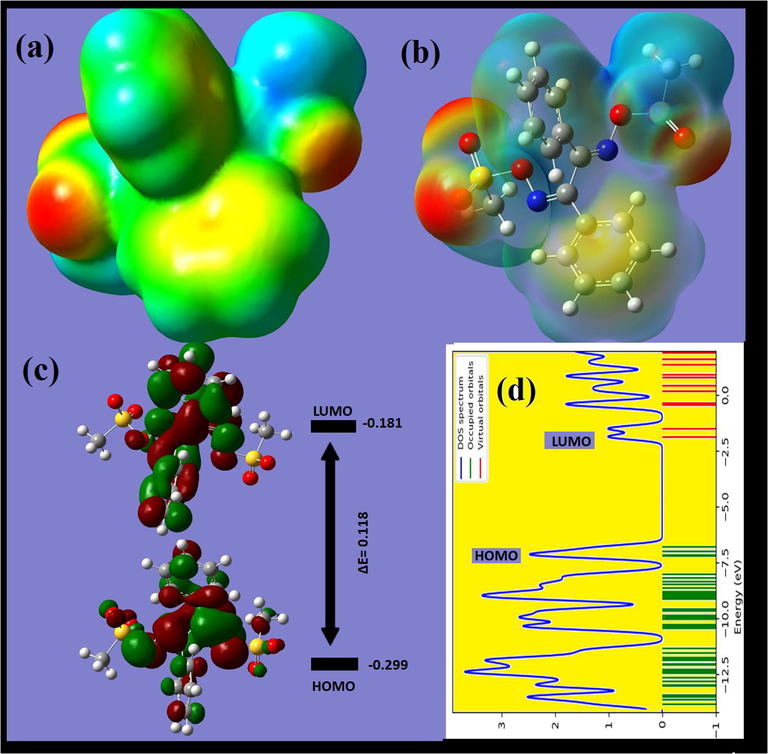
(a, b) MEP, (c) HOMO/LUMO energy gap, and (d) density of states of BDMDO.
3.9 Thermogravimetric analysis
The thermogravimetric/derivative thermogravimetric (TG/DTG) analysis of BDMDO showed one stage of decomposition pattern from 25 to 350 °C in an open-air O2 atmosphere at a heating rate of 10 °C/min. Below 105 °C, BDMDO displayed high thermal stability (Fig. 10). The compound was thermally de-structured in broad steps in the range of 110–200 °C with one TDTG = 130 °C. Therefore, BDMDO was completely decomposed, and consequently, zero residue weight was detected at T > 200 °C.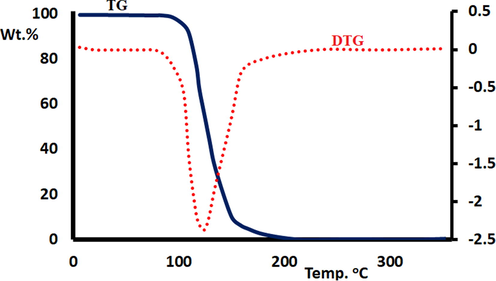
TG/DTG curves of BDMDO.
4 Conclusion
In this study, BDMDO was synthesized and fully characterized. The XRD results of BDMDO confirmed the formation of a trans-isomer. DFT studies supported the XRD results and the possibility of trans–cis isomerization as the energy between the two isomers was low. The QTS2 model reflected the TS with a N⚌C⋯C⚌N dihedral angle of ∼90°. The supramolecular assemblies supported the formation of non-covalent C—H⋯H—C and H-bond interactions in the molecular lattice; such interactions were successfully computed by MEP and HSA. The NMR spectra sufficiently matched their corresponding DFT/6-311G(d) parameters, such as HOMO–LUMO, TD-SCF, B3LYP/IR, and GIAO NMR. The optimized DFT/B3LYP/6-311G(d) structural parameters also correlated well with the experimental XRD data. The TG/DTG behavior reflected the good thermal stability of the dioxime and revealed that it decomposed via a single-step mechanism.
Acknowledgements
The authors extend their appreciation to the Deputyship for Research & Innovation, “Ministry of Education” in Saudi Arabia for funding this research work through the project number IFKSURG-1440-141.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Structural chemistry of oximes. Cryst. Growth Des.. 2013;13(6):2687-2695.
- [CrossRef] [Google Scholar]
- New mixed-valence MnII4MnIV clusters from an unusual ligand transformation. Polyhedron. 2017;122:71-78.
- [CrossRef] [Google Scholar]
- Single proton intramigration in novel 4-phenyl-3-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)-1H-1,2,4-triazole-5(4H)-thione: XRD-crystal interactions, physicochemical, thermal, Hirshfeld surface, DFT realization of thiol/thione tautomerism. J. Mol. Liq.. 2018;264:621-630.
- [CrossRef] [Google Scholar]
- Hydrophobic pocket docking, double-proton prototropic tautomerism in contradiction to single-proton transfer in thione ⇔thiol Schiff base with triazole-thione moiety: Green synthesis, XRD and DFT-analysis. J. Mol. Struct.. 2019;1180:455-461.
- [CrossRef] [Google Scholar]
- Quantum chemical insight into the molecular structure of L-chemosensor 1,3-dimethyl-5-(thien-2-ylmethylene)-pyrimidine-2,4,6-(1 H ,3 H ,5 H)-trione: Naked-eye colorimetric detection of copper(II) anions. J. Theor. Comput. Chem.. 2018;17(01):1850005.
- [CrossRef] [Google Scholar]
- Triple associates based on (oxime)Pt(II) species, 18-crown-6, and water: synthesis, structural characterization, and DFT study. J. Mol. Struct.. 2014;1068:176-181.
- [CrossRef] [Google Scholar]
- Frisch, M., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, Ga., 2009. gaussian 09, Revision d. 01, Gaussian. Inc., Wallingford CT 201.
- Synthesis, characterization, theoretical calculations and biochemical evaluation of a novel oxime ligand with complexes. J. Mol. Liq.. 2019;284:473-491.
- [CrossRef] [Google Scholar]
- One-pot synthesis of a new 2-substituted 1,2,3-triazole 1-oxide derivative from dipyridyl ketone and isonitrosoacetophenone hydrazone: Nickel(II) complex, DNA binding and cleavage properties. Bioorg. Chem.. 2017;71:325-334.
- [CrossRef] [Google Scholar]
- Entry of oximes into the brain: a review. Curr. Med. Chem.. 2008;15:743-753.
- [CrossRef] [Google Scholar]
- Dioximes: synthesis and biomedical applications. Bioorg. Chem.. 2019;82:145-155.
- [CrossRef] [Google Scholar]
- Novel oxime based flavanone, naringin-oxime: synthesis, characterization and screening for antioxidant activity. Chem. Biol. Interact.. 2014;212:40-46.
- [CrossRef] [Google Scholar]
- Potential photoacid generators based on oxime sulfonates. J. Chem. Res.. 2019;43(1-2):26-33.
- [CrossRef] [Google Scholar]
- Sittig's Handbook of Toxic and Hazardous Chemicals and Carcinogens. Elsevier; 2017. p. :336-580.
- [CrossRef]
- Sheldrick, G.M., 1997. Program for crystal-structure refinement. SHELX-97.
- Rare oxidation-state combinations and unusual structural motifs in hexanuclear Mn complexes using 2-pyridyloximate ligands. Inorg. Chem.. 2010;49(10):4388-4390.
- [CrossRef] [Google Scholar]
- 4-[5-Methyl-3-phenylisoxazol-4-yl]-benzenesulfonamide, Valdecoxib: a potent and selective inhibitor of COX-2 [4] J. Med. Chem.. 2000;43:775-777.
- [CrossRef] [Google Scholar]
- Synthesis of novel Cl2Co4L6 clusterusing 1-hydroxymethyl-3,5-dimethylpyrazole (LH) ligand: Crystal structure, spectral, thermal, Hirschfeld surface analysis and catalytic oxidation evaluation. J. Mol. Struct.. 2020;1199:126995.
- [CrossRef] [Google Scholar]
- Synthesis, characterization and antioxidant capacity of naringenin-oxime. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2012;85(1):235-240.
- [CrossRef] [Google Scholar]
- Synthesis, spectra and X-ray crystallography of dipyridin-2-ylmethanone oxime and its CuX 2 (oxime) 2 complexes: Thermal, Hirshfeld surface and DFT analysis. J. Mol. Struct.. 2018;1154:619-625.
- [CrossRef] [Google Scholar]
- Ultrasound-assisted synthesis of two novel [CuBr(diamine)2·H2O]Br complexes: Solvatochromism, crystal structure, physicochemical, Hirshfeld surface thermal, DNA/binding, antitumor and antibacterial activities. Ultrason. Sonochem.. 2018;48:1-10.
- [CrossRef] [Google Scholar]
- Crystal structure of N-[(methylsulfonyl)oxy]-/Y-((E)-2-(methyl-sulfonyl) oxy]imino-l,2-diphenylethylidene)amine, C16H16N 2O6S2. Z. Krist. – New Cryst. Struct.. 2010;225:611-612.
- [CrossRef] [Google Scholar]
- Crystal structure and magnetic properties of di-copper and di-zinc complexes with di-2-pyridyl ketone oxime. Inorg. Chem. Commun.. 2016;74:79-81.
- [CrossRef] [Google Scholar]
- Wolff, S.K., Grimwood, D.J., McKinnon, J.J., Turner, M.J., Jayatilaka, D., Spackman, M.A., 2012. Crystal explorer.
- Mononuclear Cu(II) complex of an oxime ligand derived from N-Heterocyclic hydrazide: synthesis, spectroscopy, electrochemistry, DFT calculations and catecholase activity. J. Mol. Struct.. 2019;1193:444-449.
- [CrossRef] [Google Scholar]




