

Translate this page into:
Repurposing benzimidazole and benzothiazole derivatives as potential inhibitors of SARS-CoV-2: DFT, QSAR, molecular docking, molecular dynamics simulation, and in-silico pharmacokinetic and toxicity studies
⁎Corresponding authors. mhashim@ksu.edu.sa (Mohammad Azam), veronique.seidel@strath.ac.uk (Veronique Seidel), mohnadabdalla200@gmail.com (Mohnad Abdalla)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Density Functional Theory (DFT) and Quantitative Structure-Activity Relationship (QSAR) studies were performed on four benzimidazoles (compounds 1–4) and two benzothiazoles (compounds 5 and 6), previously synthesized by our group. The compounds were also investigated for their binding affinity and interactions with the SARS-CoV-2 Mpro (PDB ID: 6LU7) and the human angiotensin-converting enzyme 2 (ACE2) receptor (PDB ID: 6 M18) using a molecular docking approach. Compounds 1, 2, and 3 were found to bind with equal affinity to both targets. Compound 1 showed the highest predictive docking scores, and was further subjected to molecular dynamics (MD) simulation to explain protein stability, ligand properties, and protein–ligand interactions. All compounds were assessed for their structural, physico-chemical, pharmacokinetic, and toxicological properties. Our results suggest that the investigated compounds are potential new drug leads to target SARS-CoV-2.
Keywords
DFT
QSAR
SARS-CoV-2 Mpro
ACE2
Molecular docking
MD simulation
Pharmacokinetic study

1 Introduction
The current COVID-19 pandemic, caused by the highly contagious newly emerged SARS-CoV-2, is a global health threat that affects millions of people worldwide, and impacts negatively on all sectors of society (Mohapatra et al., 2020a; b; Mohapatra and Rahman, 2021). Although the fatality rate for SARS-CoV-2 (∼4%) is lower compared to that of SARS-CoV (∼10%) and MERS-CoV (∼35%), its transmission rate is much higher than that of previously reported coronaviruses (CoVs) (Guo et al., 2020). In many countries, this led to lockdown measures being put into place to minimize human-to-human transmission (Mohapatra et al., 2021b). COVID-19 related deaths have now surpassed those from other epidemics caused by pathogens such as influenza A & B, Zika, Ebola and other CoVs (Mahal et al., 2021) and declared global pandemic (Dhama et al., 2020a).
The SARS-CoV-2 virus is mainly transmitted through respiratory aerosols/droplets, and to some extent via the fecal-oral route (Chan et al., 2020; Sah et al., 2021). Other means of transmission include direct and indirect contacts with contaminated objects/surfaces/fomites (Mohapatra et al., 2021a; Morawska and Cao, 2020). The SARS-CoV-2 virus primarily targets the upper and lower respiratory tract, and most people infected will experience only mild or moderate symptoms. However, in some individuals such as the elderly and/or those with pre-existing comorbidities (e.g. obesity, diabetes, cardiovascular disorders), SARS-CoV-2 infection can lead to severe complications, with multiple organ damage (Dhama et al., 2020b; Arumugam et al., 2020).
Scientists throughout the world continue to endeavor to discover and develop suitable and effective approaches to tackle this pandemic. This includes the development of vaccines such as the Covishield, Covaxin (India), AstraZeneca (UK), Moderna, Novavax (USA), Pfizer/BioNTech (Germany), Sputnik V (Russia), CanSino, Sinovac and BBIBP-CorV (China) vaccines (Madkaikar et al., 2021; Bagcchi, 2021). Vaccination programs are underway in many countries; even if clinical efficacy against the new SARS-CoV-2 variants has yet to be established for all vaccines (Callaway, 2021). Efforts of the scientific community have also been focused on the discovery of new antiviral drug leads. Several reports have highlighted the importance of targeting the Mpro of SARS-CoV-2 - an enzyme with a pivotal role in mediating viral replication and transcription - and have used molecular docking as a promising strategy to design and develop possible Mpro inhibitors (Ancy et al., 2020; Baildya et al., 2020; Mohapatra et al., 2021c). Another aim has been to target virus entry mechanisms via disruption of the interaction of SARS-CoV-2 with the human ACE2 receptor responsible for viral attachment to the host cells (Rezaei et al., 2021; Joshi et al., 2020).
In this work, we used a comprehensive in-silico approach, including molecular docking and MD simulation studies, to investigate the predictive binding affinity of four benzimidazoles and two benzothiazoles previously synthesized by our group (Mohapatra et al., 2011; 2018) towards SARS-CoV-2 Mpro and the human ACE2 receptor. We also report on the pharmacokinetic, drug-likeness, and toxicological properties of the studied compounds.
2 Density functional theory (DFT) studies
DFT is a computational modeling study used to investigate the electronic structure of molecules. The DFT analysis was carried out through GAUSSIAN inter phase. All compounds were first optimized using the Gauss View 6.0.16 program (Gauss View 6.0, 2019), and then subjected to DFT calculation using the GAUSSIAN 09 suite programs (Frisch et al., 2009). The computation was performed using the B3LYP method and exchange–correlation functional with a 6–31 + G (d, p) basis set for carbon, nitrogen, oxygen, sulfur, and hydrogen atoms. Relevant energetic properties such as the dipole moment (D) and the single point energy were calculated for each compound. Comparison of the calculated energy values revealed that compound 4 had greater single point energy compared with the other compounds (El‐ajaily et al., 2019; Sarangi et al., 2018; 2020; Mahapatra et al., 2013). Compound 2 had a lower energy value which indicated larger stability, while compound 3 possessed a greater D value compared to other compounds (Table S1).
2.1 Frontier molecular orbitals (FMO) studies
FMO studies can predict the chemical reactivity of compounds and identify their most likely reactive sites. The calculated energies EHOMO and ELUMO of compounds help to explain global reactivity descriptors such as chemical hardness, chemical potential, and electrophilicity. The stability of the investigated compounds was confirmed from the negative values obtained for their EHOMO and ELUMO (Yousef et al., 2013). The electron cloud of compound 1 was localized on the phenolic (–OH) group and azomethine nitrogen (C = N) group relating to the participating atomic sites involved in nucleophilic attack. The band energy gap correlates with the chemical reactivity and chemical stability of molecules. The energy difference [EHOMO - ELUMO] for compound 3 was found to be less than the band energy gap observed for other compounds, establishing its greater reactivity. Compound 1, on the other hand, had a greater stability. The band energy values and EHOMO - ELUMO energy variation suggest charge transfer (CT) interaction. In this work, the chemical reactivity parameters such as electronegativity (χ), chemical potential (μ), global electrophilicity index (ω), global hardness (η) and global softness (S) were calculated (Table S2). We observed that the chemical softness (S) of compound 4 was lesser than the other compounds, indicating a higher stability for compound 4. Another important parameter is the electrophilicity (
) of compounds, which assesses its ability to accept electron(s) from its surrounding. Compound 1 showed less electrophilicity than other compounds (Table S2) (Fig. 1).
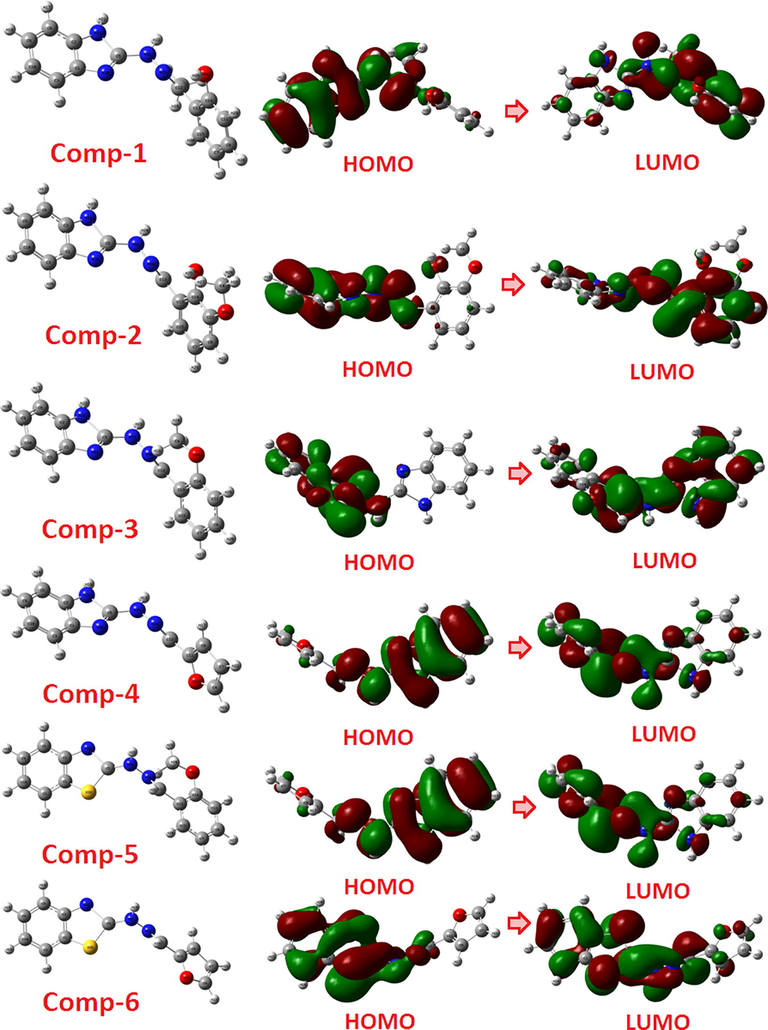
Optimized structures of compounds 1–6 using FMOs.
3 QSAR studies
QSAR modelling was used to characterize the physico-chemical properties of the investigated compounds and predict their correlations to biological activity. QSAR studies were performed using the Hyper Chem Professional 8.0.3 program. First, the structure of each compound was optimized with a semi-empirical PM3 method using a (MM+) force field, and then energy minimization was achieved using a Fletcher-Reeves conjugate gradient algorithm. A range of QSAR parameters including free energy, hydration energy, total energy, refractivity, surface area, volume, polarizability, mass, dipole moment, partition coefficient (log P), and RMS gradient were calculated. Interestingly, the log P value for compound 5 was the highest amongst all compounds, indicating the ability for this compound to efficiently permeate biological membranes (Mohapatra et al., 2021c) (Table 1).
Parameters
Comp. 1
Comp. 2
Comp. 3
Comp. 4
Comp. 5
Comp. 6
Surface area (Approx) (Å2)
386.37
422.22
373.98
354.00
392.22
373.72
Surface area (Grid) (Å2)
513.04
535.63
499.16
460.11
515.24
475.24
Volume (Å3)
820.32
859.58
796.25
710.95
820.61
735.38
Hydration energy (kcal/mol)
−13.74
−15.87
−11.85
−15.16
−10.44
−13.79
Log P
3.14
2.35
2.53
1.79
3.19
2.45
Refractivity (Å3)
21.76
23.28
21.90
15.25
26.39
19.74
Polarizability (Å3)
30.03
30.67
30.03
24.72
31.68
26.37
Mass (amu)
266.30
282.30
266.30
226.24
883.35
443.28
Total energy (kcal/mol)
135.406
23.8135
426.256
141.680
472.512
187.82
Dipole moment (Debye)
0.9822
0.3717
1.793
1.507
1.04
0.76
Free energy (kcal/mol)
135.406
23.8135
426.256
141.680
−91108
187.82
RMS gradient (kcal/Å mol)
57.39
0.09833
92.21
66.78
111.6
96.3
4 Molecular docking study
The docking analysis was executed using a receptor-oriented flexible docking protocol with the Autodock 4.2.6 software package and Python 3.8.2. The ligands were generated with MGL Tools 1.5.6 and optimized using the Avogadro program. The 3D X-ray structure for SARS-CoV-2 Mpro and the ACE2 receptor was retrieved from the Protein Data Bank ( http://www.rcsb.org/pdb) and used for the molecular docking (Adem et al., 2020; Shivanika et al., 2020; Khan et al., 2020). The active site of each enzyme was fixed with Meta Pocket 2.0 and the docking cavities were selected for the target proteins. All ligand interactions with the protein amino acid residues were visualized using the Discovery Studio 3.5 software.
The results of docking analyses were expressed in terms of scoring function (free binding energy, ΔG) for the best conformational positions of each ligand for comparative analysis. The binding affinity of the investigated compounds against SARS-CoV-2 Mpro and ACE2 is related to the favorable geometric arrangement of these ligands in the active site, hydrogen bond formation, donor–acceptor interactions and intermolecular electrostatic interactions which ultimately leads to the formation of stable complexes. The thermodynamic probability of the binding was established for each compound by the negative binding energy (ΔG) values. We observed that compounds (1,2,3) showed best results against SARS-CoV-2 Mpro and compounds (1,2,3,5) showed best results against the human ACE2 receptor (Tables S3 & S4).
The active site of Mpro (PDBID: 6LU7) is occupied by charged, polar and hydrophobic amino acid residues. A detailed description of the ligand–protein interactions within the active site of Mpro is presented in Table S3. Compound-1 formed H-bonds with Thr199 (OG1), Leu287 (O), and ionic interactions with Asp289 (OD1, OD2) (Fig. 2). Interactions with Thr199, Leu287 and Asp289 were observed for all compounds. When docked with the human ACE2 receptor (PDB ID: 6 M18), compound-1 showed ionic interactions with Asp494 (OD1), H-bonding with His493, and pi-pi interaction with Arg671 (Table S4). Interactions with His493, Asp494 and Arg671 were observed for almost all compounds.Fig. 3.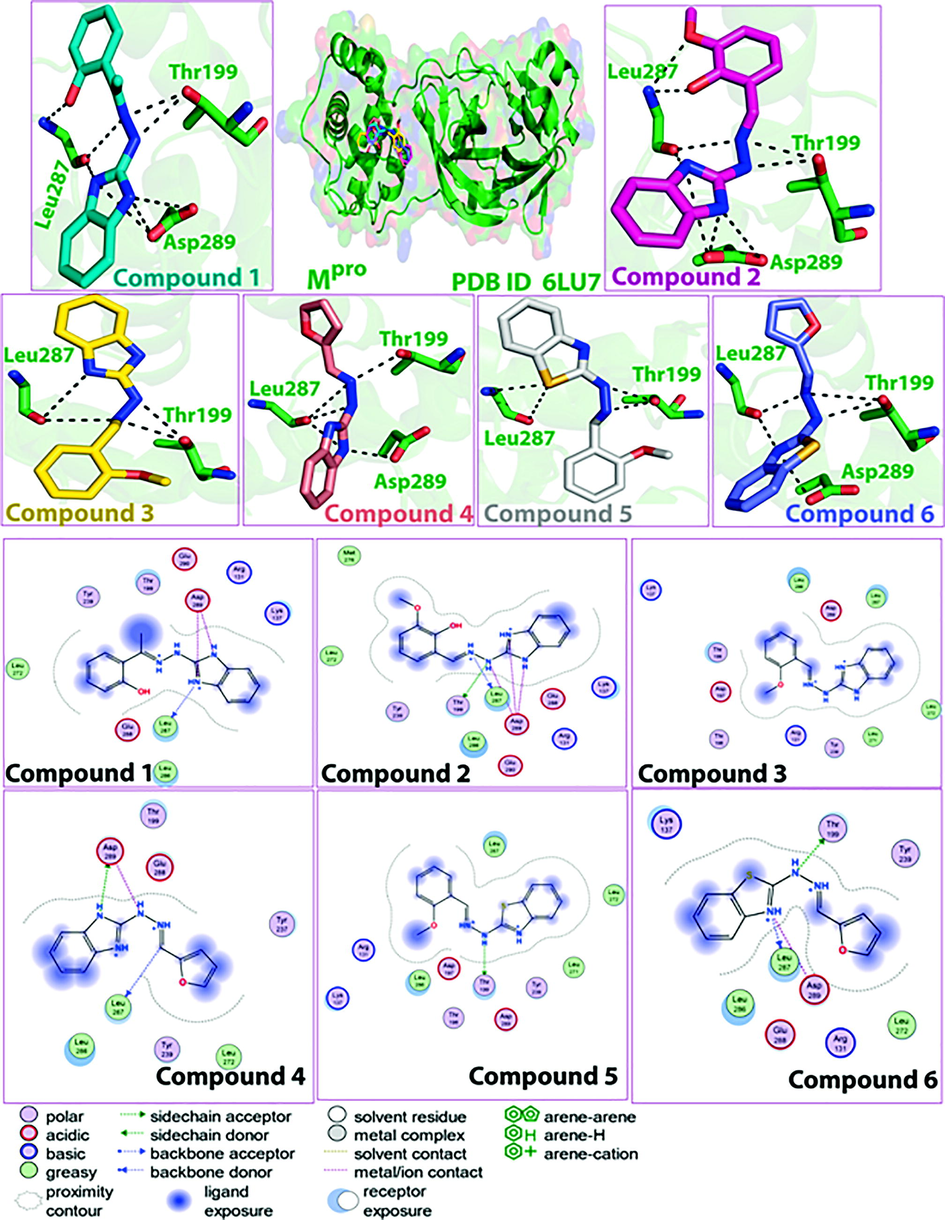
Docked pose of compounds (1–6) in the SARS-CoV-2 Mpro binding site, and corresponding 2D plots of molecular interactions.
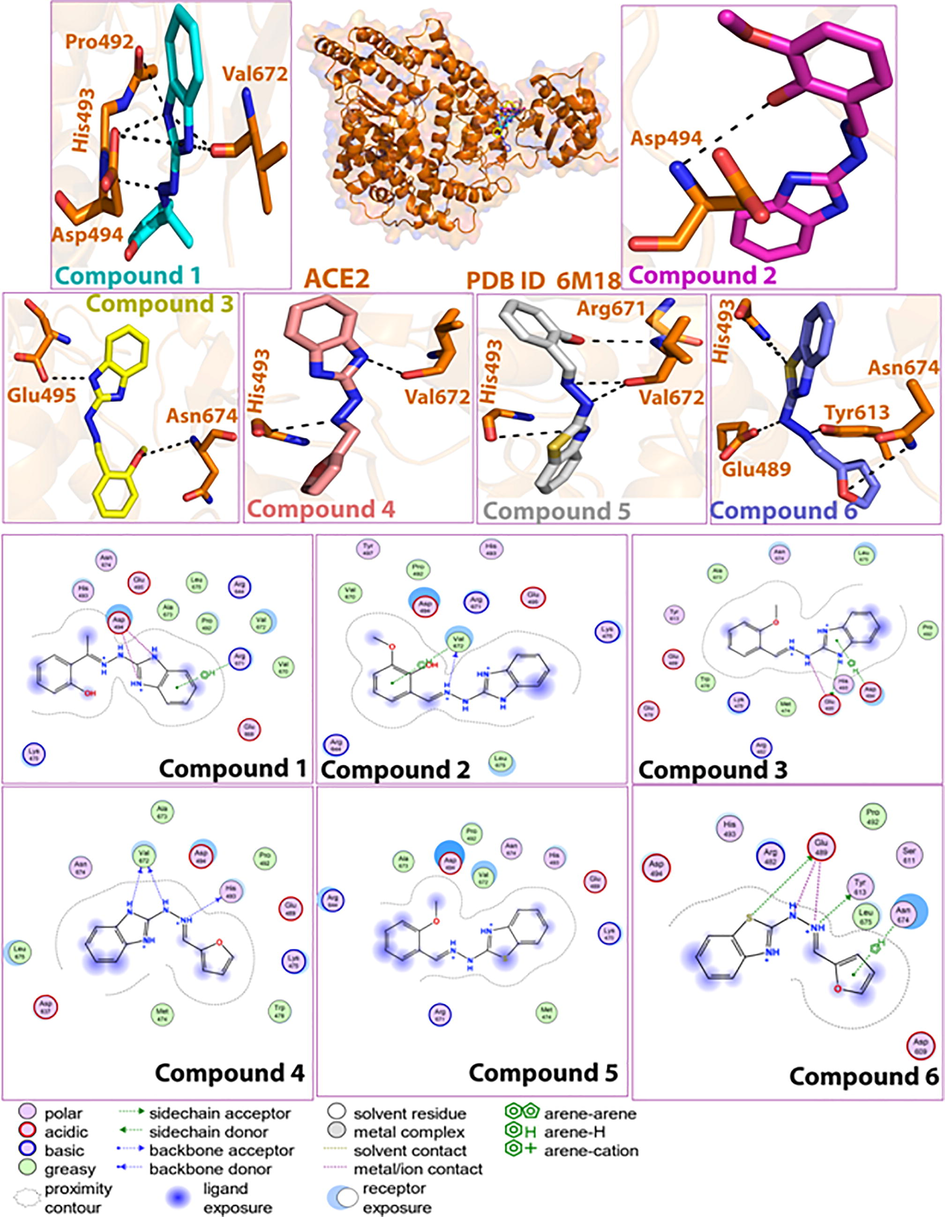
Docked pose of compounds (1–6) in the ACE2 binding site, and corresponding 2D plots of molecular interactions.
5 Molecular dynamics simulation
5.1 Protein RMSD
MD simulations were performed to study the physical movements and dynamic evolution of the (Comp.1-6LU7) and (Comp.1–6 M18) complexes (Adem et al., 2020; Shivanika et al., 2020). The MD simulation was run using the Schrödinger’s Desmond package following a previously reported procedure (Pant et al., 2020). Ligand-protein complexes were simulated for 100 ns to analyze the RMSD values, which estimate the complex stability (with lower RMSD values indicating greater stability). The protein RMSD trajectory for SARS-CoV-2 Mpro and ACE2 fluctuated up to 5 and 15 ns, respectively, before gradually reaching an equilibrium. The (Comp.1-6LU7) complex completed the 100 ns simulation without much fluctuation, suggesting a greater stability. The RMSD average value of Mpro (6LU7) and ACE2 (6 M18) at equilibrium was found to be 1.8 and 9.5 Å, respectively (Fig. 4). The deviation within 1–3 Å is acceptable for small globular protein. Based on these results, the (Comp.1-6LU7) and (Comp.1–6 M18) complexes were further analyzed for protein–ligand contacts.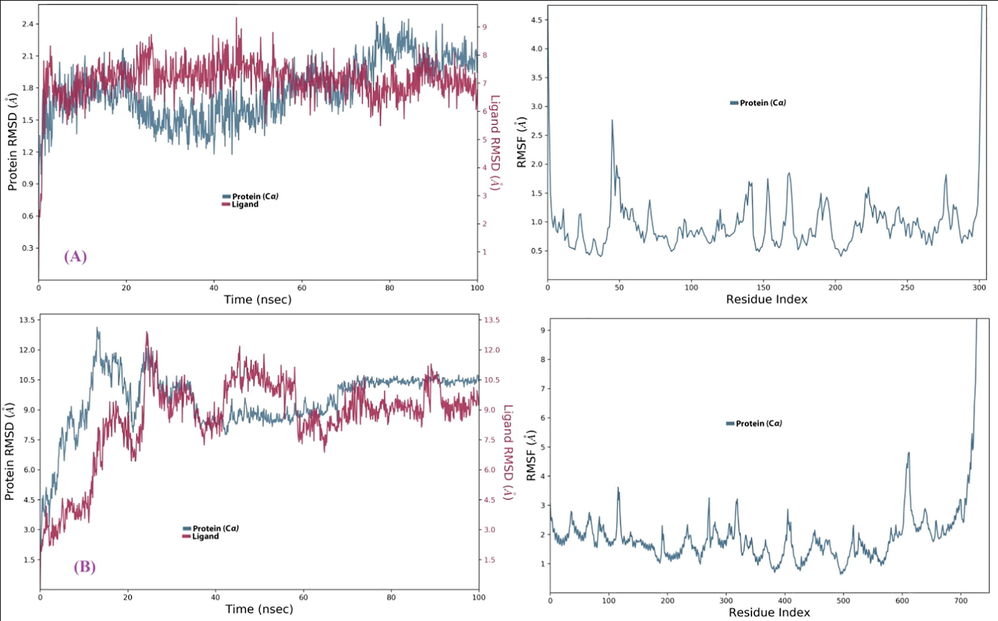
Protein RMSD trajectory of (A) (Comp.1-6LU7) and (B) (Comp.1-6 M18) complex.
5.2 Ligand properties
The ligand properties were evaluated by calculating Ligand RMSD, Molecular Surface Area (MolSA), Radius of Gyration (rGyr), Polar Surface Area (PSA) and Solvent Accessible Surface Area (SASA) values. The root-mean-square deviation (RMSD) values fluctuated until 5 and 10 ns for Comp.1-6LU7 and Comp.1–6 M18, respectively, before gradually reaching an equilibrium. The RMSD values were around 0.8 to 2.1 Å (equilibrium ∼ 1.7 Å), and 1.1 to 1.9 Å (equilibrium ∼ 1.3 Å) for Comp.1-6LU7 and Comp.1–6 M18, respectively. The rGyr values fluctuated slightly until 5 and 10 ns for Comp.1-6LU7 and Comp.1–6 M18, respectively, before gradually reaching equilibrium. The rGyr values were around 3.57 to 4.10 Å (equilibrium ∼ 3.90 Å), and 3.53 to 4.35 Å (equilibrium ∼ 3.87 Å) for Comp.1-6LU7 and Comp.1–6 M18, respectively. The MolSA values (equivalent to van der Waals surface area) were constant throughout the simulation. These were around 290 to 300 Å2 (equilibrium ∼ 295 Å2), and around 285 to 295 Å2 (equilibrium ∼ 293 Å2) for Comp.1-6LU7 and Comp.1–6 M18, respectively. The SASA values fluctuated slightly until 5 and 25 ns for Comp.1-6LU7 and Comp.1–6 M18, respectively, before gradually reaching an equilibrium. These were around 160 to 320 Å2 (equilibrium ∼ 210 Å2), and around 60 to 240 Å2 (equilibrium ∼ 80 Å2) for Comp.1-6LU7 and Comp.1–6 M18, respectively. The PSA values were constant throughout the simulation. These were around 84 to 102 Å2 (equilibrium ∼ 96 Å2), and around 88 to 112 Å2 (equilibrium ∼ 96 Å2) for Comp.1-6LU7 and Comp.1–6 M18, respectively (Figs. 5 & 6). In both cases, the ligand showed minimal fluctuation in the initial part of the simulation period, then gradually reached equilibrium which remained constant throughout the simulation, suggesting they possessed good stability in the active site of both proteins.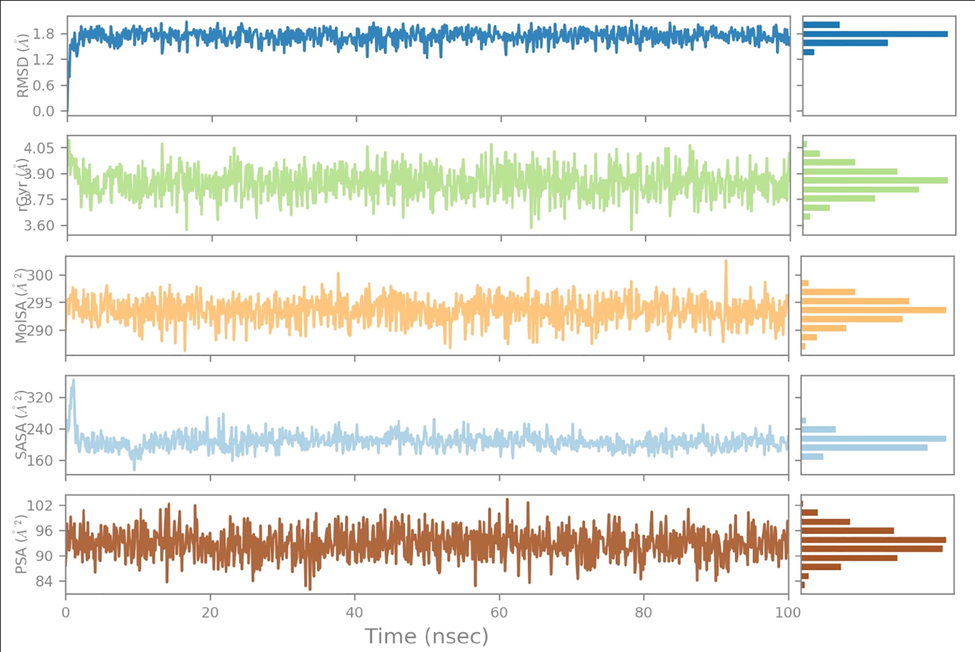
Ligand property trajectory for the (Comp.1-6LU7) complex.
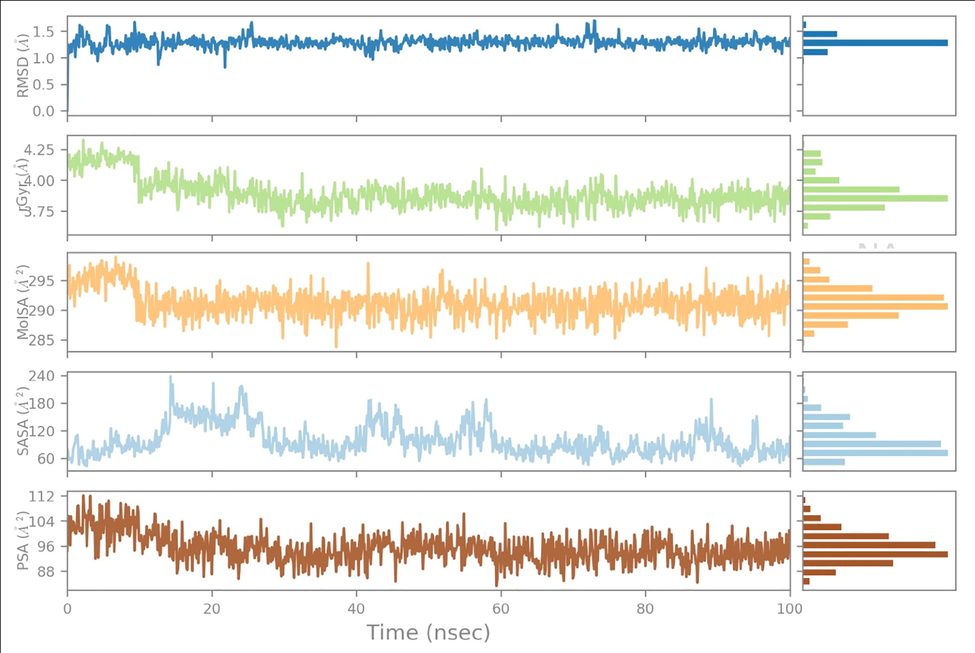
Ligand property trajectory for the (Comp.1–6 M18) complex.
5.3 Protein-ligand contacts study
The protein–ligand (P-L) contacts of the (Comp.1-6LU7) and (Comp.1–6 M18) complexes were studied using contact histograms (Dariya and Nagaraju, 2020). The P-L interactions were classified into four types such as hydrophobic, ionic, water bridges, and hydrogen bonds. The latter are commonly grouped into four sub-types such as side-chain acceptor, side-chain donor, backbone donor and backbone acceptor. They play a crucial role in ligand–protein binding and are essential to take into account when it comes to the design of new drugs. The histograms identified Tyr-239, Leu-271, Met-276, Asn-277, Leu-287, and Asp-471, Lys-475, Glu-489, Pro-492, His-493, Asp-494, Asp-637, Val-672, Asn-674, Leu-675 as the H-bond interacting amino acid residues for the (Comp.1-6LU7) and (Comp.1–6 M18) complexes, respectively. The hydrophobic contacts, involving a hydrophobic amino acid and an aromatic or aliphatic group on the ligand, were characterized as pi-cation, pi-pi and other non-specific interactions. The amino acids that showed hydrophobic interactions with the ligand included Tyr-237, Leu-272, Met-276, Leu-286, Leu-287 for 6LU7, and Trp-478, Pro-492, Met-640, Val-672, Leu-675 for 6 M18. We observed that Asp-494 showed minimal ionic interactions with the ligand for 6 M18. Most of the interacting amino acids (Tyr-239, Met-276, Asn-277, Ala-285, Leu-287 for 6LU7 and Asp-494, Leu-675, Lys-475, Val-672, Asn-674 for 6 M18) showed water bridges with the ligand (Figs. 7 & 8). An overview of all protein–ligand interactions is summarized in Fig. 7(c) & 8(c) - with the top panel showing the total number of specific protein–ligand contacts formed, and the bottom panel showing the nature of the interacting amino acid residues over the course of the trajectory. The residues that made a number of specific contact with the ligand in a particular trajectory frame are represented by a darker shade of orange. The number of P-L contacts varied between 0 and 6 for 6LU7 and between 0 and 12 for 6 M18. The receptor-ligand complex showed darker bands for the Leu-287, Met-276, Tyr-239 row for Comp.1-6LU7 and the Leu-675 and Asp-494 row for Comp.1–6 M18, suggesting that these amino acids have more interactions with the ligand in all possible orientations, in agreement with the histogram results.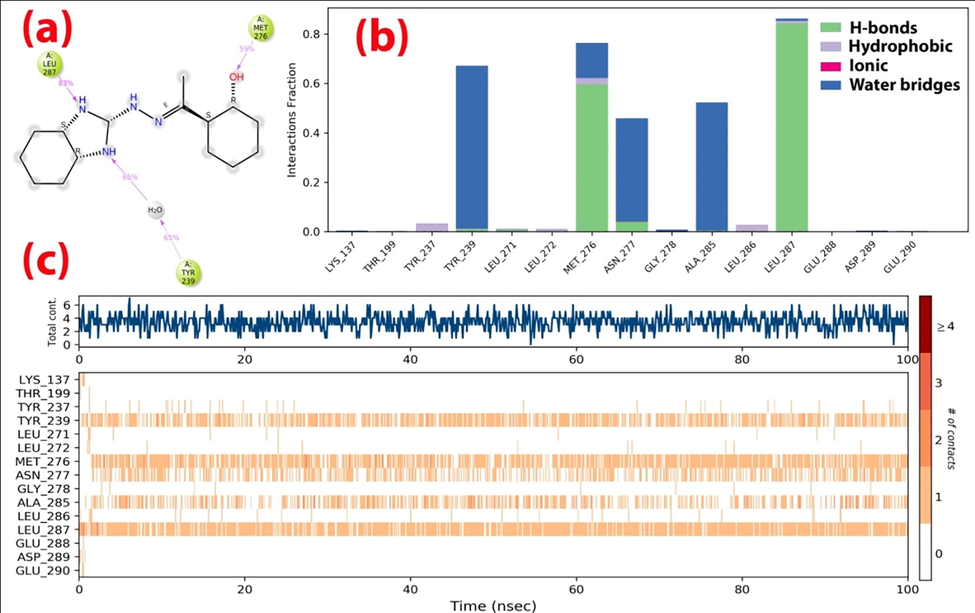
Contact plots and ligand–protein interaction residues for the (Comp.1-6LU7) complex.
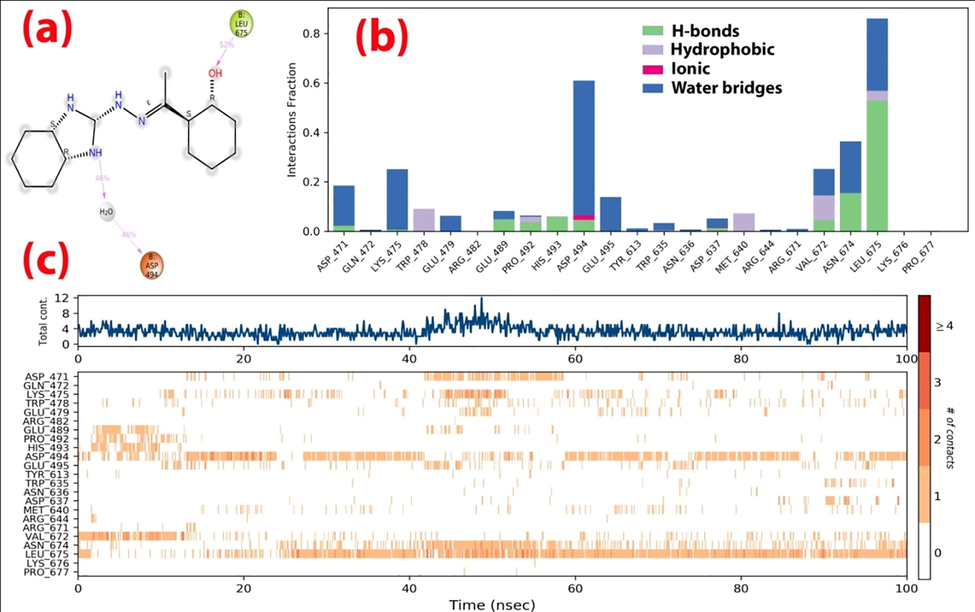
Contact plots and ligand–protein interaction residues for the (Comp.1-6 M18) complex.
6 Pharmacokinetics, drug-likeness, and toxicological properties
Knowledge of the pharmacokinetic and drug-likeness properties of newly designed ligands for specific targets is essential to predict the absorption, distribution, metabolism, and excretion of potential new drug leads (Shivanika et al., 2020; Savale et al., 2021). In this study, pharmacokinetic properties predictions for the investigated compounds (Fig. 9) were carried out using the admetSAR (versions 1 and 2) and SwissADME software. Their Lipinski’s properties were determined using physico-chemical data retrieved from the PubChem database. We observed that all compounds obeyed the Lipinski’s rule of five, with molecular weights < 500 g/mol, number of hydrogen bond donors and acceptors < 5, logP value < 5 and molar refractivity < 140 (Lipinski, 2000, 2004). The Topological Polar Surface Area (TPSA) of all compounds was < 90 Å2, indicating their ability to permeate biological membranes (Shivanika et al., 2020; Dariya and Nagaraju, 2020). Moreover, all compounds showed good solubility and high gastrointestinal absorption. Their low number of rotatable bonds (3 or 4) suggested a good flexibility. The synthetic accessibility value of all compounds was ≤ 3, which indicated the feasibility of synthesis (Tables S5-S11). The toxicity of the investigated compounds was also evaluated using the pkCSM software tool (http://structure.bioc.cam.ac.uk/pkcsm) (Table S10). We observed that the compounds were neither hepatotoxic nor carcinogenic, and that compound 4 and 6 showed better toxicity results.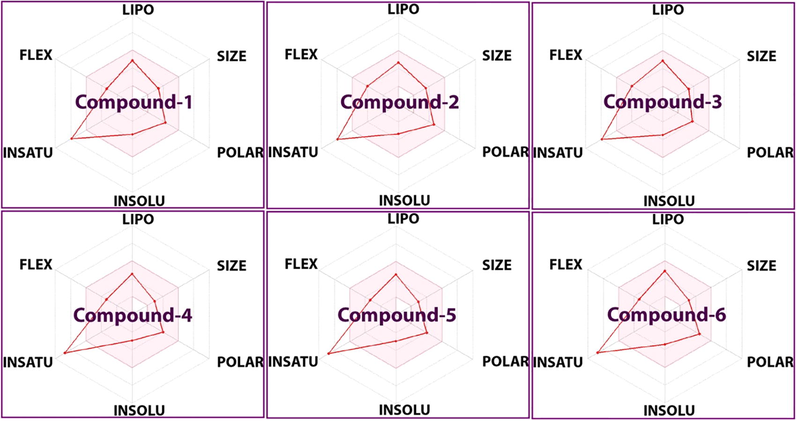
Pharmacokinetic and drug-likeness properties of the investigated compounds (LIPO: lipophilicity, INSOLU: insolubility, INSATU: insaturation, and FLEX: flexibility).
7 Conclusion
Four benzimidazole and two benzothiazole derivatives were investigated for their binding affinity for Mpro and ACE2 receptor using a molecular docking approach. Compounds 1, 2, and 3 were found to bind with equal affinity to both targets, which may be more effective in clinical practice. One of the benzimidazoles (compound 1) was further subjected to molecular dynamics simulation to explain protein–ligand complex stability, ligand properties, and protein–ligand contacts. We established that all compounds obeyed the Lipinski’s rule of five and showed good solubility, high gastrointestinal absorption, good ability to permeate biological membranes, good synthetic accessibility, and low toxicity. Taken altogether, these data suggest that the investigated compounds have the potential to combat SARS-CoV-2. We hope that this work will provide scope for further studies to establish the potential of benzimidazole and benzothiazole derivatives as new Mpro and/or ACE2 receptor inhibitors.
Acknowledgement
The authors acknowledge the financial support through Researchers Supporting Project number (RSP-2021/147), King Saud University, Riyadh, Saudi Arabia. This work was supported by Shandong University postdoctoral fellowship to Mohnad Abdalla. All authors are thankful to their respective Institutions for the support they received.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Caffeic acid derivatives (CAFDs) as inhibitors of SARS-CoV-2: CAFDs-based functional foods as a potential alternative approach to combat COVID-19. Phytomedicine. 2020;85:153310.
- [CrossRef] [Google Scholar]
- Possibility of HIV-1 protease inhibitors-clinical trial drugs as repurposed drugs for SARS-CoV-2 main protease: a molecular docking, molecular dynamics and binding free energy simulation study. J. Biomol. Struct. Dyn.. 2020;39(15):5368-5375.
- [CrossRef] [Google Scholar]
- COVID-19 and the World with Co-Morbidities of Heart Disease, Hypertension and Diabetes. J Pure Appl Microbiol.. 2020;14(3):1623-1638.
- [Google Scholar]
- The world’s largest COVID-19 vaccination campaign. Lancet Infect. Dis.. 2021;21(3):323.
- [CrossRef] [Google Scholar]
- Inhibitory activity of hydroxychloroquine on COVID-19 main protease: an insight from MD simulation studies. J. Mol. Struct.. 2020;1219:128595.
- [CrossRef] [Google Scholar]
- Could new COVID variants undermine vaccines? Labs scramble to find out. Nature. 2021;589(7841):177-178.
- [Google Scholar]
- A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395(10223):514-523.
- [Google Scholar]
- Understanding novel COVID-19: Its impact on organ failure and risk assessment for diabetic and cancerpatients. Cytokine Growth Factor Rev.. 2020;53:43-52.
- [Google Scholar]
- An update on SARS-CoV-2/COVID-19 with particular reference to its clinical pathology, pathogenesis, immunopathology and mitigation strategies. Travel Med. Infect. Dis.. 2020;37:101755.
- [CrossRef] [Google Scholar]
- Transition Metal Complexes of (E)-2((2-hydroxybenzylidene) amino-3-mercaptopropanoic acid: XRD, Anticancer, Molecular modeling and Molecular Docking Studies. ChemistrySelect.. 2019;4(34):9999-10005.
- [Google Scholar]
- GAUSIAN 09. Wallingford CT: Gaussian Inc; 2009.
- GaussView 6.0, (Gaussian Inc., Wallingford, CT, USA) 2019.
- The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak– an update on the status. Mil. Med. Res.. 2020;7:11.
- [Google Scholar]
- In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking. Eur. Rev. Med. Pharmacol. Sci.. 2020;24(8):4529-4536.
- [Google Scholar]
- Antifungal screening and molecular docking simulation of silica supported synthesized sitosteryl hydrogen phthalate using microwave irradiation. Hem. Ind.. 2020;74(6):377-388.
- [Google Scholar]
- Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000;44(1):235-249.
- [Google Scholar]
- Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol.. 2004;1(4):337-341.
- [Google Scholar]
- Recent progress in chemical approaches for thedevelopment of novel neuraminidase inhibitors. RSC Adv.. 2021;11(3):1804-1840.
- [Google Scholar]
- Synthesis, characterisation, XRD, molecular modelling and potential antibacterial studies of Co(II), Ni(II), Cu(II), Zn(II), Cd(II) and Hg(II) complexes with bidentate azodye ligand. J. Saudi Chem. Soc.. 2013;20(6):635-643.
- [Google Scholar]
- Synthesis, spectral, thermal, kinetic and antibacterial studiesof transition metal complexes with benzimidazolyl-2-hydrazones of o-hydroxyacetophenone, o-hydroxybenzophenone and o-vanillin. Bull. Chem. Soc. Ethiop.. 2018;32(3):437-450.
- [Google Scholar]
- Challenges in controlling COVID-19 in migrants in Odisha, India. Diabet. Metab. Syndrome: Clin. Res. Rev.. 2020;14(6):1593-1594.
- [Google Scholar]
- Infection capability of SARS-CoV-2 on different surfaces. Egypt. J. Basic Appl. Sci.. 2021;8(1):75-80.
- [Google Scholar]
- Mohapatra, R.K., Mishra, S., Azam, M., Dhama, K., 2021b. COVID-19, WHO guidelines, pedagogy, and respite. Open Med. 16, 491–493.
- Synthesis and characterization of transition metal complexes with benzimidazolyl-2-hydrazones of o-anisaldehyde and furfural. J. Korean Chem. Soc.. 2011;55(6):926-931.
- [Google Scholar]
- Computational investigations of three main drugs and their comparison with synthesized compounds as potent inhibitors of SARS-CoV-2 main protease (Mpro): DFT, QSAR, molecular docking, and in silico toxicity analysis, Journal of King Saud University –. Science. 2021;33(2):101315.
- [CrossRef] [Google Scholar]
- The recent challenges of highly contagious COVID-19; causing respiratory infections: symptoms, diagnosis, transmission, possible vaccines, animal models and immunotherapy. Chem. Biol. Drug Des.. 2020;96(5):1187-1208.
- [Google Scholar]
- Is it possible to control the outbreak of COVID-19 in Dharavi, Asia’s largest slum situated in Mumbai? Anti-Infective Agents. 2021;19(4)
- [CrossRef] [Google Scholar]
- Airborne transmission of SARS-CoV-2: The world should face the reality. Environ. Int.. 2020;139:105730.
- [CrossRef] [Google Scholar]
- Peptide-like and small-molecule inhibitors against Covid-19. J. Biomol. Struct. Dyn.. 2020;39(8):2904-2913.
- [Google Scholar]
- ACE2: Its potential role and regulation in severe acute respiratory syndrome and COVID-19. J. Cell. Physiol.. 2021;236(4):2430-2442.
- [Google Scholar]
- The COVID-19 vaccination campaign in Nepal, emerging UK variant and futuristic vaccination strategies to combat the ongoing pandemic. Travel Med. Infect. Dis.. 2021;41:102037.
- [CrossRef] [Google Scholar]
- Synthesis and characterization of some binuclear metal complexes with a pentadentate azodye ligand: an experimental and theoretical study. Appl. Organometal. Chem.. 2020;34(8)
- [CrossRef] [Google Scholar]
- Synthesis and characterization of tetranuclear metal complexes with an octadentate azodye ligand. Chem. Afr.. 2018;1(1-2):17-28.
- [Google Scholar]
- Pharmacoinformatics approach based identification of potential Nsp15 endoribonuclease modulators for SARS-CoV-2 inhibition. Arch. Biochem. Biophys.. 2021;700:108771.
- [CrossRef] [Google Scholar]
- Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2020
- [CrossRef] [Google Scholar]
- Structural, spectral analysis and DNA studies of heterocyclic thiosemicarbazone ligand and its Cr(III), Fe(III), Co(II) Hg(II), and U(VI) complexes. J. Mol. Struct.. 2013;1045:145-159.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2021.101637.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




