

Translate this page into:
Synthesis, biological evaluation and molecular docking of pyrimidine and quinazoline derivatives of 1,5-benzodiazepine as potential anticancer agents
⁎Corresponding authors. djaya@banasthali.ac.in (Jaya Dwivedi)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A new series of Pyrimidine (A, D and F) and quinazoline (B, C and E) analogues of 1,5-benzodiazepines were prepared via its nitrile-derived amidoximes in and nitrilium ions, respectively using one pot Domino reaction with DMAD in presence of DABCO catalyst and benzanilide in presence of Tf2O and 2-chloropyridine. The prepared molecules were examined for their biological property namely apoptotic and antiproliferative effects through cell cycle arrest using breast cancer cell line of human (MCF-7). Receptor-ligand interactions were studied on human epidermal evolution factor receptor (HER-2) with the help of molecular docking using Autodock 4.2.6 molecular modeling software. All the compounds demonstrated inhibitory effects on cell proliferation in a concentration dependent fashion (20–100 μg/mL). Notably, compound C exhibited highest inhibitory activity and caused inhibition of S and G2 phase in cell cycle arrest via caspase dependent apoptotic pathway in MCF-7 cells lines.
Keywords
Pyrimidine
Quinazoline
1,5-Benzodiazepines
Domino reaction
Anticancer agent

1 Introduction
Recently, molecules of increasing structural complexity have been synthesized using improved chemo-, disastereo- enantio and regioselective methods (Mayer et al., 2001). The possibility to join two or more molecules through organo catalytic reactions in one process has become a major challenge for chemists, to avoid the expensive protecting groups, long reaction time and tedious purification procedures involved in each synthetic step. Synthesis of complex molecular complexity with diversity using simple substrates imposes a huge challenge in contemporary organic chemistry at industrial and academic level. To achieve this, domino reactions have been adopted for the efficient disastereo and enantio selective preparation of complex compounds from the simple precursors through simple processes (Alba et al., 2009; Chen et al., 2017).
Pyrimidines have been widely used as anticancer (Addepalli et al., 2018), anti-arrhythmic, anti-inflammatory, analgesic, anti-HIV, serotonin receptor (5-HT6) antagonist, antimicrobial, antiviral, herpes simplex virus type-1 (HSV-1) inhibitor and hepatitis-A virus (HAV) etc. Pyrimidine analogs have also been documented for antileishmanial activity, antimalarial, anti-Parkinson activities and platelet aggregation inhibitors (Kumar and Narasimhan, 2018).
Quinazolines are well known for their versatile and important role in various biological activities including anti-inflammatory, antimicrobial, antihyperlipidemic, antihypertensive, anticonvulsant activities etc. (Ajani, 2016). Benzodiazepine acts as a key nucleus whose derivatives have displayed wide range of therapeutic activities namely anticonvulsant, antianxiety, analgesic, sedatives, anti-depressant, hypnotic, anti-HIV agents, anticancer etc. (Kaur Gill et al., 2014; Misra, et al., 2018).
Since last decades a tremendous rise in breast cancer incidences has been recorded, worldwide. In India, 25.8 per 100,000 women with mortality 12.7/ 100,000 women due to breast cancer (Malvia et al., 2017). Even more, development of resistance to available therapeutic agents has further worsened the situation. These statistics are alarming and requires exhaustive efforts of Our strategy for the synthesis of quinazoline derivatives (B, C and E) was based on the use of nitrile as coupling partner with an activated benzanilide (9) (activation of which was achieved by its reaction with Tf2O followed by 2-chloropyridine). The resulting nitrilium ion set the stage ready for its cyclocondensation with the π-electron system of the vinyl or are part of the 1,5-benzodiazepine (8, 11 and 18) and delivered quinazolines (B, C and E) in one step.
Notably, Pyrimidine (Kaur et al., 2015), quinazoline (Abuelizz et al., 2017), benzodiazepine (Kaur Gill et al., 2014) and their analogues have been chronicled for beneficial effects in variety of cancers (Das et al., 2019; Kolawole and Benjamin, 2019; Scherakov et al., 2018). In light of these facts, efforts have been made to synthesized newer series of novel hybrid derivatives of quinazoline, pyrimidine and 1,5-benzodizepine. Further these derivatives were examined for in-vitro anti-cancer activity against breast cancer lines (MCF-7) employing cell cycle arrest assay. In addition, docking study of the newly prepared molecules was also performed against human epidermal production factor receptor (HER-2) using Autodock 4.2.6molecular modeling software.
2 Experimental
Oven dried glass apparatus were used in entire study. Perkin-Elmer FT-IR RXI spectrophotometer was used to record infrared spectra of synthesized compounds. Bruker Ascend 400 MHz spectrometer was operated at 400 MHz for 1H; and for 13C, 100 MHz was used to record 1H NMR and 13C NMR spectra of synthesized compounds. In 1H NMR Tetramethylsilane of δ 0.00 ppm was functioned as an internal standard, while CDCl3 of δ 77.23 ppm was used in 13C NMR. Chemical shifts are presented in δ ppm. JEOL MS route (600H) instrument was used to obtain high-resolution electron impact mass spectra (HR-EIMS). Precoated TLC silica gel plates (60F-254, Merck) were used to check reactions. All chemicals and reagents were procured from Sigma Aldrich.
2.1 Preparation of (Z)-2-benzoyl-3-(dimethyl-amino) acrylonitrile (2)
The mixture of benzoylacetonitrile 1 (10.0 mmol) and DMF.DMA (15 mL) was heated through refluxing set up for 6 h and was concentrated under reduced pressure. The residue solution was triturated with hexane and filtered followed by washed with pure hexane to give 2 as a yellow solid.
2.2 Preparation of 4-phenyl-1H-benzo[b][1,4]diazepine-3-carbonitrile (4)
Dimethylaminomethylene ketone derivative 2 (10.0 mmol) and o-phenylenediamine 3 (10.0 mmol) were mixed in ethanol and refluxed for 11 h. To achieve product excess of solvent was removed by rotary evaporator followed by addition of ice-cold water. Product was re-crystallized from chloroform (CHCl3) and dried over Na2SO4 to give 4 as a brown solid.
2.3 Preparation of (Z)-N′-hydroxy-4-phenyl-1H-benzo[b][1,4]diazepine-3-carboximidamide (5)
A solution of compound 4 (29.1 mmol) was prepared in 25 mL ethanol was mixed with solution of sodium carbonate (29.1 mmol) and 29.1 mmol hydroxylamine hydrochloride in 25 mL water and irradiated the mixture solution with an ultrasound probe for the time of 15–30 min at 55 °C. Resulting mixture was further concentrated under the influence of reduced pressure followed by dissolving in 50 mL of dichloromethane, dried over Na2SO4. Residue filtered, dried and recrystallized from mixture of chloroform-hexane to afford compound 5.
2.4 Preparation of 5,6-dihydroxy-2-(4-phenyl-1H-benzo[b][1,4]diazepin-3-yl)-pyrimidine-4-carboxylic acid methyl ester (A)
To a solution of DABCO (0.09 mmol), amidoxime 4 (0.9 mmol) and dioxane at −10 °C, DMAD was added and resulted reaction mixture stirred for 20 min. Reaction mixture was heated gradually under graded temperature up to 37 °C. Microwave assisted heating was provided to reaction mixture in biphasic mode, in first phase it was heated at 80 °C for 8–10 min and second phase it was exposed to a temperature 120 °C for 20 min. Finally, volatile components were removed at reduced pressure and residue to afford compound A as a dark brown solid.
2.5 Preparation of (Z)-3-(dimethylamino)acryloyl cyanide (7)
The mixture containing DMA (15 mL), DMF and pyruvonitrile 6 (10.0 mmol) was heated under reflux set up for 6 h and concentrated under reduced pressure. The residue was made fine with hexane and filtered followed by washed with hexane to give 7 as a dark blue solid.
2.6 Preparation of 5H-Benzo[b][1,4]diazepine-2-carbonitrile (8)
Dimethylaminomethylene ketone derivative (7) (10.0 mmol) and o-phenylenediamine 3 (10.0 mmol) were mixed in ethanol and refluxed for 11 h. To achieve product excess of solvent was removed by rotary evaporator followed by addition of ice-cold water. Product was re-crystallized from chloroform and dried over Na2SO4 to give 8 as a brown solid.
2.7 Preparation of 4-(2-phenylquinazolin-4-yl)-1H-benzo[b][1,4]diazepine (B)
A solution of benzanilide (10.0 mmol), 2-chloropyridine (20.0 mmol) and dichloromethane (50 mL) was stirred for time of 30 min. The temperature of the current reaction was maintained to −78 °C (using dry ice and acetone) and Tf2O (trifluoromethane sulfonic anhydride) (20.0 mmol) was added into it dropwise. After 30 min the mixture was kept in an ice bath and gently heat to a room temperature. Then compound 8 was added and reaction mixture was heated to 45 °C in an oil bath for 5 h. It was then cooled to normal room temperature and the trifluoromethane sulfonate salts were neutralized by triethylamine. The volatiles were separated by rotary evaporation and the product was purified by preparative TLC method to give quinazoline derivative B as a yellow solid.
2.8 General procedure of 2-benzoyl-3,3-bis(methylthio)acrylonitrile (10)
A mixture of t-BuOK (1.34 g, 12.0 mmol), DMF (3.0 mL) and dry toluene was sufficiently stirred in ice bath. To this benzoylacetonitrile 1 (6.00 mmol) was added. After 30 min of stirring, CS2 (1.00 mL, 6.00 mmol) was added. In next step, methyl iodide (2.0 mL, 0.012 mol) was added after 2 h of continuous stirring and external cooling. This reaction mixture was poured into ice water after 2 h of further stirring and 3 h of reflux. Obtained product was extracted with the help of toluene and water, dried over Na2SO4 and was concentrated by removal of solvent using rotary to give [10] as a black solid.
2.9 Preparation of 2-methylsulfanyl-4-phenyl-1H-benzo[b][1,4]diazepine-3-carbonitrile (11)
Oxoketene dithioacetal derivative 10 (10.0 mmol) and o-phenylenediamine 3 (10.0 mmol) were mixed in ethanol and refluxed for 11 h. To achieve product excess of solvent was removed by rotary evaporator followed by addition of ice-cold water. Product was re-crystallized from chloroform and dried over Na2SO4 to give 11 as a brown solid.
2.10 Preparation of 2-(methylthio)-4-phenyl-3-(2-phenylquinazolin-4-yl)-1H-benzo[b][1,4]diazepine (C)
A solution of benzanilide (10.0 mmol), 2-chloropyridine (20.0 mmol) and dichloromethane (50 mL) solution was stirred for the time of 30 min. The temperature of the reaction mixture was maintained to −78 °C (using dry ice and acetone) and Tf2O (trifluoromethane sulfonic anhydride) (20.0 mmol) was added into it dropwise. After 30 min, the reaction mixture was kept in an ice bath and warmed to room temperature. Then compound 11 was added and reaction mixture was heated to 45 °C in an oil bath for 5 h. It was then cooled to room temperature and the trifluoromethane sulfonate salts were neutralized by triethylamine. The volatiles were separated by rotary evaporation and the product was purified by preparative TLC method to give quinazoline derivative C as a brown solid.
2.11 Preparation of 2,4-dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepine-7-carbonitrile (14)
A mixture of malonic ester 12 (10.0 mmol), 3,4-diaminobenzonitrile 13 (10.0 mmol), ethanol and piperidine (4–5 drops) was heated for 6 h under reflux. Using rotary evaporator, the resulting solution mixture was concentrated and stored at room temperature. The finally obtained crystalline compound was thus filtered followed by washing with cold aqueous ethanol solution and dried to give 14 as a dark blue solid.
2.12 Preparation of (Z)-N′-hydroxy-2,4-dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepine-7-carboximidamide (15)
A solution of 29.1 mmol compound 14 was prepared in 25 mL ethanol and mixed with solution of sodium carbonate (29.1 mmol) and hydroxylamine hydrochloride (29.1 mmol) in 25 mL water and irradiated with an ultrasound probe for 15–30 min at 55 °C. Resulting mixture was concentrated under lower pressure and dissolved in 50 mL of dichloromethane, dried over Na2SO4. Residue filtered, dried and recrystallized using chloroform-hexane to afford compound 15.
2.13 Preparation of methyl-2-(2,4-dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-7yl)-5,6-dihydroxypyrimidine-4-carboxylate (D)
To a solution containing DABCO (0.09 mmol), amidoxime 15 (0.9 mmol) and dioxane, DMAD was added at −10 °C with continuous stirring for 20 min. The reaction temperature was then increased to 37 °C. Microwave assisted heating was provided to reaction mixture in biphasic mode, in first phase it was heated at 80 °C for 8–10 min and second phase, it was exposed to a temperature 120 °C for 20 min. Finally, volatile fractions were removed under reduced pressure and residue obtained was recrystallized to afford compound D as a brown solid.
2.14 Preparation of (E)-ethyl-2-cyano-3-phenylacrylate (17)
Ethyl cyanoacetate 16 (10.0 mmol), benzaldehyde (10.0 mmol) and fused sodium acetate (15.0 mmol) were mixed in glacial acetic acid. After 5 h of reflux, the reaction mixture was transferred into ice water and the resulting solid components was filtered, washed with water, dried over Na2SO4 and recrystallized to give compound 17 as a yellow solid.
2.15 Preparation of 2-oxo-4-phenyl-2,34,5-tetrahydro-1H-benzo[b][1,4]diazepine 3-carbonitrile (18)
α,β-unsaturated ketone derivative (17) (0.01 mol) and o-phenylenediamine 3 (10.0 mmol) were mixed in ethanol and piperidine (4–5 drops) was refluxed for 6 h. About half portion of the solvent was evaporated by rotary evaporation and the final mixture was allowed to cool at room temperature. The crystalline compound thus formed was filtered followed by washing with cold aqueous ethanol solution and dried to give 18 as a brown solid.
2.16 Preparation of 4-phenyl-3-(2-phenylquinazolin-4-yl)-4,5-dihydro-1H-benzo[b] [1,4]diazepine-2(3H)-one (E)
A solution of benzanilide (10.0 mmol), 2-chloropyridine (20.0 mmol) and dichloromethane (50 mL) was stirred for 30 min. The temperature of the reaction was maintained to −78 °C (using dry ice and acetone) and Tf2O (trifluoromethane sulfonic anhydride) (20.0 mmol) was added into it dropwise. After 30 min the reaction mixture was kept in an ice water bath and warmed to room temperature. The compound 18 was added and reaction mixture was heated to 45 °C in an oil bath for 5 h. It was then cooled to room temperature and the trifluoromethane sulfonate salts were neutralized by triethylamine. The volatiles were separated by rotary evaporation and the product was purified by preparative TLC method to give quinazoline derivative E as a reddish brown solid.
2.17 Preparation of (Z)-N′-hydroxy-2-oxo-4-phenyl-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepine-3-carboximidamide (19)
A solution of compound 18 of 29.1 mmol was prepared in 25 mL ethanol and mixed with solution of sodium carbonate (29.1 mmol) and hydroxylamine hydrochloride (29.1 mmol) in 25 mL water and irradiated under the influence of ultrasound probe for 15–30 min at 55 °C. Resulting mixture was concentrated under reduced pressure and dissolved in 50 mL of dichloromethane, dried over Na2SO4. Residue filtered, dried and recrystallized using chloroform-hexane solution to afford compound 19.
2.18 Preparation of 5,6-Dihydroxy-2-(2-oxo-4-phenyl-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)-pyrimidine-4-carboxylic acid methyl ester (F)
DMAD was added to a 0.09 mmol solution of DABCO, 0.9 mmol amidoxime 19 and dioxane at −10 °C and reaction mixture was then stirred for 20 min. The reaction temperature was then increased to 37 °C. The heating of reaction mixture in microwave was made two times, first time for 8–10 min at 80 °C and second time for 20 min at 120 °C. The volatile components were removed at reduced pressure and the residue materials was recrystallized to afford compound F as a dark yellow solid.
2.19 Biological activity
2.19.1 Cell lines and culture condition
MCF-7, human breast cancer cell line was procured from the National Centre for Cell Science (NCCS), Pune, India and were preserved in Dulbecco’s modified Eagle medium containing a high concentration of glucose (4.5 g/L), 10% fetal bovine serum with100 mg/mL and 100 U/mL penicillin streptomycin incubated at 37 °C. Pre-counted cells were placed in each well in each set of experiment before the addition of test compounds. All the experimental samples were solubilized in DMSO (sterile filtered) prior their mixing to the culture media.
2.19.2 Antiproliferative activity
Cell proliferation inhibitory activity of test compounds was investigated on MCF-7 cell lines using3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay. Briefly, 100 μl of test compound (20–100 μg/mL) was added to 24 h cultured cells in triplicate wells and incubated for 24 h. Further, 20 μl of MTT dye (5 mg/ml) was added to each well followed by further incubation for 4 h and absorbance was recorded at 550 nm (Gupta et al., 2018). The cell % inhibition was calculated using the following formula: where: As – absorbance of sample; Ac – absorbance of control.
2.19.3 Morphological study
Morphological observations of MCF-7 cells treated with different compounds for cytotoxicity were done to determine the changes induced by the standard and the test compounds. Cells were equally seeded in flat bottom of a plate previously treated with tissue culture and then treated with different concentrations of test compounds. After 24 h of treatment, the morphology of cell was investigated under inverted microscope and images were captured at magnification using digital microscopy 10.
2.19.4 Caspase-3 activity assay
The caspase-3 influencing ability of test compound and standard in the cell lysate was examined usingcaspase-3 colorimetric assay kit (Sigma-Aldrich USA). Assay was performed as per manufacturer’s protocol (Ajji et al., 2017).
2.19.5 Cell cycle study
MCF-7 cells (5 × 105 cells) were incubated at (37 °C, 5% CO2) in each well with 100 μg/ml test compound or DMSO vehicle as control media for 24 h. Following trypsinzation, cells were washed followed by centrifugation at 2000g for 10 min and the pellet was resuspended in PBS (0.5 mL). Fixation was accomplished by addition of 1.2 mL of 70% cold ethanol for 2 h. The fixed cells were then washed with PBS followed by centrifugation at 2000g for 10 min. After suspending cells in 0.3 mL PBS solution, 20 mg/ml RNAase was added and incubated for the time of 1 h. After adding 0.5 mg/ml propidium iodide, cells were further incubated at 4 °C for 30 min. Finally, the cells were investigated for cell cycle using Beckman Coulter flow cytometer where the excitation and emission wavelength were used at 488 nm and 670 nm (Ajji et al., 2017; Gupta et al., 2018).
2.19.6 Docking study
The docking studies of compound A–F were performed on the crystal structure of the kinase domain of human (PDB ID: 3PP0) using Autodock 4.2.6 molecular modeling software. Protein was retrieved from protein data bank and was refined by water molecules removal, co-crystallized ligand, by adding kollmann charges and polar hydrogens. In order to achieve maximum interaction between all residues with the ligand, a grid box with X, Y and Z axis with 40, 42 and 42 points was generated using AutoGrid4.0 respectively. Conformation of energy minimization of all ligands (pdb format) was carried out using ChemBio3D Ultra (version 12) and was used to calculate of Gasteiger–Huckel charges and was saved in Autodock default format. Autodock was used to determine 20 possible binding conformations i.e., 20 runs for every docking using LGA search. Default protocol was utilized with initial 150 randomly placed individuals and a maximum number of 2.5 × 105 energy evaluations and 2.7 × 104 generations. A mutation and crossover rate of 0.02 and 0.8 were used, respectively.
The binding affinity of the test ligands with the receptors is expressed in terms of dock score, which indicate the estimated binding free energy (kcal/mol) in negative terms, corresponding to this enzyme inhibition constant (Ki) value is expressed. Greater value of estimated binding free energy in negative terms or lower value of enzyme inhibition constant indicate the greater binding affinity between the ligand and receptor(Zhou et al., 2017).
3 Results and discussion
3.1 Chemistry
The synthesis of desired 1, 5-benzodiazepines (4, 8, 11 and 18), (Schemes 1 and 2) was achieved by the cyclocondensation reaction of o-phenylenediamine (3) with the nitrile bearing (i) oxoketene dithioacetal (10) (ii) dimethylamino methylene ketone (2, 7) and (iii) α, β-unsaturated ketone (17) derivatives. Synthons 2, 7, 10 and 17were obtained from benzoylacetonitrile (1), pyruvonitrile (6) and cyanoacetic ester (16) from their reaction with (i) CS2 + CH3I (in presence of base), (ii) dimethylformamide dimethyl acetal and (iii) with benzaldehyde (in presence of a base) respectively as shown in Schemes 1 and 2. The 1, 5-benzodiazepines 14 (bearing a nitrile function on position 7) were formed directly by the cyclocondensation of 3,4-diaminobenzonitrile (13) with diethyl malonate (12).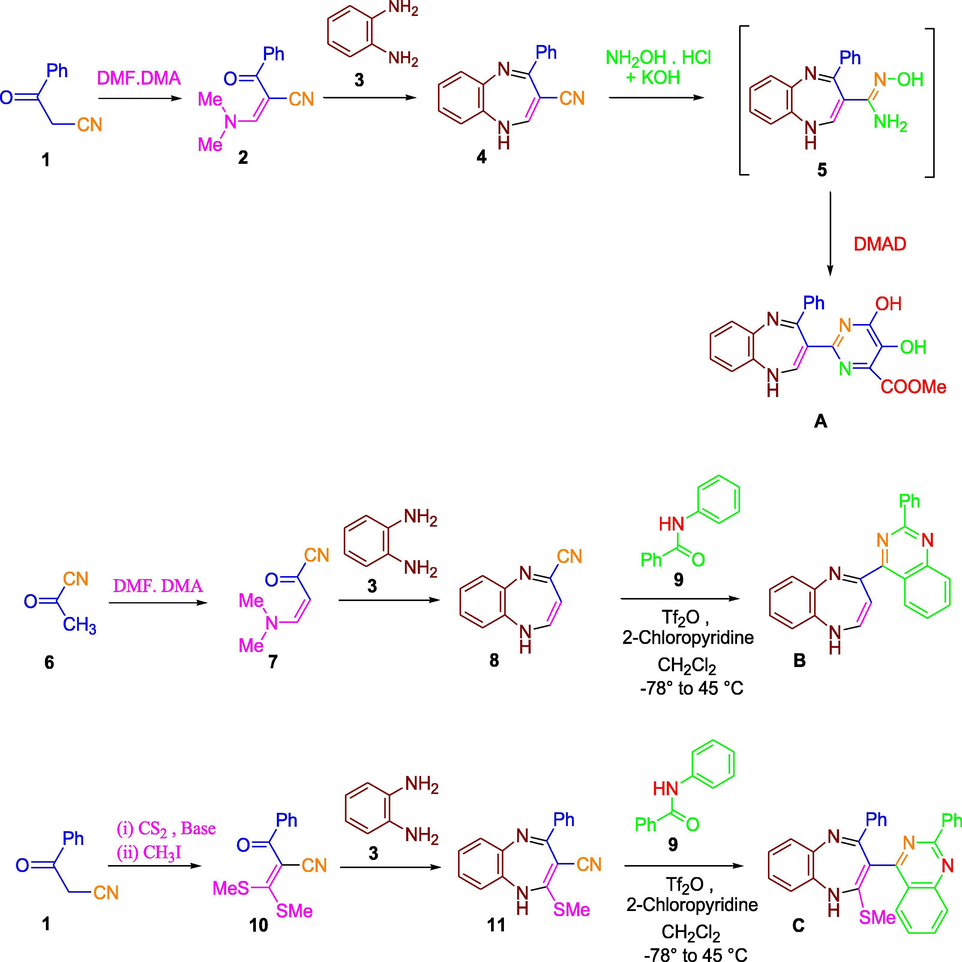
Synthesis of Compounds A, B and C.
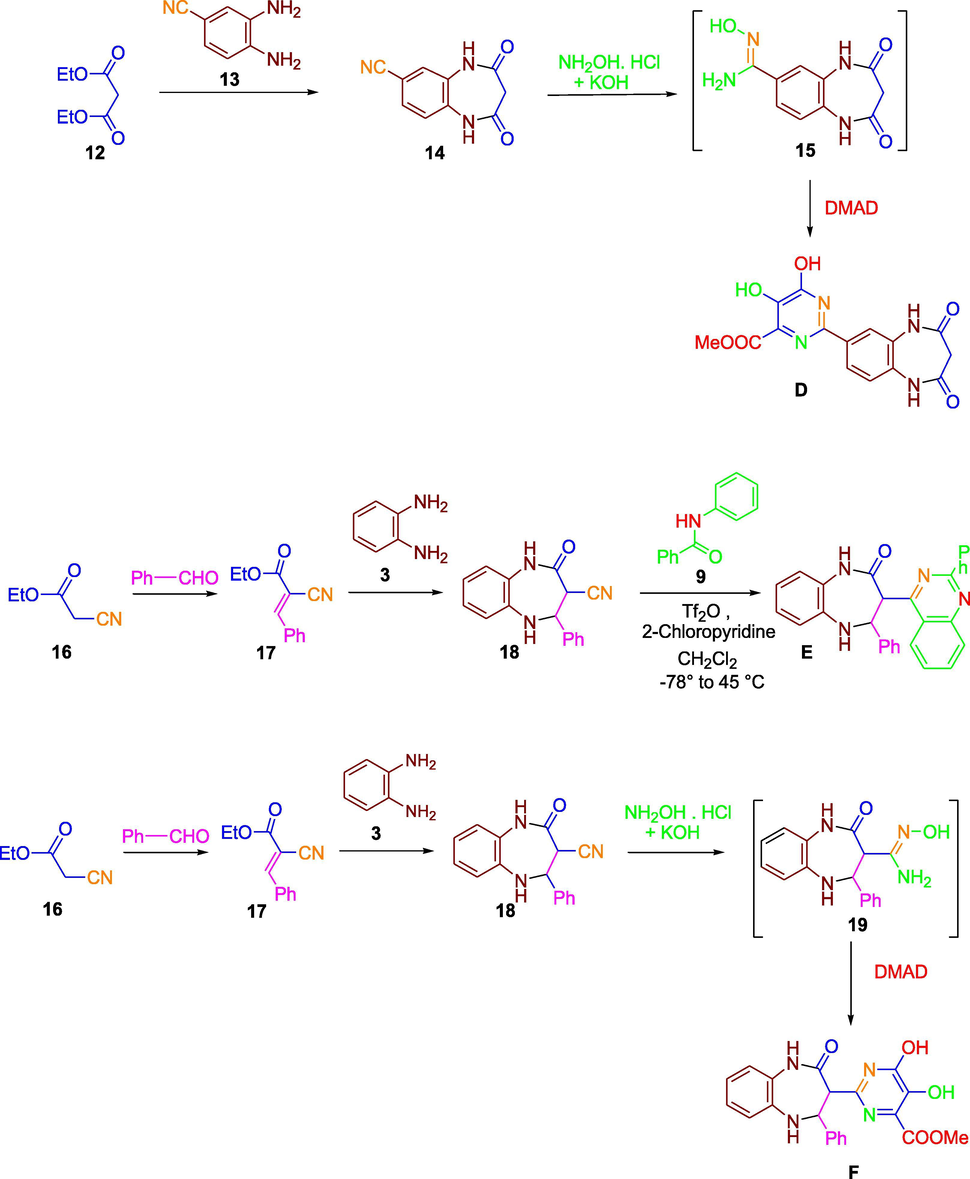
Synthesis of Compounds D, E and F.
Amidoximes 5, 15 and 19 were obtained from the reaction yields of hydroxylamine hydrochloride with a base and 1, 5-benzodiazepines 4, 14 and 18. The cycloaddition of amidoximes 5, 15 and 19 with dimethyl acetylene dicarboxylate (DMAD) introduced a series of tandem C-O and C-N coupling sequence prior to a concurrent cyclo-condensation of the produced intermediate to give pyrimidine derivatives A, D and F as outlined in Schemes 1 and 2. The catalyst 1,4-diazabicyclo[2.2.2]octane (DABCO) was used in these to increase the yield of the reaction product (Humphrey et al., 2010; Ngwerume and Camp, 2010).
3.2 Biological activity
3.2.1 Antiproliferative activity
The antiproliferative activities of test compounds toward the growth of Human breast cancer cell line (MCF-7) in vitro are presented in Fig. 1. The antiproliferative effect of optimum dose (0.1 mg/ml) to inhibit the cell growth. MCF-7 cells were treated with increasing concentration of test compounds and standard lapatinib (20–100 μg/mL), and cell viability was analyzed after 24 h of treatment. IC50 of each test compound was determined in-vitro, lower IC50 value were suggestive of higher anti-proliferative activity. All the test compounds caused impressive cell proliferation inhibition in dose-dependent fashion. Amongst all, compound C showed best antiproliferative activity with lowest IC50 (1.89 μg/mL) on experimented cancer cells.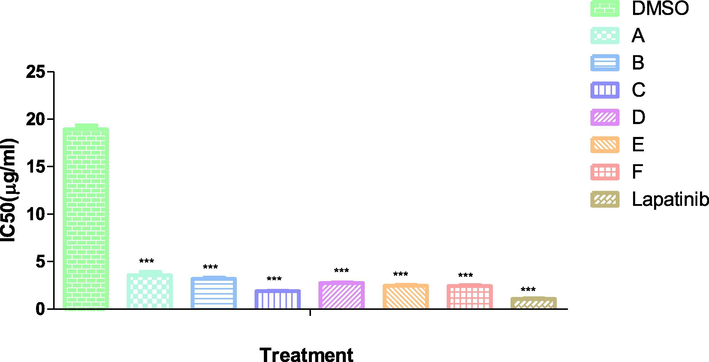
Inhibitory concentration (IC50) of test compound and standard.
3.2.2 Morphological study
Morphological investigation of MCF-7 cells treated with different compounds for cytotoxicity was carried out to determine the changes induced by standard and the test compounds. The control cells were found to be well adhered demonstrating the normal morphology of cells. In contrast, most of standard and test compounds treated cells showed changes in their morphology as cells converted to shrunken, round shape, membrane blebbing and presence of apoptotic bodies. These observations clearly indicated involvement of apoptotic mechanism for cell death. It was found that cells could not be attached to the walls and floating in the medium (Fig. 2).
Morphological changes and micro-scopic analysis of MCF-7 cells after 24 h treatment with test compounds A-F (100 μg/ml), Lapatinib (Positive control) and DMSO (control).
3.2.3 Caspase 3 activity assay
Activation cascade of caspases is responsible for execution of apoptosis phenomena in cell. In particular, caspase-3 acts as a key mitochondrial enzyme for regulating cellular apoptotic pathway. Treatment of MCF7 cell lysate with compound C (100 μg/ml) caused a significant (P < 0.05) increase incaspase-3 activation compared to lapatinib and control. Results of the study clearly suggested the involvement of caspase-3 pathway induction of mitochondrial dependent apoptosis by compound C.
3.2.4 Cell cycle distribution study
Cell proliferation inhibition was further confirmed by determining cell cycle distribution of in presence and absence of compound C, after 24 h of exposure. Finding of the study clearly indicated that compound C causes arrest of MCF-7 cells in S and G2 phase with and 50% cells death via apoptosis (Figs. 3 and 4).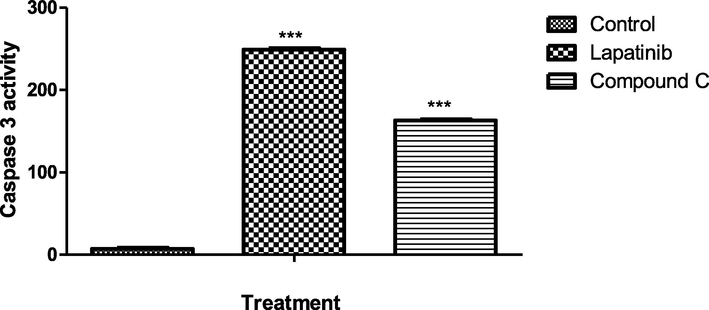
Effects of compound C and lapatinib on caspase-3 activity.
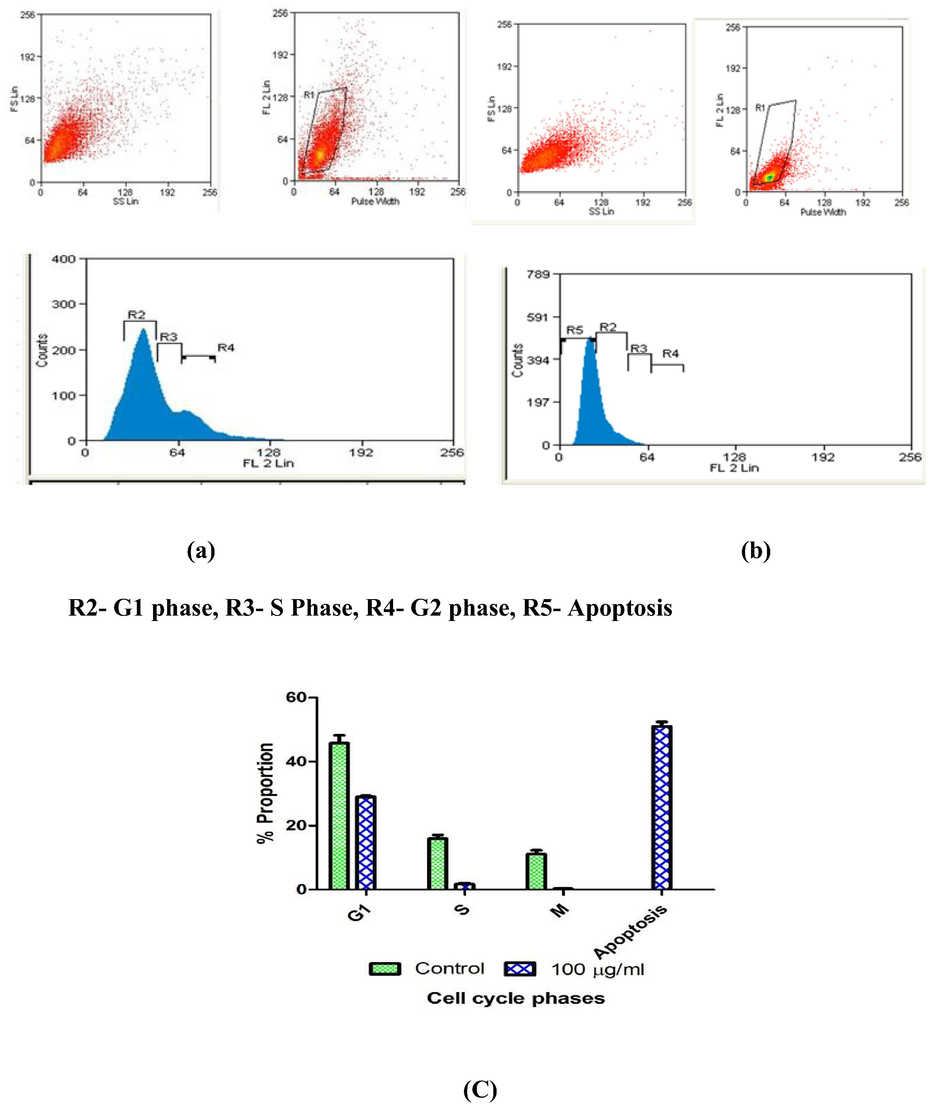
Induction of cell cycle arrest and apoptosis in Compound C treated MCF-7 cells. Cell cycle analysis output of MCF-7 (a) cells after 24 h of incubation (b) cells after 24 h of incubation with Compound C (100 μg/ml). Proportion of MCF-7 (c) cells in apoptosis, G1, S, G2 and apoptosis phases after 24 h of treatment with Compound C (100 μg/ml).
3.2.5 Docking study
Human epidermal growth factor receptor (HER) tyrosine kinase family encompasses diverse receptors of homologous epidermal growth factor (EGF) including HER2/neu/ErbB2, EGFR/ErbB1/HER1, HER4/ErbB4 and HER3/ErbB3. Among this, HER2 is a glycoprotein (185 kDa) acts as promising target in breast cancer therapy. About, 30% of breast and ovarian tumors have shown overexpression of HER2/neugene and associated proteins and recorded as key factor in precipitation of metastatic cancer.
Drugs targeting HER2 act at its extracellular or intracellular region, for example, humanized monoclonal antibody trastuzumab (Herceptin) directly act at extracellular domain while other small kinase inhibitors like neratinib and lapatinib target the intracellular tyrosine kinase domain of HER2. Researchers have now initiated developing novel therapeutic agents against HER2 for effective management of variety of cancers.
Drug lapatinib consist of pyrimidine ring, which is also present in compounds (A-E), based upon this it was envisioned that compounds might show activity against HER2, subsequently compounds were in-silico tested via docking studies. Molecular docking results depicted favorable receptor-ligand interactions for compounds A-F with human epidermal growth factor receptor (HER 2) with high lib dock score (−11.96 to −7.99) and good binding poses. (Table1 & Fig. 5).
Compound
Structure
Docking score
Ki
A
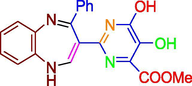
−8.32
800.44 nM
B
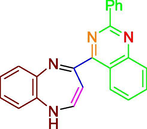
−10.35
25.87 nM
C
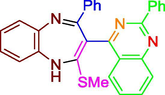
−11.96
1.72 nM
D
−7.99
1.39 µM
E
−11.46
3.99 nM
F
−11.46
3.39 M
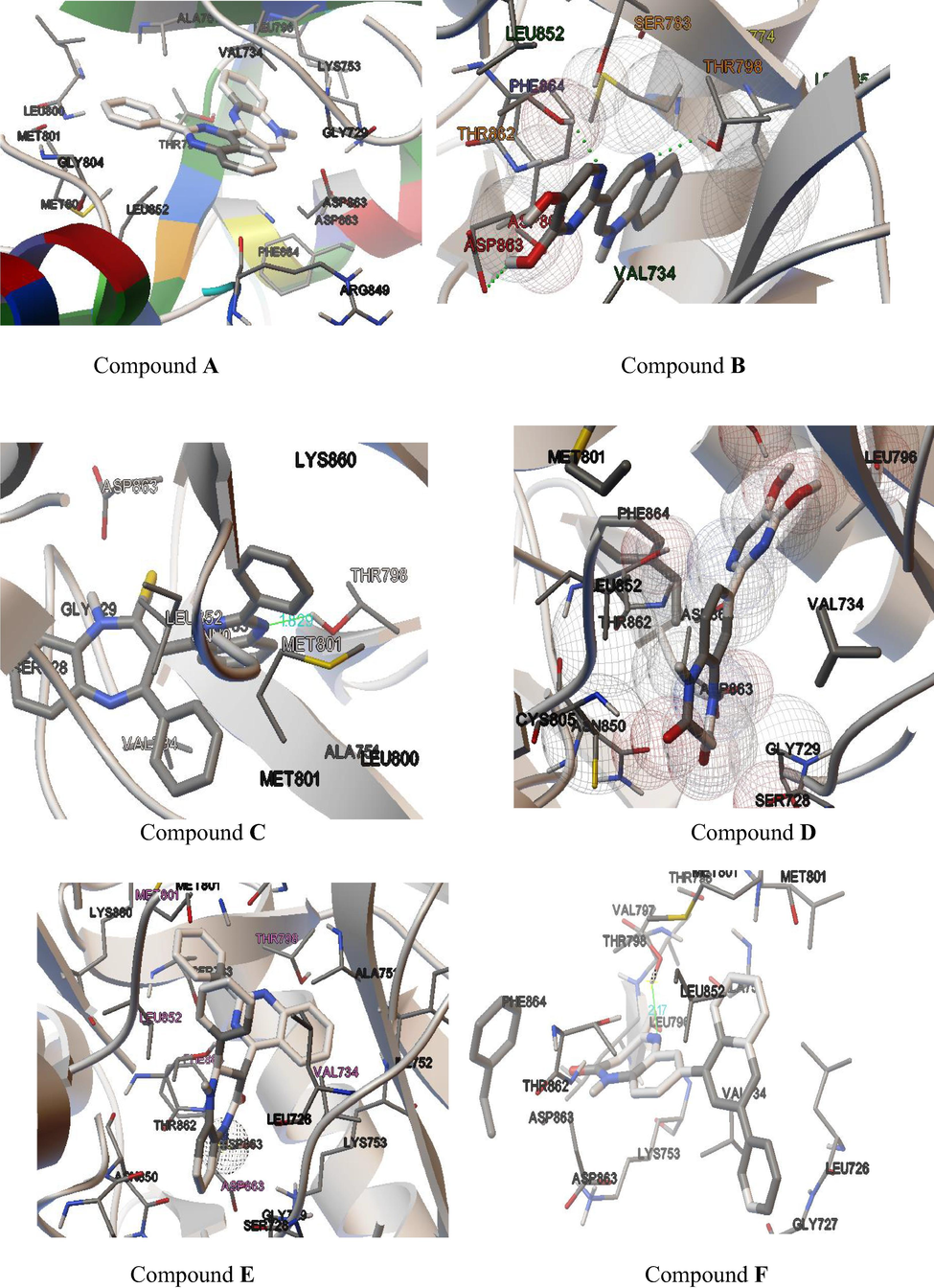
Docked pose of test compound A–F inside the 3PP0.
4 Conclusion
In summary, one-pot domino approach in an ecofriendly manner was adopted to prepared potentially bioactive pyrimidine and quinazoline derivatives (A–F) of 1, 5-benzodiazepines. The IC50 for the compounds demonstrated the strong cytotoxic property against breast cancer cell line MCF-7 of human. Morphological observation of cancer cells clearly indicated membrane damaging effects of compounds A–F. Cell cycle distribution and caspase 3 assay study showed that compound C causes arrest of MCF-7 cells in S and G2 phase probably via activation of the mitochondrial caspases cell death pathway. In addition, molecular docking results depicted favorable receptor-ligand interactions for compounds A-F with human epidermal receptor (HER 2) with high lib dock score and good binding poses. Hence, it can be concluded that anticancer activity of test compound may be attributed to two distinct mechanisms i.e. cell cycle arrest with the mitochondrial cell death pathway during activation, and inhibition of HER 2 enzyme in MCF7 cells. Our findings indicate that test compound holds substantial caliber to be categorized as anticancer agents and could be used to prevent and treat breast cancer. Nevertheless, further studies are required to evaluate their toxicity and efficacy at clinical level in alleviating breast cancer.
Acknowledgements
The authors are deeply grateful to Department of Science and Technology (DST), New Delhi, India for financial support under their CURIE (Consolidation of University Research for Innovation and Excellence in Women Universities) Scheme (SR/CURIE-III/01/2015(G) and Young Scientist Scheme (YSS/2015/001838), India. Authors are grateful to the Researchers Supporting Project No. (RSP-2019/1) King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis and anticancer activity of new quinazoline derivatives. Saudi Pharm. J.. 2017;25(7):1047-1054.
- [Google Scholar]
- Synthesis and anticancer activity evaluation of novel azacalix[2]arene[2]pyrimidines. Eur. J. Med. Chem.. 2018;151:214-225.
- [Google Scholar]
- Quinazoline pharmacophore in therapeutic medicine. Bangladesh J. Pharmacol.. 2016;11(3):716-733.
- [Google Scholar]
- Balsamin induces apoptosis in breast cancer cells via DNA fragmentation and cell cycle arrest. Mol. Cell. Biochem.. 2017;432(1–2):189-198.
- [Google Scholar]
- Synthesis of 2,4,5-triarylated imidazoles via three-component domino reaction under catalyst-free conditions. J. Saudi Chem. Soc.. 2017;21(1):76-81.
- [Google Scholar]
- Discovery of new quinazoline derivatives as irreversible dual EGFR/HER2 inhibitors and their anticancer activities- part 1. Bioorg. Med. Chem. Lett.. 2019;29:591-596.
- [Google Scholar]
- Chickpea lectin inhibits human breast cancer cell proliferation and induces apoptosis through cell cycle arrest. Protein Peptide Lett.. 2018;25(5):492-499.
- [Google Scholar]
- Development of a second-generation, highly efficient manufacturing route for the HIV integrase inhibitor raltegravir potassium. Org. Process Res. Dev.. 2010;15(1):73-83.
- [Google Scholar]
- Recent development in [1, 4] benzodiazepines as potent anticancer agents: a review. Mini Rev. Med Chem.. 2014;14(3):229-256.
- [Google Scholar]
- Anti-cancer pyrimidines in diverse scaffolds: a review of patent literature. Recent Pat. Anti-canc. Drug Dis.. 2015;10(1):23-71.
- [Google Scholar]
- Theoritical bio-significance evalution of quinazoline analogues. Int. Res. J. Pure App. Chem.. 2019;18(1):1-8.
- [Google Scholar]
- Therapeutic potential of heterocyclic pyrimidine scaffolds. Chem. Cent. J.. 2018;12(1):38.
- [Google Scholar]
- Epidemiology of breast cancer in Indian women. Asia-Pac. J. Clin. Oncol.. 2017;13(4):289-295.
- [Google Scholar]
- Synthesis and molecular docking of pyrimidine incorporated novel analogue of 1, 5-benzodiazepine as antibacterial agent. J. Chem. Sci.. 2018;130(3):31.
- [Google Scholar]
- Synthesis of highly substituted pyrroles via nucleophilic catalysis. J. Org. Chem.. 2010;75(18):6271-6274.
- [Google Scholar]
- Steroidal pyrimidines and dihydrotriazines as novel classes of anticancer agents against hormone-dependent breast cancer cells. Front. Pharmacol.. 2018;8(979)
- [CrossRef] [Google Scholar]
- Design, synthesis, docking studies and biological evaluation of novel dihydro-1, 3, 5-triazines as human DHFR inhibitors. Eur. J. Med. Chem.. 2017;125:1279-1288.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2019.12.002.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




