

Translate this page into:
Structural and functional insights into the major mutations of SARS-CoV-2 Spike RBD and its interaction with human ACE2 receptor
⁎Corresponding author. arunbgurung@gmail.com (Arun Bahadur Gurung)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Coronavirus Disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly spread around the world jeopardizing the global economy and health. The rapid proliferation and infectivity of the virus can be attributed to many accumulating mutations in the spike protein leading to continuous generation of variants. The spike protein is a glycoprotein that recognizes and binds to cell surface receptor known as angiotensin-converting enzyme 2 (ACE2) leading to the fusion of the viral and host cell membranes and entry into the host cells. These circulating variants in the population have greatly impacted the virulence, transmissibility, and immunological evasion of the host. The present study is aimed at understanding the impact of the major mutations (L452R, T478K and N501Y) in the receptor-binding domain (RBD) of spike protein and their consequences on the binding affinity to human ACE2 through protein–protein docking and molecular dynamics simulation approaches. Protein-protein docking and Molecular mechanics with generalised Born and surface area solvation (MM/GBSA) binding free energy analysis reveal that the spike mutants-L452R, T478K and N501Y have a higher binding affinity to human ACE2 as compared to the native spike protein. The increase in the number of interface residues, interface area and intermolecular forces such as hydrogen bonds, salt bridges and non-bonded contacts corroborated with the increase in the binding affinity of the spike mutants to ACE2. Further, 75 ns all-atom molecular dynamics simulation investigations show variations in the geometric properties such as root mean square deviation (RMSD), radius of gyration (Rg), total solvent accessible surface area (SASA) and number of hydrogen bonds (NHBs) in the mutant spike:ACE2 complexes with respect to the native spike:ACE2 complex. Therefore, the findings of this study unravel plausible molecular mechanisms of increase in binding affinity of spike mutants (L452R, T478K and N501Y) to human ACE2 leading to higher virulence and infectivity of emerging SARS-CoV-2 variants. The study will further aid in designing novel therapeutics targeting the interface residues between spike protein and ACE2 receptor.
Keywords
Spike mutations
RBD
ACE2
COVID-19
SARS-CoV-2
SARS-CoV-2 variants

1 Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a member of the Coronaviridae family, the Betacoronavirus genus, and the Sarbecovirus subgenus, with a 29.9-kb linear single-stranded positive-sense RNA genome (Lu et al., 2020; Wang et al., 2020). Spike (S), envelope (E), membrane (M), and nucleocapsid (N) are among the four structural proteins encoded by SARS-CoV-2 genome, along with 16 non-structural proteins (Nsp1 to Nsp16) (Wang et al., 2020). The spike glycoprotein is a homotrimer found on the coronavirus surface that aids in the recognition of the human host cell surface receptor angiotensin converting enzyme 2 (ACE2) (Mercurio et al., 2021). This recognition is necessary for the fusion of the viral and host cellular membranes, which allows the viral nucleocapsid to be transferred into the host cells (Zhang et al., 2020). The coronavirus disease 2019 (COVID-19), which is caused by SARS-CoV-2, has a massive impact on world health and the economy (Li et al., 2021). The epicentre of the current pandemic was initially detected in Wuhan, Hubei province, China. Since then, the disease has spread rapidly around the world, affecting millions of individuals and causing unprecedented number of deaths (Wu et al., 2020). Several studies have shown that SARS-CoV-2 is closely linked to bat SARS-like-CoVs, but the virus's origin and intermediate host species remain unknown (Sun et al., 2020; Zhou and Shi, 2021). The viral genome undergoes numerous changes in the spike proteins in order to be able to leap species and infect a new mammalian host (Guruprasad, 2021).
The spike protein is made up of two subunits, S1 and S2, which aid in cellular receptor Angiotensin-converting enzyme 2 (ACE2) affinity and membrane fusion, respectively (Satarker and Nampoothiri, 2020). The receptor-binding domain (RBD) of the S1 unit may directly bind to the ACE2 receptor and is also the primary target of SARS-CoV-2 neutralizing antibodies (Ab) (Kadam et al., 2021; Souza et al., 2021). S1 is thus thought to be a hotspot for mutations of clinical significance in terms of virulence, transmissibility, and immunological evasion of the host (Shang et al., 2020; Yi et al., 2020). In SARS-CoV-2, the PRRA sequence motif between the S1 and S2 subunits serves as a furin cleavage site (Ou et al., 2020). SARS-CoV-2 has evolved into numerous co-circulating variants since its discovery in Wuhan in 2019 (Jia and Gong, 2021). The binding ability of the circulating variants' spike protein to human ACE2 has considerably enhanced, resulting in a considerable increase in its replication and transmission (Korber et al., 2020; Thomson et al., 2021). In the spike, there are 3698 nucleic acid mutations (frequency: 0.968%) and 2746 amino acid mutations (frequency: 2.157%). Substitution mutations like D614G, N501Y, Y453F, N439K/R, P681H, K417N/T, and E484K, as well as deletion mutations like ΔH69/V70 and Δ242–244 are the most common in the spike protein (Li et al., 2021). The D614G mutation enhanced viral proliferation and transmission as compared to wild-type viruses (Daniloski et al., 2021). Due to the E484K substitution, the 501Y.V2 variant is more resistant to multiple monoclonal antibodies, convalescent plasma, and vaccine sera, whereas the N501Y substitution enhanced the affinity to human ACE2 and infectivity (Cele et al., 2021; McCormick et al., 2021; Noh et al., 2021). A large number of SARS-CoV-2 variants have emerged as a result of the virus's global transmission, particularly emerging strains recently discovered in the United Kingdom (variant 20I/501Y.V1, lineage B.1.1.7), South Africa (variant 20H/501Y.V2, lineage B.1.351), and Brazil (variant 20 J/501Y.V3, and lineage P.1). These variations all have the N501Y mutation in the SARS-CoV-2 spike (S) protein, which is the target of most COVID-19 vaccines (Jia and Gong, 2021). The emerging variants have been classified into Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1) and Kappa and Delta (B.1.617.1 and B.1.617.2) (https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/). The Alpha variant features a N501Y mutation as well as a K417N mutation. The E484K mutation is seen in an emerging variant variation derived from B.1.1.7. Other than N501Y, both Beta and Gamma variants harbours additional substitutions. The E484K mutation is present in the Beta variants, whereas the E484K and K417T mutations are present in the Gamma variants. In India's second COVID-19 wave, the newest significant variants, Delta and Kappa, were discovered to share two mutations: E484Q and L452R. Delta variant additionally has a unique mutation, T478K in addition to the two mutations listed above (Khateeb et al., 2021).
SARS-CoV-2 may evade human immune response by continuous genomic evolution, such as substitution in the viral RBD and/or deletion and insertion in the viral spike's N-terminal domain loops, particularly in immunocompromised hosts (Li et al., 2021). The best way to prevent SARS-CoV-2 infection is to be vaccinated. There has been more than eight COVID-19 vaccines approved for vaccination among priority groups under an Emergency Use Authorization (EUA), including the Moderna mRNA-1273 vaccine, the Pfizer-BioNtech BNT162b2 vaccine, China's CoronaVacTM and Sinopharm's COVID-19 vaccines, and Russia's Sputnik V and EpiVacCorona vaccines, AstraZeneca's ChAdOx1 novel coronavirus 2019 (nCoV-19) and Janssen's Ad26.COV2.S (Jia and Gong, 2021). The protective rate and efficacy of each vaccines against different variants, however, varies. Because growing evidence suggests that SARS-CoV-2 Variants B.1.351 and B.1.1.7 are resistant to neutralizing antibodies in convalescent plasma and vaccinee sera, it is uncertain if these vaccines are still efficacious against SARS-CoV-2 mutants continually produced in the community (Chen et al., 2021; Wang et al., 2021). In the present studies, we have modelled the mutant structures of Spike protein-L452R, T478K and N501Y (Khateeb et al., 2021) through in silico mutagenesis technique and evaluated their binding interactions with human ACE2 with respect to the native spike protein using protein–protein docking method. We have characterized the interface regions of native and mutant spike:ACE2 complexes. The stability of native and mutant spike models were investigated using all-atom molecular dynamics simulations.
2 Materials and methods
2.1 Retrieval of native ACE2 and native Spike RBD complex structure
The crystal structure of the receptor-binding domain (RBD) of SARS-CoV-2 spike protein bound to the cell receptor ACE2 was retrieved from protein data bank (http://www.rcsb.org/) using accession ID: 6M0J. The structure is solved through the X-ray diffraction method at a resolution of 2.45 Å (Lan et al., 2020). The structure complex was split into ACE2 assigned as chain A and spike RBD assigned as chain B using UCSF Chimera tool (Pettersen et al., 2004).
2.2 In silico mutagenesis of Spike protein
Three mutant models of Spike protein-L452R, T478K and N501Y were generated using the mutagenesis program of PyMOL Molecular Graphics System software version 2.3.3 Schrödinger, LLC. The rotamer was selected using the highest frequency of occurrence.
2.3 Protein-protein docking
The interaction of ACE2 with the three mutant models of Spike (L452R, T478K and N501Y) were studied using High Ambiguity Driven protein–protein DOCKing (HADDOCK) version 2.4 program which uses an information-driven flexible protein–protein docking approach (Van Zundert et al., 2016). The binding sites of ACE2 and Spike mutant were defined based on the interaction profile of crystal structure of ACE2-spike (PDB ID: 6M0J). The different conformations of the protein–protein complexes were ranked based on the HADDOCK score which is computed using the following scoring function (equation 1). HADDOCK Score = 0.2 × Electrostatic energy + 1.0 × Van der Waals energy + 1.0 × Desolvation energy + 0.1 × Restraints violation energy (1)
The best structures of the conformers were downloaded in PDB format.
2.4 MM/GBSA energy calculation:
Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) program of the HawkDock web server (Weng et al., 2019) was used to predict binding free energies of the protein–protein complexes as well as per residue-free energy contributions.
2.5 Characterization of protein–protein interface and hot spot residues
The interface residues, interface area and molecular interactions were evaluated using PDBSum program (Laskowski, 2009) and the hot spot residues in the interface region were determined through alanine scanning mutagenesis program of DrugScorePPi webserver (Kruger and Gohlke, 2010).
2.6 Molecular dynamics simulation:
The native spike:ACE2 and mutant spike:ACE2 complexes were subjected to 75 ns MD simulation studies using GROningen MAchine for Chemical Simulations (GROMACS) 2019.2 software package (Hess et al., 2008). The topology files of the complexes were prepared using GROMOS96 43a1 force field. The complexes were prepared for MD simulation within a water-filled 3-D cube of 1 Å spacing using a three-point water-model (SPC216) with periodic boundary conditions. Newton's equations of motion were integrated using a leap-frog time integration algorithm. The complexes were neutralized by the addition of 0.15 M NaCl and energy minimization was performed using the steepest descent method. The temperature was set at 300 K and the complexes were subjected to equilibration under 100 ps in NVT (Number of particles, Volume and Temperature) ensemble and another 100 ps under NPT ensemble (Number of particles, Pressure and Temperature). The complexes were subjected to production MD run for 75 ns in NPT ensemble after heating and equilibration. The geometrical properties were computed using defined programs of GROMACS 2019.2 software. MD simulations data was plotted using Xmgrace plotting tool.
3 Results
We have selected three major mutations-L452R, T478K and N501Y that occur in the receptor-binding domain (RBD) of spike protein (Fig. 1). The binding interactions between human ACE2 and these three mutant models of Spike (L452R, T478K and N501Y) were studied using protein–protein docking method. The binding affinity between the protein partners were ranked according to the HADDOCK score (Table 1). The HADDOCK scores for Native Spike:ACE2, L452R Spike:ACE2, T478K Spike:ACE2 and N501Y Spike:ACE2 complexes were −137.6 ± 2.8, −140.5 ± 10.2, −141.3 ± 1.0 and −140.3 ± 1.3 respectively. In each of the docked complexes, electrostatic energy contributes significantly to the binding followed by van der Waals energy and desolvation energy. Further, the MM/GBSA binding free energy of the complexes- Native Spike:ACE2, L452R Spike:ACE2, T478K Spike:ACE2 and N501Y Spike:ACE2 complexes were found to be −73.06 kcal/mol, −76.59 kcal/mol, −75.0 kcal/mol and −75.42 kcal/mol (Table 2). The top 5 residues contributing significantly to the binding between ACE2 and native Spike protein includes Tyr41 (−3.18 kcal/mol), Gln42 (−3.07 kcal/mol), Thr27 (−3.02 kcal/mol), Gln24 (−3.0 kcal/mol) and Lys353 (−2.54 kcal/mol) of ACE2 and Gln498 (−4.63 kcal/mol), Gln493 (−4.54 kcal/mol), Tyr505 (−4.47 kcal/mol), Lys417 (−4.25 kcal/mol) and Phe486 (−4.17 kcal/mol). The key residues majorly contributing towards the interaction between ACE2 and Spike L452R mutant includes Tyr41 (−4.27 kcal/mol), Thr27 (−3.04 kcal/mol), Leu45 (−2.94 kcal/mol), Tyr83 (−2.83 kcal/mol) and Gln24 (−2.77 kcal/mol) of ACE2 and Tyr505 (−6.45 kcal/mol), Lys417 (−4.72 kcal/mol), Phe486 (−4.62 kcal/mol), Gln493 (−4.2 kcal/mol) and Leu455 (−4.05 kcal/mol). The residues such as Tyr83 (−4.16 kcal/mol), Tyr41 (−3.19 kcal/mol), Thr27 (−3.01 kcal/mol), Gln24 (−3.0 kcal/mol) and Lys31 (−2.76 kcal/mol) of ACE2 and Phe486 (−5.01 kcal/mol), Tyr505 (−4.41 kcal/mol), Gln493 (−4.26 kcal/mol), Lys417 (−4.21 kcal/mol) and Tyr489 (−4.17 kcal/mol) contribute significantly to the interaction between ACE2 and Spike T478K mutant. The top 5 residues contributing considerably to the binding between ACE2 and Spike N501Y mutant includes Tyr41 (−5.08 kcal/mol), Gln24 (−3.11 kcal/mol), Thr27 (−2.96 kcal/mol), Gln42 (−2.75 kcal/mol) and Lys31 (−2.64 kcal/mol) of ACE2 and Tyr501 (−7.18 kcal/mol), Tyr505 (−5.01 kcal/mol), Tyr449 (−4.72 kcal/mol), Gln493 (−4.47 kcal/mol) and Phe486 (−4.22 kcal/mol). Thus, the L452R, T478K and N501Y mutations in the spike increased binding affinity with ACE2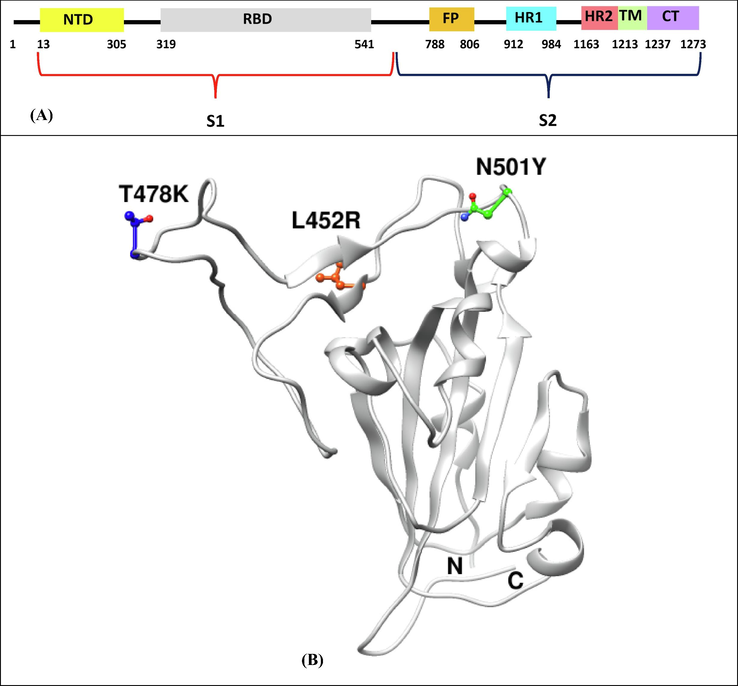
Schematic diagram of spike protein domains- N-terminal domain (NTD), receptor-binding domain (RBD), fusion peptide (FP), heptapeptide repeat sequence 1 (HR1), heptapeptide repeat sequence 2 (HR2), transmembrane region (TM) and cytoplasm domain (CT), and structural positions of mutations (T478K, L452R and N501Y on spike RBD region.
Parameters
Native Spike:ACE2
L452R Spike:ACE2
T478K Spike:ACE2
N501Y Spike:ACE2
HADDOCK score
−137.6 ± 2.8
−140.5 ± 10.2
−141.3 ± 1.0
−140.3 ± 1.3
Cluster size
200
200
200
200
RMSD from the overall lowest-energy structure (Å)
0.6 ± 0.4
0.8 ± 0.5
0.5 ± 0.3
0.8 ± 0.5
Van der Waals energy (kcal/mol)
−63.1 ± 4.2
−63.9 ± 8.4
−66.9 ± 5.3
−61.0 ± 0.6
Electrostatic energy (kcal/mol)
−260.2 ± 7.8
−273.5 ± 8.7
−264.9 ± 20.4
−284.9 ± 4.2
Desolvation energy (kcal/mol)
–23.3 ± 2.3
–22.6 ± 2.8
–22.4 ± 2.7
–22.9 ± 2.2
Restraints violation energy (kcal/mol)
8.9 ± 1.4
7.6 ± 2.9
10.0 ± 1.3
5.7 ± 0.8
Buried surface area (Å2)
1891.0 ± 60.1
1986.3 ± 95.1
1990.5 ± 45.8
1973.5 ± 47.9
Z-score
0.0
0.0
0.0
0.0
Complexes
MM-GBSA Binding free energy (kcal/mol)
Top 5 residues contributing majorly to the binding energy
ACE2
Spike
Native Spike:ACE2
−73.06
Tyr41 (−3.18), Gln42 (−3.07), Thr27 (−3.02), Gln24 (−3.0), Lys353 (−2.54)
Gln498 (−4.63), Gln493 (−4.54), Tyr505 (−4.47), Lys417 (−4.25), Phe486 (−4.17)
L452R Spike:ACE2
−76.59
Tyr41 (−4.27), Thr27 (−3.04), Leu45 (−2.94), Tyr83 (−2.83), Gln24 (−2.77)
Tyr505 (−6.45), Lys417 (−4.72), Phe486 (−4.62), Gln493 (−4.2), Leu455 (−4.05)
T478K Spike:ACE2
−75.00
Tyr83 (−4.16), Tyr41 (−3.19), Thr27 (−3.01), Gln24 (−3.0), Lys31 (−2.76)
Phe486 (−5.01), Tyr505 (−4.41), Gln493 (−4.26), Lys417 (−4.21), Tyr489 (−4.17)
N501Y Spike:ACE2
−75.42
Tyr41 (−5.08), Gln24 (−3.11), Thr27 (−2.96), Gln42 (−2.75), Lys31 (−2.64)
Tyr501 (−7.18), Tyr505 (−5.01), Tyr449 (−4.72), Gln493 (−(−4.47), Phe486 (−4.22)
To explain the differences in the binding affinity of the native and mutant spike proteins with ACE2, their docked complexes were next subjected to interface statistics analysis using PDBSum (Table 3). The interface residues and molecular interactions between ACE2 and spike in native and mutant complexes are shown in Fig. 2. The complex between ACE2 and native Spike protein shows an interface area of 952:984 Å2, number of 19:18 interface residues, 1 salt bridge, 10 hydrogen bonds and 133 non-bonded contacts. In the ACE2 and L452R spike mutant model complex, there are 20:23 interface residues, an interface area of 1085:1059 Å2, 2 salt bridges, 11 hydrogen bonds and 177 non-bonded contacts. The complex between ACE2 and T478 Spike mutant shows 19:21 interface residues, 977:987 Å2 area, 2 salt bridges, 10 hydrogen bonds and 156 non-bonded contacts. The interface between ACE2 and N501Y spike mutant has 19:19 residues, an interface area of 962:1020 Å2, 1 salt bridge, 13 hydrogen bonds and 143 non-bonded contacts. The increase in the binding affinity of L452R spike mutant to ACE2 can be attributed to an increase in the number of interface residues, interface area, salt bridges, hydrogen bonds and non-bonded contacts. The increased binding affinity of T478K spike mutant to ACE2 can be explained in terms of increase in the number of interface residues, interface area, number of salt bridges and number of non-bonded contacts. The increased binding affinity of N501Y spike mutant to ACE2 can be explained in terms of an increase in the number of interface residues, interface area, number of hydrogen bonds and number of non-bonded contacts. The total number of hotspot residues in the interface region of Native Spike:ACE2, L452R Spike:ACE2, T478K Spike:ACE2 and N501Y Spike:ACE2 were 5 (Asp30, Tyr41, Tyr83 in ACE2 and Tyr489, Tyr505 in native spike protein) (Suppl. Table 1), 6 (Asp38, Tyr41, Tyr83 in ACE2 and Tyr449, Tyr489, Tyr505 in L452R spike mutant) (Suppl. Table 2), 6 (Asp30, Asp38, Tyr41, Tyr83 in ACE2 and Tyr489, Tyr505 in T478K spike mutant) (Suppl. Table 3) and 6 (Asp38, Tyr41, Tyr83 in ACE2 and Tyr489, Tyr501, Tyr505 in N501Y spike mutant) (Suppl. Table 4) respectively.
Systems
Chain
No. of interface residues
Interface Area (Å2)
No. of salt bridges
No. of disulphide bonds
No. of hydrogen bonds
No. of non-bonded contacts
Native Spike:ACE2
A
19
952
1
–
10
133
B
18
984
L452R Spike:ACE2
A
20
1085
2
–
11
177
B
23
1059
T478K Spike:ACE2
A
19
977
2
–
10
156
B
21
987
–
13
143
N501Y Spike:ACE2
A
19
962
1
B
19
1020
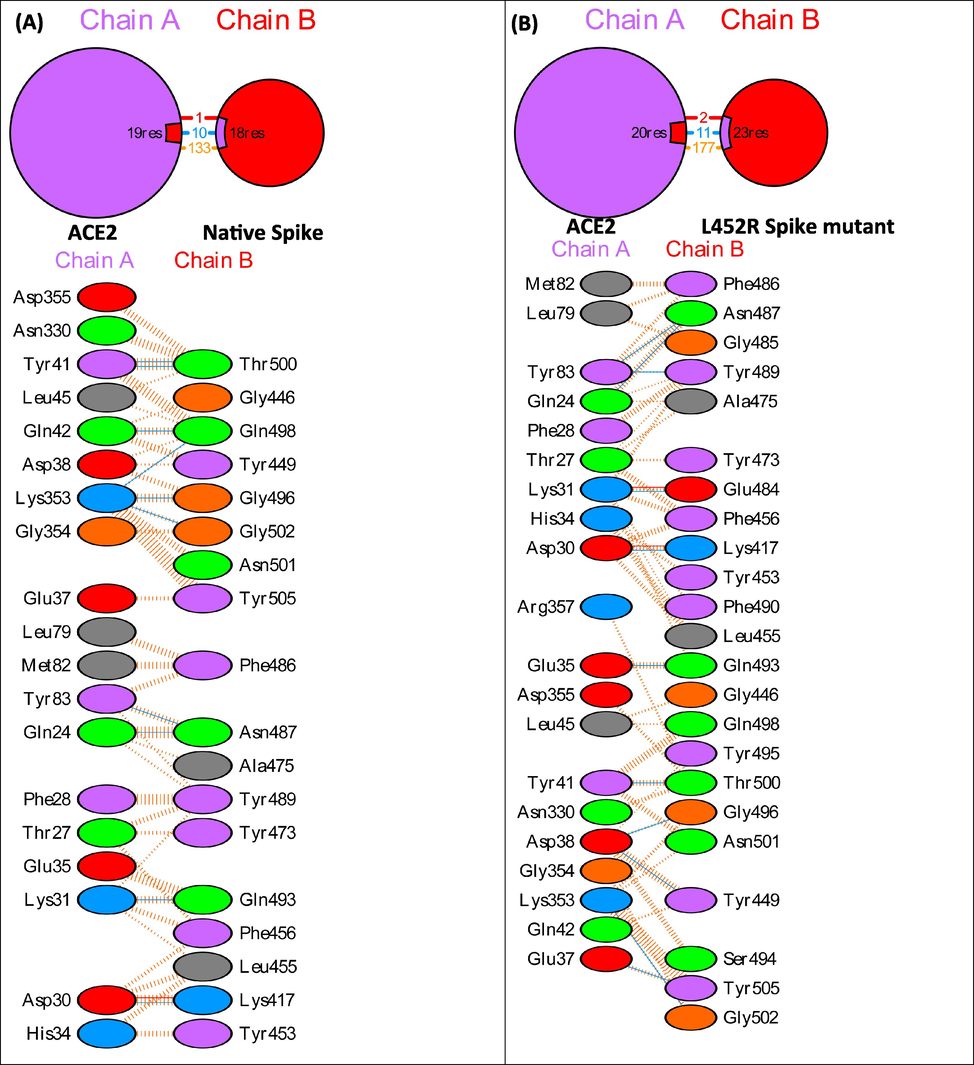
PDBSum interface analysis showing interface residues and molecular interactions in native and mutant Spike:ACE2 complexes.
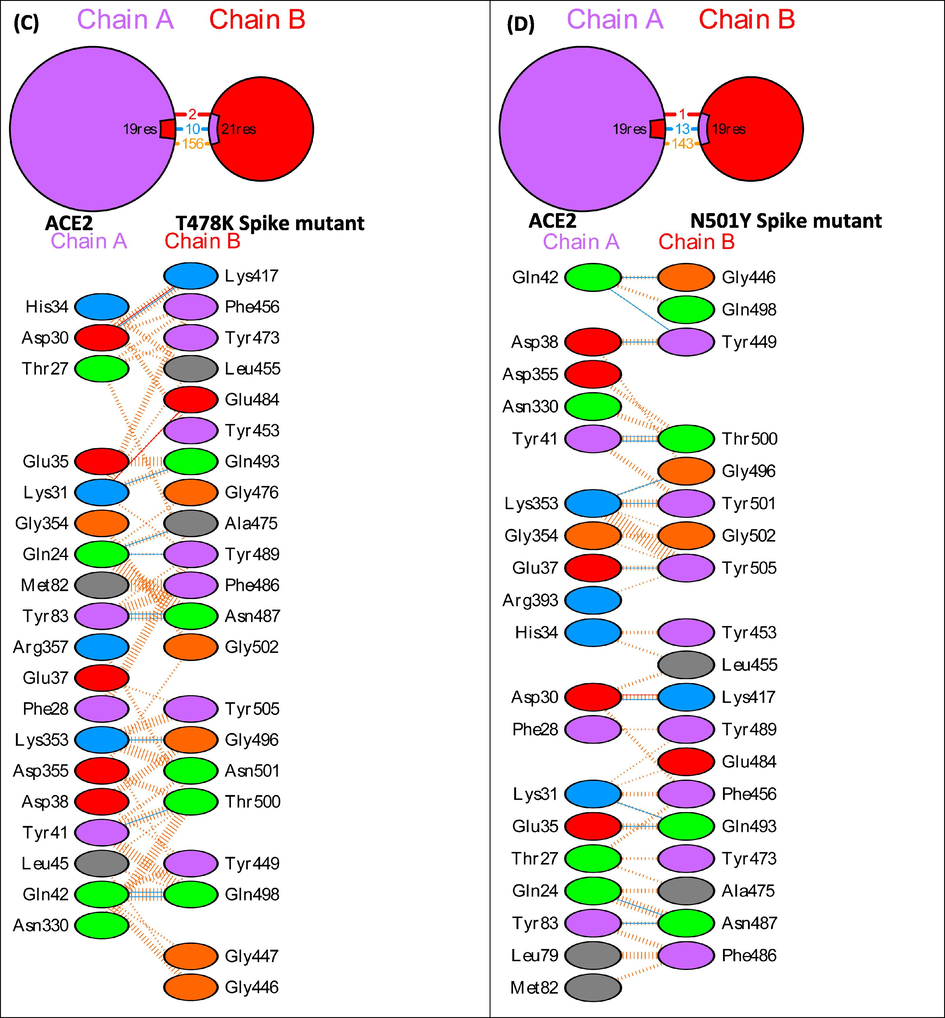
PDBSum interface analysis showing interface residues and molecular interactions in native and mutant Spike:ACE2 complexes.
Systems
RMSD (nm)
Rg (nm)
Total SASA (nm2)
No. of H-bonds
Native Spike:ACE2
0.465537598 ± 0.086582959
3.116693675 ± 0.020677384
322.5054567 ± 6.129235
636.1970706 ± 14.63797953
L452R Spike:ACE2
0.378618486 ± 0.052506812
3.033101278 ± 0.030052667
323.1132277 ± 6.044326
631.5473 ± 15.02665
T478K Spike:ACE2
0.423504386 ± 0.075685
3.035738 ± 0.034536
326.1164 ± 5.607122
625.00 ± 14.01999
N501Y Spike:ACE2
0.493888 ± 0.06266
3.027204 ± 0.041179
322.6669 ± 6.344532
630.6005 ± 15.20264
Further, the Native Spike:ACE2, L452R Spike:ACE2, T478K Spike:ACE2 and N501Y Spike:ACE2 complexes were subjected to 75 ns MD simulations study in an aqueous solution. We made a comparative assessment of the geometric parameters such as RMSD, Rg, SASA and the number of hydrogen bonds between the mutant and native spike complexes (Table 4). Root mean square deviation (RMSD) is the average distance between the backbone atoms of starting structure with simulated structures. The RMSD plot of backbone atoms of the Native and mutant spike in complex with ACE2 are shown in Fig. 3. The average RMSD values of the Native Spike:ACE2, L452R Spike:ACE2, T478K Spike:ACE2 and N501Y Spike:ACE2 complexes were 0.465537598 ± 0.086582959 nm, 0.378618486 ± 0.052506812 nm, 0.423504386 ± 0.075685 nm and 0.493888 ± 0.06266 nm respectively (Table 4). The decrease in the RMSD values in the mutant spike:ACE2 complex except for N501Y spike:ACE2 seems to suggest that the spike RBD mutations caused a decrease in the conformational stabilities in the protein–protein complexes.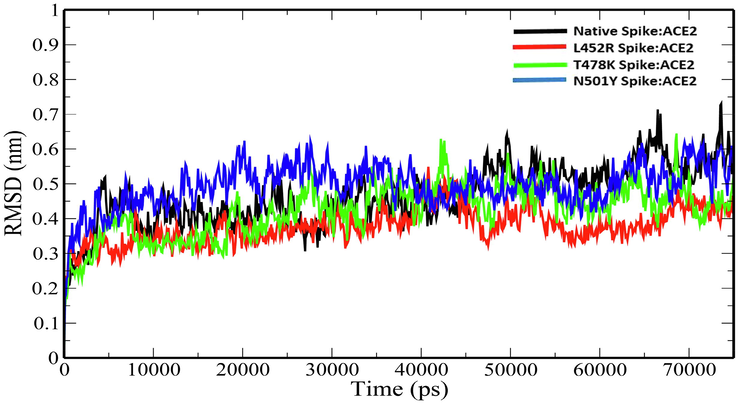
Plot of backbone RMSD versus time (ps) for native and mutant Spike:ACE2 complexes.
Radius of gyration (Rg) explains the structural compactness of a protein which is calculated along the x-, y- and z-axes, as a function of time. The Rg plot for the native and mutant spike:ACE2 complexes show stable fluctuations throughout the simulation time. (Fig. 4). The average Rg values of Native Spike:ACE2, L452R Spike:ACE2, T478K Spike:ACE2 and N501Y Spike:ACE2 complexes were 3.116693675 ± 0.020677384 nm, 3.033101278 ± 0.030052667 nm, 3.035738 ± 0.034536 nm and 3.027204 ± 0.041179 nm (Table 4). The slight decrease in the Rg values of the mutant spike:ACE2 complexes indicates that the mutations increase the overall compactness of the protein–protein complexes. Solvent-accessible surface area (SASA) is an approximate surface area of a protein that is accessible to solvent molecules. The average total SASA values of Native Spike:ACE2, L452R Spike:ACE2, T478K Spike:ACE2 and N501Y Spike:ACE2 complexes were 322.5054567 ± 6.129235 nm2, 323.1132277 ± 6.044326 nm2, 326.1164 ± 5.607122 nm2 and 322.6669 ± 6.344532 nm2 respectively (Table 4). The increase in total SASA for mutant spike:ACE2 complexes (Fig. 5) causes alteration in the folding pattern of the protein–protein complex. On account of the increase in the SASA of the protein upon mutations, there is a decrease in the number of hydrogen bonds in the protein–protein complex (Fig. 6). The average number of hydrogen bonds in Native Spike:ACE2, L452R Spike:ACE2, T478K Spike:ACE2 and N501Y Spike:ACE2 complexes were 636.1970706 ± 14.63797953, 631.5473 ± 15.02665, 625.00 ± 14.01999 and 630.6005 ± 15.20264 respectively (Table 4).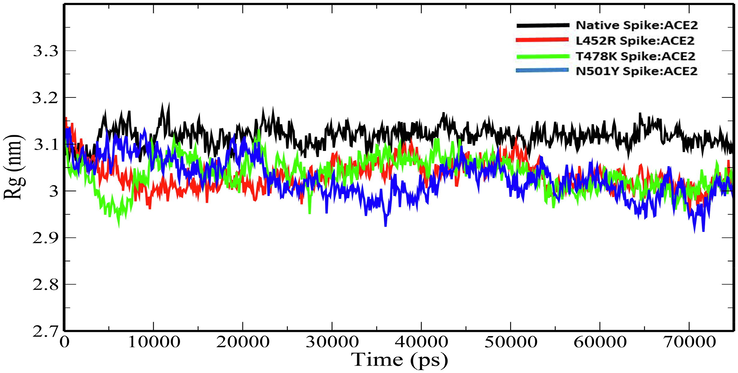
Plot of Rg versus time (ps) for for native and mutant Spike:ACE2 complexes.
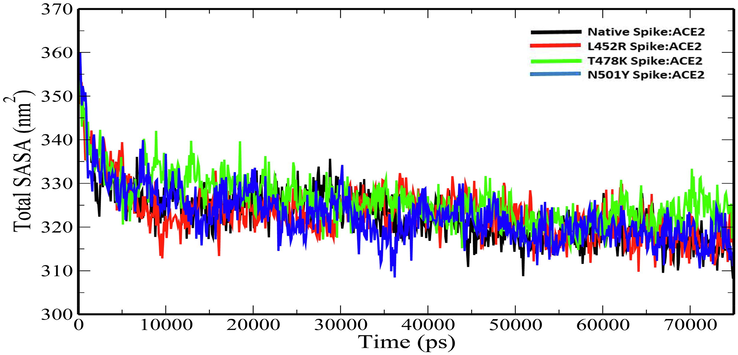
Plot of total SASA versus time (ps) for for native and mutant Spike:ACE2 complexes.
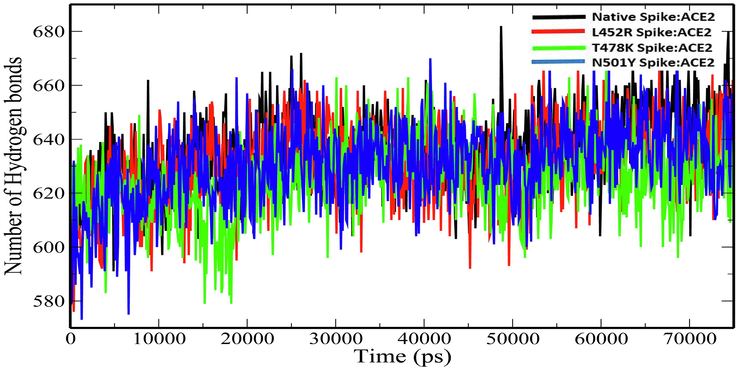
Plot of number of hydrogen bonds versus time (ps) for for native and mutant Spike:ACE2 complexes.
4 Discussion
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a member of the Coronaviridae family with a 29.9-kb linear single-stranded positive-sense RNA genome (Lu et al., 2020; Wang et al., 2020). The virus uses the spike glycoprotein found on the coronavirus surface that aids in the recognition to ACE2 and the fusion of the viral and host cellular membranes (Zhang et al., 2020). The coronavirus disease 2019 (COVID-19), which is caused by SARS-CoV-2, has caused a massive impact on world health and the economy (Li et al., 2021). The spike protein is made up of two subunits, S1 and S2, which aid in cellular receptor Angiotensin-converting enzyme 2 (ACE2) affinity and membrane fusion, respectively (Satarker and Nampoothiri, 2020). S1 is considered a hotspot for mutations of significant clinical significance in terms of virulence, transmissibility, and immunological evasion of the host (Shang et al., 2020; Yi et al., 2020). SARS-CoV-2 has evolved into numerous co-circulating variants since its discovery in Wuhan in 2019 (Jia and Gong, 2021). The emerging variants have been classified into Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1) and Kappa and Delta (B.1.617.1 and B.1.617.2). The binding ability of the circulating variants' spike protein to human ACE2 has considerably enhanced, resulting in a considerable increase in its replication and transmission (Korber et al., 2020; Thomson et al., 2021). In the present study, we have investigated the binding interactions of native and three major mutant spike proteins-L452R, T478K and Y501Y to human ACE2 and provided plausible mechanisms of variations in the binding affinity using protein–protein docking and molecular dynamics simulation approaches. The HADDOCK docking scores and HawkDock MM/GBSA binding energy analysis show that the mutant spike proteins have a higher binding affinity to ACE2. Interface statistics show that the increase in the binding affinity of the mutant spike to ACE2 is correlated with the increase in the number of interface residues, interface area, hydrogen bonds, salt bridges and non-bonded contacts. The hotspot residues which cause a substantial increase in the binding free energy of at least 2.0 kcal/mol when mutated to alanine mutation (Thorn and Bogan, 2001) were computed for the native as well as mutant spike:ACE2 complexes. Molecular dynamics simulations studies reveal compelling changes in the geometric parameters such as RMSD, Rg, total SASA and number of hydrogen bonds. These data provide useful insights into the molecular aspects of the enhanced binding affinity of spike mutants to ACE2 receptor and may help in designing therapeutics targeting the interface region. While the functional impact of a single mutation alone on the spike protein has been investigated through our studies, it would be fascinating to explore the cumulative effects of all such mutations on the binding interactions of spike protein to ACE2 receptor. Further, deciphering the structure of mutant spike protein in complex with ACE2 through experimental techniques such as X-ray crystallography will provide detailed mechanistic insights into their binding interactions.
5 Conclusion
The emerging SARS-CoV-2 variants have posed a threat to the world’s healthcare systems due to their high transmissibility, virulence, infectivity, and their ability to neutralize the serum antibodies. This rapid infectivity of the virus can be attributed to the large accumulating mutations in the spike protein, a glycoprotein that helps the virus gain entry into the human cells via binding to cell surface ACE2 receptor. Using computational simulation approaches, we presented the probable mechanisms of increase in the binding affinity of spike mutants (L452R, T478K, and N501Y) to human ACE2 receptor. Our studies will help in developing novel therapeutics against emerging SARS-CoV-2 variants by blocking the interface region of ACE2:Spike complex.
Acknowledgments
The authors would like to extend their sincere appreciation to the Researchers Supporting Project number (RSP-2021/306), King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Escape of SARS-CoV-2 501Y. V2 from neutralization by convalescent plasma. Nature. 2021;593:142-146.
- [Google Scholar]
- Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med.. 2021;27:717-726.
- [Google Scholar]
- The Spike D614G mutation increases SARS-CoV-2 infection of multiple human cell types. Elife. 2021;10:e65365
- [Google Scholar]
- Human SARS CoV-2 spike protein mutations. Proteins Struct. Funct. Bioinforma.. 2021;89:569-576.
- [Google Scholar]
- GRGMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput.. 2008;4:435-447.
- [CrossRef] [Google Scholar]
- Jia, Z., Gong, W., 2021. Will Mutations in the Spike Protein of SARS-CoV-2 Lead to the Failure of COVID-19 Vaccines? J. Korean Med. Sci. 36.
- SARS-CoV-2, the pandemic coronavirus: Molecular and structural insights. J. Basic Microbiol.. 2021;61:180-202.
- [Google Scholar]
- Emerging SARS-CoV-2 variants of concern and potential intervention approaches. Crit. Care. 2021;25:1-8.
- [Google Scholar]
- Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812-827.
- [Google Scholar]
- Kruger D.M., Gohlke, H., 2010. DrugScorePPI webserver: fast and accurate in silico alanine scanning for scoring protein--protein interactions. Nucleic Acids Res. 38, W480--W486.
- Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215-220.
- [Google Scholar]
- Li, X., Zhang, L., Chen, S., Ji, W., Li, C., Ren, L., 2021. Recent progress on the mutations of SARS-CoV-2 spike protein and suggestions for prevention and controlling of the pandemic. Infect. Genet. Evol. 104971.
- Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565-574.
- [Google Scholar]
- Protein structure analysis of the interactions between SARS-CoV-2 spike protein and the human ACE2 receptor: from conformational changes to novel neutralizing antibodies. Cell. Mol. Life Sci.. 2021;78:1501-1522.
- [Google Scholar]
- SARS-CoV-2 mutations, vaccines, and immunity: implication of variants of concern. Signal Transduct. Target. Ther.. 2021;6:1-2.
- [Google Scholar]
- Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun.. 2020;11:1-12.
- [Google Scholar]
- UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem.. 2004;25:1605-1612.
- [CrossRef] [Google Scholar]
- Structural proteins in severe acute respiratory syndrome coronavirus-2. Arch. Med. Res.. 2020;51:482-491.
- [Google Scholar]
- Souza, P.F.N., Mesquita, F.P., Amaral, J.L., Landim, P.G.C., Lima, K.R.P., Costa, M.B., Farias, I.R., Lima, L.B., Montenegro, R.C., 2021. The human pandemic coronaviruses on the show: The spike glycoprotein as the main actor in the coronaviruses play. Int. J. Biol. Macromol.
- COVID-19: epidemiology, evolution, and cross-disciplinary perspectives. Trends Mol. Med.. 2020;26:483-495.
- [Google Scholar]
- Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell. 2021;184:1171-1187.
- [Google Scholar]
- ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics. 2001;17:284-285.
- [Google Scholar]
- The HADDOCK2. 2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol.. 2016;428:720-725.
- [Google Scholar]
- Wang, M.-Y., Zhao, R., Gao, L.-J., Gao, X.-F., Wang, D.-P., Cao, J.-M., 2020. SARS-CoV-2: structure, biology, and structure-based therapeutics development. Front. Cell. Infect. Microbiol. 10.
- Increased resistance of SARS-CoV-2 variant P. 1 to antibody neutralization. Cell Host Microbe. 2021;29:747-751.
- [Google Scholar]
- HawkDock: a web server to predict and analyze the protein–protein complex based on computational docking and MM/GBSA. Nucleic Acids Res.. 2019;47:W322-W330.
- [Google Scholar]
- A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265-269.
- [Google Scholar]
- Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell Mol Immunol. 2020;17:621-630.
- [Google Scholar]
- Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med.. 2020;46:586-590.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2021.101773.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




