

Translate this page into:
Synthesis, anticancer and molecular docking studies of new class of benzoisoxazolyl-piperidinyl-1, 2, 3-triazoles
⁎Corresponding authors. mahalins@srmist.edu.in (Sakkarapalayam M. Mahalingam), anatarajan@ksu.edu.sa (Natarajan Arumugam)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A small library of hitherto unexplored novel 5-fluorobenzoisoxazolyl-piperidinyl-1, 2, 3-triazole derivatives has been synthesized from 2-azido-fluorobenzoisoxazolyl piperidinyl ethanone and various alkynes in good to excellent yields through a click chemistry approach. Compounds thus synthesized were evaluated for their cytotoxicity against HepG-2 and A549 cancer cells. Interestingly, compounds 4c, 4d, 4e and 4h displayed significant cytotoxicity against HepG-2 and A549 cancer cells. Toxicity study of active compounds was compared with human normal lung IMR-90 cells. Molecular docking has also been investigated for 4a-i with Chk1 protein and the compounds 4c and 4h displayed reasonable molecular interactions with good docking scores.
Keywords
1,2,3-Triazoles
Click chemistry reaction
Anticancer activity
Molecular docking studies

1 Introduction
Multifunctional hybrid heterocycles comprising more than one biologically active entity, wherein each distinct active unit exerts various modes of action, can be useful in the treatment of complex diseases such as cancer (Suresh Kumar et al., 2020). Among the heterocyclic pharmacophore, 1, 2, 3-triazoles occupy a prominent place in drug discovery ascribable to their wide range of biological profiles including antibacterial (Malah et al., 2020; Kushwaha et al., 2014) and anticancer activities (Lakkkula et al., 2019; Sambasiva Rao et al., 2014; Zhang et al., 2014). Triazole is unique structural motif and due to their stability, high selectivity to metabolic degradation and the ability of hydrogen bonding that can make interactions with binding sites more easily in target proteins in the biological system (Bozorov et al., 2019). It was recognized that various types of chemical ties at C-4 position of the 1, 2, 3-triazole core eliminated the planarity that makes better solubility, an essential property in the process of drug development (Zhang et al., 2014). Compound embedded with 1, 2, 3-triazole units displayed significant anticancer activities. For instance, novobiocin analog A (Fig. 1) showed significant cytotoxic activity (Devanesan et al., 2017; AlSalhi et al., 2016) against breast cancer cell lines which possesses 1, 2, 3-triazole as an active unit (Peterson and Blagg, 2010). Similarly, coumarin conjugated 1, 2, 3-triazole derivatives B and C (Fig. 1) exhibited anticancer activity (Shakeel-u-Rehman et al., 2014). It is pertinent to note that pharmaceuticals may develop impurities at various stages of their development which makes the pharmaceutical risky to be administered thus they must be identified and quantitated. In this context, analytical instrumentation and methods (Siddiqui et al., 2017; Al Othman et al., 2013; Rahman et al., 2006) play an essential role for assessing the quality of drug in the process of drug development.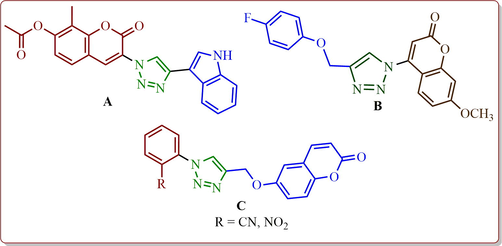
Representatives of triazole heterocyclic hybrids as potent anticancer leads.
Piperidine is another important class of pharmacophore entity (Rubiralta et al., 1991) as this scaffold is present in many natural products and these analogs possess multifarious pharmaceutical activities viz. anti-inflammatory, antibacterial, antihypertensive, anticonvulsant and antimalarial activity (Lawson et al., 2020; Suresh Kumar et al., 2018). Piperidine derivatives are active blocker for the growth of farnesyl transferase (FT) enzyme, which is found to be active in various forms of cancer (Ocasio-Malavé et al., 2020; Nara et al., 2003). In this context, our research group has recently reported the piperidine comprising heterocyclic hybrids which displayed potent anticancer activity (Almansour et al., 2016, 2018). Likewise, benzoisoxazole is also an important class of heterocycle as its derivatives possesses potent Hsp90 inhibitory properties of cancer target (Gopalsamy et al., 2008). Due to biological precedents of the aforementioned heterocycles, we reasoned that the combination of triazole comprising piperidine and benzoisoxazole motifs in a single molecule would be of great interest in the context of anticancer drug discovery. Herein, we wish to report the synthesis of a small library of 5-fluorobenzoisoxazolyl-piperidinyl-1, 2, 3-triazole derivatives and evaluation for their cytotoxicity. In addition, molecular docking studies with human Chk1, which plays an essential role in cell cycle regulation and DNA damage response has also been performed, and the possible interactions of the synthesized compounds with Chk1 protein are predicted.
2 Materials and methods
Synthesis triazole tethered benzoisoxazole piperidine hybrid heterocycles 4a-i
The alkyne (1 mol), and azide 3 (1 mol), CuSO4 (0.4 mol) followed by sodium ascorbate (0.6 mol) in mixture of solvents THF: MeOH: H2O (1:1:1:5 v/v) and the reaction mixture was stirred at 20–25 °C for 10 hrs. Completion of the reaction (TLC), the reaction mixture was distilled out under vacuum. The organic layer was dried over anhydrous Na2SO4 and distilled under vacuum. The crude product was purified by column chromatography using hexane/ethylacetate as an eluent.
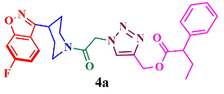
Compound (4a): Yield 89.2%; Mp: 68–70 °C; IR (KBr, cm−1): 3082, 2928, 1733, 1661, 1616, 1271, 1218, 1121; 1H NMR (CDCl3, 400 MHz): δ 0.85–0.87 (t, J = 8.0 Hz, 3H), 1.13–1.81 (m, 4H), 1.97–2.17 (q, 2H), 3.02–3.08 (m, 1H), 3.39–3.48 (m, 3H), 4.01–4.03 (t, J = 8 Hz, 1H), 4.51–4.55 (m, 1H), 5.20 (s, 2H), 5.30 (s, 2H), 7.20–7.68 (m, 8H), 7.69 (s, 1H) ; 13C NMR: δ 11.8, 26.1, 29.6, 33.7, 42.1, 45.0, 52.8, 58.6, 61.5, 97.6, 97.9, 112.6, 112.8, 117.0, 122.2, 122.3, 126.1, 126.7, 129.1, 131.7, 132.9,137.8, 142.6, 159.6, 163.2, 163.6, 166.8; MS: m/z 506.1; Anal. Calcd for C27H28FN5O4: C, 64.15; H, 5.58; N, 13.85; Found C, 64.26H, 5.70 N, 13.94.
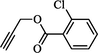
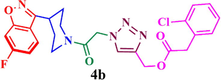
Compound (4b): Yield 91.0%; Mp: 54–56 °C; IR (KBr, cm−1): 3078, 2926,1730, 1663, 1615, 1271, 1217, 1121; 1H NMR (CDCl3, 400 MHz): δ 1.91–2.17 (m, 4H), 3.01–3.07 (m, 1H), 3.30–3.44(m, 2H), 4.03–4.06 (q, 1H), 4.51–4.54 (q, 1H), 5.36 (s, 2H), 5.49 (s, 2H), 7.08 (s, 1H), 7.24–7.26 (m, 2H), 7.41 (m, 2H), 7.61 (s, 1H), 7.82–7.83 (d, J = 4 Hz, 1H). 7.94 (s, 1H); 13C NMR: δ 29.7, 30.2, 33.8, 42.1, 45.0, 51.1, 58.6, 97.5, 97.8, 112.7, 112.9, 117, 122.2, 122.3, 126.0, 126.7, 129.3, 131.1, 131.7, 132.9, 133.9, 142.7, 159.9, 163.0, 163.4, 163.8, 164.0, 165.3, 165.5; MS: m/z 498; Anal. Calcd for C24H21ClFN5O4: C, 57.89; H, 4.25; N, 14.07 Found C, 58.03; H, 4.34 N, 14.16.
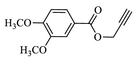
Compound (4c): Yield 87.8%; Mp: 71–73 °C; IR (KBr, cm−1): 3082, 2935,1710, 1664, 1615, 1271, 1219, 1123; 1H NMR (CDCl3, 400 MHz): δ 1.84–1.96 (m, 4H), 2.95–3.05 (m, 1H), 3.30–3.37 (m, 2H), 3.85 (s, 6H), 3.96–3.99 (m, 1H), 4.44–4.47 (m, 1H), 5.24 (s, 2H), 5.40 (s, 2H), 6.77 (s, 1H), 6.79–7.03 (m, 1H), 7.18–7.21 (m, 1H), 7.45 (s, 1H), 7.53–7.60 (m, 2H), 7.84 (s, 1H); 13C NMR: δ29.9, 30.3, 33.9, 42.2, 45.2, 51.1, 56.2, 58.0, 97.6, 97.9, 110.4, 112.1, 112.8, 113.1, 113.1, 117.1, 122.2, 122.3, 124.0, 126.0, 143.5,148.7, 153.3, 159.9, 163.1, 163.4, 164.0, 165.6, 166.8; MS: m/z 524.2; Anal. Calcd for C26H26FN5O6: C, 59.65; H, 5.01; N, 13.38; Found: C, 59.75; H, 5.12 N, 13.46.
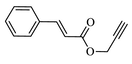
Compound (4d): Yield 88.4; Mp: 65–67 °C; IR (KBr, cm−1): 3079, 2927,1716, 1664, 1612, 1273, 1218, 1121; 1H NMR (CDCl3, 400 MHz): δ 1.30 (s, 9H), 1.90–2.16 (m, 4H), 2.99–3.05 (m, 1H), 3.37–3.43 (m, 2H), 4.03–4.06 (m, 1H), 4.48–4.51 (m, 1H), 5.36 (s, 2H), 5.48 (s, 2H), 7.07 (s, 1H), 7.23–7.25 (d, J = 8 Hz, 1H), 7.40–7.42 (d, J = 8 Hz, 2H), 7.63 (s, 1H), 7.93–7.95 (m, 3H); 13C NMR: δ 29.7, 30.2, 31.6, 33.8, 35.1, 42.1, 45.0, 51.1, 58.0, 97.5, 97.9, 112.7, 113.0, 117.0, 122.3, 122.4, 125.4, 125.9, 126.9, 129.7, 129.9, 143.3, 157.0, 160.0, 163.0, 163.5, 163.9, 164.0, 165.5, 166.3; EI-MS: m/z 520; Anal. Calcd for C28H30FN5O4: C, 64.73; H, 5.82; N, 13.48; Found: C, 64.84; H, 5.91 N, 13.57.
Compound (4e): Yield 86.5%; Mp: 117–119 °C; IR (KBr, cm−1): 3080, 2924,1712, 1662, 1637, 1615, 1271, 1218, 1121; 1H NMR (CDCl3, 400 MHz): δ 1.92–1.98 (m, 4H), 3.02–3.08 (m, 1H), 3.38–3.45 (m, 2H), 4.04–4.07 (m, 1H), 4.52–4.55 (m, 1H), 5.35 (s, 2H), 5.38 (s, 2H), 6.41–6.43 (d, J = 8 Hz, 1H), 7.06–7.08 (d, J = 8 Hz, 1H), 7.10 (s, 1H), 7.24–7.25 (d, 3H), 7.37–7.49 (m, 2H), 7.62–7.72 (m, 2H), 7.91 (s, 1H); 13C NMR: δ29.8, 30.3, 33.8, 42.1, 45.1, 51.2, 57.7, 97.5, 97.8, 112.7, 113.0. 117.0. 117.5, 122.2, 122.3, 128.2, 129.0, 130.5, 134.2, 145.6, 159.9, 163.0, 163.4, 163.9, 164.0, 165.5, 166.8; EI-MS: m/z 490.1; Anal. Calcd for C26H24FN5O4: C, 63.80; H, 4.94; N, 14.31; Found: C, 64.91; H, 5.02; N, 14.40.
Compound (4f):Yield 90.2%; Mp: 66–68 °C; IR (KBr, cm−1): 3079, 2927,1725, 1663, 1615, 1272, 1218, 1122; 1H NMR (CDCl3, 400 MHz): δ 1.84–2.16 (m, 4H), 2.97–3.02 (m, 1H), 3.32–3.40 (m, 2H) 3.98–4.01(m, 1H), 4.44–4.47 (m, 1H), 5.28 (s, 2H), 5.46 (s, 2H), 6.99–7.03 (m, 1H), 7.17–7.19 (d, J = 8 Hz, 1H), 7.55–7.56 (d, J = 4 Hz, 1H), 7.88 (s, 1H), 8.11–8.18(m, 4H); 13C NMR: δ 29.9, 30.3, 33.8, 42.2, 45.2, 51.2, 58.9, 97.6, 97.9, 112.8, 113.1, 117.1, 122.1, 122.3, 123.7, 126.2, 131.1, 135.2, 142.5, 150.7, 159.9, 163.1, 163.4, 164.0, 165.6, 166.8; EI-MS: m/z 509.2; Anal. Calcd for C24H21FN6O6: C, 56.69; H, 4.16; N, 16.53; Found: C, 56.80; H, 4.27 N, 16.65.
3 Results and discussion
3.1 Chemistry
In the present investigation, the synthesis of new class of triazole tethered benzoisoxazole piperidine hybrid heterocycles has been achieved for the first time in three good yielding steps employing Huisgen 1, 3-dipolar cycloaddition reaction via a click chemistry approach. The synthetic strategy planned for the preparation of desired target 4 is summarized in Scheme 1. Our synthetic methodology commenced with commercially available isoxazole 1, which was subjected to the sequence of transformation as outlined in Scheme 2. Thus, isoxazole derivative 1 was treated with chloroacetyl chloride in presence of Et3N in CH2Cl2 at 0–5 °C to furnish N-acylated derivative 2 in good yield. Compound 2 was further reacted with NaN3 in mixture of solvents (water: acetone, 1:3) at ambient temperature to afford azide 3.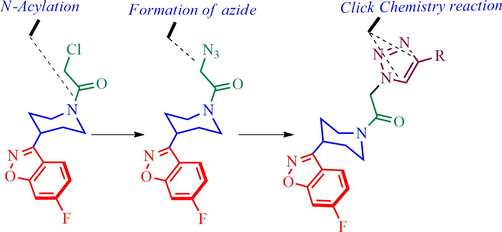
Synthetic plan implemented in this article.
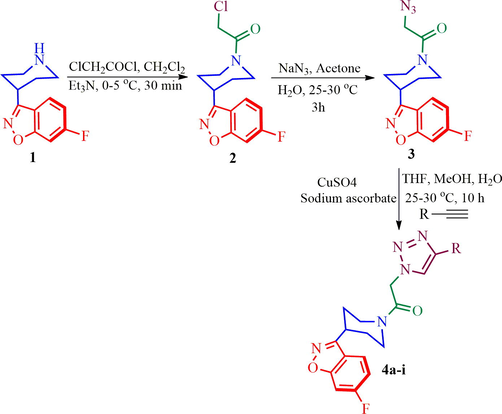
Synthesis of novel triazole derivatives 4a-i.
Having synthesized azide 3 in good yield, the click chemistry reaction was performed for the synthesis of a series of heterocyclic hybrids 4a-i. Thus, the azide 3 was reacted with various alkynes in the presence of CuSO4, sodium ascorbate in mixture of solvent (THF, water and methanol (1: 1: 1) to obtain the targeted 6-flurobenzo[d]isoxazol containing triazoles 4a-i in good yields as described in Scheme 2. The HPLC purity of newly synthesized compounds 4a-i was determined and given in Table.
Entry
Alkynes
Product
Yield%
HPLC purity
1
89.2
98.5
2
91.0
97.8
3
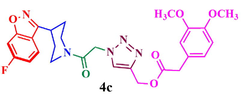
87.8
99.5
4
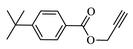
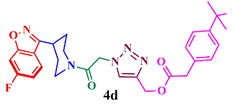
88.4
99.1
5
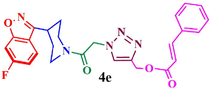
86.5
98.0
6
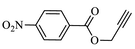
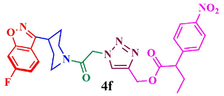
90.2
98.8
7
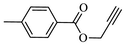
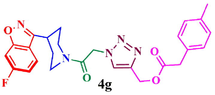
85.4
98.2
8
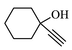
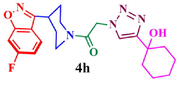
81.0
99.1
9
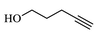
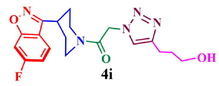
80.3
97.9
The structure of triazole tethered benzoisoxazole derivatives is in complete agreement with the IR and NMR spectral data as described for a representative case 4a. In the IR spectrum, the bands at 1661 and 1616 cm−1 were due to amide carbonyl and C = N groups, respectively. The bands at 1271, 1218 and 1121 cm−1 confirm the availability of C-F, C-N and C-O groups, respectively. In the 1H NMR spectrum, the singlet at 7.69 belongs to the triazole ring hydrogen. A singlet at δ 5.30 ppm was assigned to the methylene group adjacent to the amide carbonyl group. The multiplets in the region between 1.13 and 5.20 ppm were due to piperidinyl ring hydrogens and aliphatic side chain hydrogens. In 13C NMR spectrum of 4a, the signal at δ 166.8, 163.6 ppm are assigned to the amide and adjacent to benzyl carbonyls respectively. The signals at δ 11.8 and 26.1 ppm indicates the methyl and methylene carbons attached to aryl ring, respectively. Further, the presence of molecular ion peak at m/z 506.1 confirms the formation of triazole derivative 4a. HPLC purity for this compound was found to be 98.5%. The other compounds 4a-i were analyzed by the similar straightforward consideration. Finally, the structure of compound 4i was unambiguously ascertained by a single crystal X-ray diffraction analysis [CCDC No.1869222] (Fig. 2).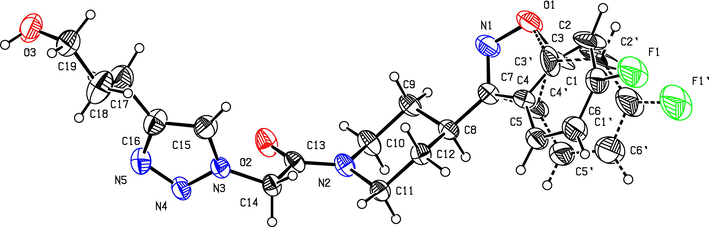
ORTEP diagram of 4i.
The plausible pathway for the construction of triazole tethered piperidinyl benzoisoxazole derivative 4 is described in Scheme 3. The nitrogen lone pair of piperidinyl benzoisoxazole 1 attacks the chloroacetyl chloride furnishing the N-acylated piperidinyl benzoisoxazole 2 which was then treated with sodium azide to afford the azide derivative 3. Simultaneously, the copper (I) generated in situ π-complex with terminal alkyne 5 in the presence of a base, the terminal hydrogen of alkyne is deprotonated and consequently initiation of the reaction to form copper(I) acetylide 7 and then the azide 3 coordinates to copper acetylide to form copper complex 8 followed by formation of regioselective 5-cuprated triazole 9. Finally, compound 9 was protonated to furnish the desired triazole product 4 and the active catalyst was also regenerated.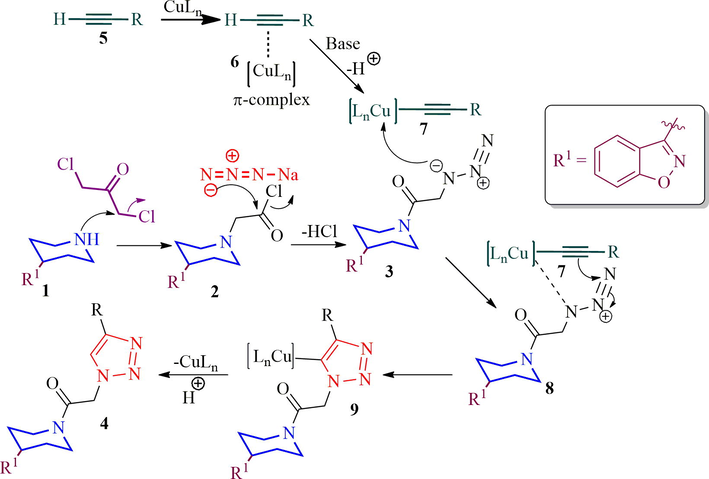
The formation of triazole tethered piperidinylbenzoisoxazole, 4.
3.2 Cytotoxicity studies
The synthesized triazole derivatives 4a-i were examined for their cytotoxic properties against human A549 and HepG-2 cells (Fig. 3), compounds 4a, 4c and 4 h displayed promising activity against HepG-2 cells with IC50 value of 59.8, 47.1 and 66.8 µg/mL, respectively when compared to standard drug cisplatin. In addition, compound 4e showed potent activity against A549 cells with IC50 value of 66.5 µg/mL than other tested compounds (Table 2). Rest of the compounds showed less cytotoxic properties against tested cells. Moreover, cytotoxic study of active compounds such as 4a, 4b, 4c, 4d, 4e and 4 h were examined against human normal lung IMR-90 cells. Among the active compounds, 4c did not show toxicity against normal cells with IC50 value of > 200 µg/mL than other active compounds (Table 2). Therefore, based on the toxicity results 4c might be used as an anticancer agent against cancer cells because it was demonstrated that 4c showed good cytotoxic activity against cancer cells and no toxic against normal cells up to > 200 µg/mL. nt: not tested.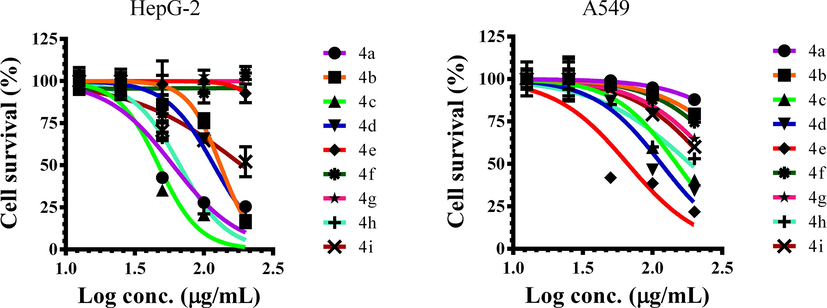
Cytotoxic activities of synthesized compounds 4a-i against HepG-2 and A549 cells for 24 h incubation. Data is calculated by mean ± SD of three independent experiments using Graphpad Prism 6.
Compound
HepG-2 (µg/mL)
A549 (µg/mL)
IMR-90 (µg/mL)
4a
59.8
>200
>200
4b
131
>200
>200
4c
47.1
149
>200
4d
118
115
>200
4e
>200
66.5
>200
4f
>200
>200
nt
4 g
>200
>200
nt
4 h
66.8
187
173
4i
189
>200
nt
Cisplatin
22.5
19.3
–
3.3 Molecular docking
3.3.1 In silico docking studies of ligands 4a-i with Chk1
Chk1 is conserved in most of the species including humans and is one of the essential proteins in cell cycle regulation at various stages of the cell cycle including S phase, G2/M transition and M Phase following DNA damage (Sanchez et al., 1997; Zhang et al., 2014; Zhang and Hunter, 2014). The human Chk1 protein is made up of 467 amino acids, and its N-terminal Chk1-Kd has 265 residues (Chen et al., 2000). Chk1 has a two-lobe fold with ATP-binding site located in between the two lobes (Fig. 4A). Comparison of the various homologue structures of Chk1 reveals that the active site of Chk1-KD is highly conserved and the secondary structure adopts similar conformation including most of the side chains positions (Chen et al., 2000). Moreover, triazole derivatives are identified to be one of the novel inhibitors for Chk1 (Oza et al., 2010). Based on this literature evidence, the Chk1-KD domain was chosen to predict the efficacy of the compounds 4a-i using molecular docking studies, and these synthesized ligands are docked to the ATP-binding pocket of the Chk1-KD protein.![Docking results from AutoDock Vina is shown for compounds 22 K (5-[(1R,3S)-3-aminocyclohexyl]-6-bromo-3-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a] pyrimidin-7-amine) co-crystallized with Chk1 kinase domain (A), 4a (B) and 4c (C). ChK1 is shown in cartoon and colour cyan. The ligand molecules are shown stick model and carbon are colored yellow, oxygen are shown in red and nitrogen are shown in blue. The hydrophobic and hydrogen bonding residues are shown in ball and stick and colored as green (carbon), blue (nitrogen) and red (oxygen). The hydrogen bonds are shown in dotted lines and distances (Å) between amino acids and corresponding ligand atoms are marked. The inset shows the close view of ligand molecules with the binding pocket of Chk1 residues surrounded by hydrophobic contacts and hydrogen bonds.](/content/185/2020/32/8/img/10.1016_j.jksus.2020.09.012-fig7.png)
Docking results from AutoDock Vina is shown for compounds 22 K (5-[(1R,3S)-3-aminocyclohexyl]-6-bromo-3-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a] pyrimidin-7-amine) co-crystallized with Chk1 kinase domain (A), 4a (B) and 4c (C). ChK1 is shown in cartoon and colour cyan. The ligand molecules are shown stick model and carbon are colored yellow, oxygen are shown in red and nitrogen are shown in blue. The hydrophobic and hydrogen bonding residues are shown in ball and stick and colored as green (carbon), blue (nitrogen) and red (oxygen). The hydrogen bonds are shown in dotted lines and distances (Å) between amino acids and corresponding ligand atoms are marked. The inset shows the close view of ligand molecules with the binding pocket of Chk1 residues surrounded by hydrophobic contacts and hydrogen bonds.
Initial docking study was conducted with the control molecule 22 K (5-[(1R,3S)-3-aminocyclohexyl]-6-bromo-3-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidin-7-amine) co-crystallized with Chk1 kinase domain (PDB ID:3OT3) (Fig. 4A) (Labroli et al., 2011). The best docking score (−8.4 kcal/mol) as well the top scoring pose superimposed with the co-crystallized structure less than Root Mean Square Deviation (RMSD) 1.0 Å indicated that the control molecule 22 K was successfully docked with the target protein Chk1-KD (Table S1, vide supplementary data). The docking studies suggest that the 21 residues of Chk1 protein surround the compounds 4a-i. Out of 21 residues, nine residues of Chk1 (Leu15, Val23, Ala36, Lys38, Val68, Leu84, Leu137, Asp148, Leu151), make hydrophobic contacts with the drug molecules. In which, Val23 of Chk1 involved in hydrophobic contacts with all ligand molecules followed by Leu137, which makes hydrophobic contacts with all compounds 4a-h except 4i molecule (vide supplementary data). The Ala36, Leu84, and Leu15 are the other residues majorly contribute to making hydrophobic contacts with ligand molecules. Similarly, Glu17, Ala19, Gly21, Cys87, Asp130, Lys132, Glu134, Asn135, Ser147, Asp148, Gly150, and Leu151 residues are involved in N–H…N, O–H…N and O–H…O hydrogen bonds with the ligands. In which, Lys132 and Cys87 are frequent amino acids interacting with the ligands, followed by Asn135, Ser147, and Asp148. In control, Val23, Ala36, and Leu137 residues involved in hydrophobic contacts. In the current docking experiments, all three residues (Val23, Ala36, and Leu137) are making significant contributions. Similarly, in control Cys87 and Glu134 amino acids are making strong hydrogen bonds. In the current study, Cys87 is one of the common amino acids making strong hydrogen bonds (Table S1 in supplementary data & Fig. 4C). In addition to that Asn135, Ser147, and Asp148 residues interact with a 22 K inhibitor (Labroli etl., 2011) as well as a UCN-01 inhibitor (Zhao et al., 2002) via well-refined water molecules. In our docking experiment, the carbonyl moieties make such hydrogen bonds. For example, the molecule 4a, the carbonyl groups linked with the triazole moiety involved in strong hydrogen bonds with Asn135 and Ser147 (Table S1 in supplementary data, Fig. 4B). Similarly, compounds 4e, 4f, 4 g and 4i (Table S1, supplementary data) makes strong hydrogen bonds with one of these residues. In other hands, the hydroxyl groups introduced in the 4 h and 4i molecules also makes significant hydrogen bonds with Asp130, Lys132, and Asp148 of Chk1-KD. All the top scored poses of the compounds 4a-i in the binding pocket of Chk1-KD suggest that the molecules are sandwiched in between the hydrophobic core of the Chk1-KD. Leu15, Val23, Ala36, Lys38, and Leu84 forms hydrophobic core from one side, and the remaining amino acids of Chk1-KD Val68, Leu137, Asp148 and Leu151 on the other side. Whereas in the hydrogen bond interactions except for Glu17, Ala19, and Gly21, all other nine residues are located on the C-terminal of the molecule (vide supplementary data). The similar environment is also observed with the other Chk1-KD inhibitors such as UCN-01, staurosporine, and SB-218078 (Trott and Olson, 2010) Six out of the nine docking results, the halogen atoms are oriented towards N-terminal lope of Chk1-KD (vide supplementary data), whereas 4 h and 4i molecule the F atoms are oriented towards C-terminal lope. The compounds 4a-b and 4e make π-cation interactions with the Lys38 of Chk1-KD and 4b makes a reasonable salt bridge with the same residue.
In cytotoxic results, the compounds 4a, 4c, 4 h performed better against HepG-2 cells and compound 4e showed potent activity against A549 cells. In comparison with cytotoxic results, compound 4a (Fig. 4B), 4c (Fig. 4C), 4 h and 4e (vide supplementary data) showed reasonable docking score −9.1 kcal/mol. In all four cases, Val23, Leu137 residues have common interactions with Chk1-KD domain. Among the four, 4c and 4 h have a better binding affinity −1.5 kcal/mol, and −1.7 kcal/mol respectively. Interestingly, the interface area also more or less similar (168.7 Å2 for 4c and 169.4 Å2 for 4 h) for these two compounds. Thus, the cytotoxic results are consistent with docking results. Overall, the hydrophobic and hydrogen bonding interactions obtained from the docking experiments provide a structural evident for the potency of compounds, especially 4c, and 4 h against Chk1-KD, and may motivate to explore further the possibilities of designing more specific inhibitors in the future rounds of drug design.
4 Conclusion
A series of novel triazole tethered benzoisoxazoles 4a-i were designed and synthesized by regioselective copper catalyzed 1, 3-dipolar cycloaddition reaction via a click chemistry approach. The synthesized compounds were obtained in good to excellent yields (80–91%) and purity (97–99.5%). Compounds thus synthesized were assayed for their cytotoxicity against HepG-2 and A549 cancer cells. Interestingly, compounds 4c, 4d, 4e and 4 h displayed significant cytotoxicity against HepG-2 and A549 cancer cells. Reasonable binding interactions of 4c and 4 h with Chk1-KD correlates well with the cytotoxic activity and hold great promise as a potential drug against cancer.
Acknowledgement
The project was supported by Researchers Supporting Project number (RSP-2020/231), King Saud University, Riyadh, Saudi Arabia. Dr. Abdul Ajeees Abdul Salam acknowledges the intramural research grant provided by MAHE (MAHE/DREG/PhD/IMF/2019).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Almansour, A.I., Arumugam, N., Suresh Kumar R., Periasamy, V.S., Alshatwi, A.A., Ghabbour, H.A., 2016. Anticancer compounds US patent US9,486,444B1.
- Almansour, A.I., Arumugam, N., Suresh Kumar R., Periasamy, V.S., Alshatwi, A.A., Athinarayanan, J., (2018) Anticancer compounds US patent US9,873,699B1.
- Review on pharmaceutical impurities, stability studies and degradation products. Rev. Adv. Sci. Eng.. 2013;2:155-166.
- [Google Scholar]
- Green synthesis of silver nanoparticles using Pimpinella anisum seeds: antimicrobial activity and cytotoxicity on human neonatal skin stromal cells and colon cancer cells. Int. J. Nanomed.. 2016;11:4439-4449.
- [Google Scholar]
- 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem.. 2019;27:3511-3531.
- [Google Scholar]
- The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: Implications for Chk1 regulation. Cell. 2000;100:681-692.
- [Google Scholar]
- Rapid biological synthesis of silver nanoparticles using plant seed extracts and their cytotoxicity on colorectal cancer cell lines. J. Clust. Sci.. 2017;28:595-605.
- [Google Scholar]
- Discovery of benzisoxazoles as potent inhibitors of chaperone heat shock protein 90. J. Med. Chem.. 2008;51:373-375.
- [Google Scholar]
- Design and synthesis of novel 2H-chromen-2-one derivatives bearing 1,2,3-triazole moiety as lead antimicrobials. Bioorg. Med. Chem. Lett.. 2014;24:1795-1801.
- [Google Scholar]
- Discovery of pyrazolo[1,5-a]pyrimidine-based CHK1 inhibitors: A template-based approach-Part 2. Bioorg. Med. Chem. Lett.. 2011;21:471-474.
- [Google Scholar]
- Synthesis and anticancer activity of 1,2,3-triazole fused N-arylpyrazole derivatives. Russ. J. Gen. Chem.. 2019;89:831-835.
- [Google Scholar]
- Dispiropyrrolidine tethered piperidone heterocyclic hybrids with broad-spectrum antifungal activity against Candida albicans and Cryptococcus neoformans. Bioorg. Chem.. 2020;100:103865
- [Google Scholar]
- Design, synthesis, and antimicrobial activities of 1,2,3-triazole glycoside clickamers. Molecules. 2020;25:790.
- [Google Scholar]
- Discovery and structure-activity relationships of novel piperidine inhibitors of farnesyltransferase. J. Med. Chem.. 2003;46:2467-2473.
- [Google Scholar]
- Synthesis of novel 4-Boc-piperidone chalcones and evaluation of their cytotoxic activity against highly-metastatic cancer cells. Bioorg. Med. Chem. Let.. 2020;30:126760
- [Google Scholar]
- Discovery of a novel class of triazolones as checkpoint kinase inhibitors-hit to lead exploration. Bioorg. Med. Chem. Lett.. 2010;20:5133-5138.
- [Google Scholar]
- Click chemistry to probe Hsp90: Synthesis and evaluation of a series of triazole-containing novobiocin analogues. Bioorg. Med. Chem. Lett.. 2010;20:3957-3960.
- [Google Scholar]
- The importance of impurity analysis in pharmaceutical products: An integrated approach. Accred. Qual. Assur.. 2006;11:69-74.
- [Google Scholar]
- Rubiralta, M., Giralt, E., Diez, E., Structure, preparation, reactivity and synthetic applications of piperidine and its derivatives, Elsevier, Amsterdam, 1991.
- Synthesis of novel 1,2,3-triazole substituted-N-alkyl/aryl nitrone derivatives, their anti-inflammatory and anticancer activity. Eur. J. Med. Chem.. 2014;80:184-191.
- [Google Scholar]
- Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science (New York, N.Y.). 1997;277(5331):1497-1501.
- [Google Scholar]
- Synthesis and biological evaluation of novel isoxazoles and triazoles linked 6-hydroxycoumarin as potent cytotoxic agents. Bioorg. Med. Chem. Lett.. 2014;24:4243-4246.
- [Google Scholar]
- Analytical techniques in pharmaceutical analysis: A review. Arabian J. Chem.. 2017;10:S1409-S1421.
- [Google Scholar]
- In vitro mechanistic exploration of novel spiropyrrolidine heterocyclic hybrids as anticancer agents. Front. Chem.. 2020;8:465.
- [Google Scholar]
- Suresh Kumar, R., Antonisamy, P., Almansour, A.I., Arumugam, N., Periyasami, G., Altaf, M., Kim, H.R., Kwon, K.B., 2018. Functionalized spirooxindole-indolizine hybrids: Stereoselective green synthesis and evaluation of anti-inflammatory effect involving tnf-α and nitrite inhibition. 152, 417–423.
- AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comp. Chem.. 2010;31:455-461.
- [Google Scholar]
- Synthesis and biological evaluation of 4-(1,2,3-triazol-1-yl)coumarin derivatives as potential antitumor agents. Bioorg. Med. Chem. Lett.. 2014;24:799-807.
- [Google Scholar]
- Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer. 2014;134:1013-1023.
- [Google Scholar]
- Structural basis for Chk1 inhibition by UCN-01. J. Biol. Chem.. 2002;277:46609-46615.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2020.09.012.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




