

Translate this page into:
Non covalent interactions and molecular docking studies on morphine compound
⁎Corresponding authors. issaoui_noureddine@yahoo.fr (Noureddine Issaoui), omar@ksu.edu.sa (Omar Al-Dossary)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
The (5α,6α)-7,8-didehydro-4,5-epoxy-17-methylmorphinan-3,6-diol (morphine)molecule has been studied using the density functional theory and molecular docking methods and non covalent interactions. The conformational analysis of the molecule at the B3LYP/6−311++G** and HF/6−311++G** levels has been made. The comparison of the structural parameters computed using the B3LYP function with the experimental data has revealed their good agreement. The weak intermolecular interactions in the morphine structure have been analyzed using several techniques. The Hirshfeld surface study has been carried out to identify the diverse intermolecular interactions (mainly hydrogen bonds) and the … π stacking interactions. The analysis of the topological (AIM, ELF, LOL) and non covalent (RDG, IRI, DORI) interactions has revealed different categories of inter- and intramolecular contacts on the basis of the electron localization density and color scale indicator, respectively. The molecular docking study has been carried out to examine the possibility of biological application of the title conformer using the 1DLO (cancerous), 2BK3 (Parkinson), 3LN1 (inflammatory), 4HOE (microbial), and 5 K95 (schizophrenia) enzymes. The analysis has shown that the morphine structure can be used not only in analgesia, but also in the treatment of diseases. The investigated compound has shown good results with monoamine oxidase B (MOAB) at a score of −105.04 kcal/mol.
Keywords
Morphine
Hirschfield surface
Localized-orbital locator
Non-covalent interactions
Docking molecular

1 Introduction
Heterocyclic molecules, including carbon, and heterogeneous atoms (oxygen, nitrogen, and sulfur) has caught much interest of manufacturers owing to their agrochemicals and pharmaceutical properties (Lamberth and Dinges, 2012; Saleh et al., 2019). The modification of the heterocyclic conformation strongly affects the chemical reactivity of a complex (Dua et al., 2011). The (5α,6α)-7,8-didehydro-4,5-epoxy-17-methylmorphinan-3,6-diol alkaloid (morphine) with the chemical formula C17H19NO3is one of the most important aromatic molecules. It is commonly known as the most famous anesthetic, which represents an alkaloid molecule extracted from opium clinically used for pain relief (Wu et al., 2021). However, the excessive usage of morphine and other opioids can changes synaptic neuroplasticity, including the neuron density, as well as the postsynaptic sites and dendritic terminals (Beltran-Campos et al., 2015).
In this study, the structure of morphine was characterized using the topological assay, which included the theory of Atoms in Molecule (AIM), electron localization function (ELF), and localized-orbital locator (LOL), as well as the analysis of weak interactions and docking. The literature survey revealed a lack of quantum calculations, non covalent contacts examination, and pharmaceutical research for morphine. The molecular optimization at the B3LYP/6−311++G** and HF/6−311++G** levels for our conformation was made. The bond lengths and angles were determined by both methods and the root-mean-square deviation (RMSD) was calculated. Comparison of the experimental and theoretical parameters showed that the B3LYP DFT calculation yields a smaller RMSD value. In terms of the self-consistent field (SCF) energy and RMSD value, the B3LYP hybrid functional with the 6−311++G** basis set appeared to be more reliable. Using this method, we attempted to provide a detail description. In particular, the B3LYP functional yielded good outcomes for aromatic molecules (Sagaama et al., 2020b; Karrouchi et al., 2020; Gatfaoui et al., 2020; ISSa et al., 2020). In addition, the Hirshfeld surface (HS) analysis of shape indices dnorm, di, and de and curvedness was made. Using the topological and weak interaction analysis, the type and strength of the covalent and non covalent molecular contacts in morphine were predicted. The topological study was based on the AIM, ELF, and LOL approaches. The covalent and non covalent contacts established in the investigated structure can be found by the reduced density gradient (RDG), interaction region indicator (IRI), and density overlap region indicator (DORI) techniques. The docking simulation was aimed at clarifying the possibility of the simultaneous pain relief and disease treatment. For this purpose, the docking simulation of the morphine molecule with five proteins was performed. The human immunodeficiency, monoamine oxidase B (MOAB), COX-2, candida albicans, and phosphodiesterase enzymes were related to the cancerous, Parkinson, inflammatory, microbial, and schizophrenia diseases, respectively.
2 Computational details
The crystallographic structure of the compound under study was taken from the Cambridge Crystallographic Data Centre (CCDC), code CCDC 920204. The molecular modeling of (5α,6α)-7,8-didehydro-4,5-epoxy-17-methylmorphinan-3,6-diol (morphine)was performed in the GAUSSIAN 09 program (Frisch et al., 2009) at the DFT and Hartree–Fock levels with the 6−311++G(d; p)basis set. Then, the optimized geometry was visualized using the Gauss View 6.0.16 (GaussView). The colored molecular graphics of the isolated morphine geometry and crystal structure was generated using the Chemcraft software (chemcraftprog). The noncovalent interactions in the morphine crystal geometry were investigated by different techniques, including the RDG and DORI, using the Multiwfn 3.8 software (Lu and Chen, 2012). Basing on the experimental input file (.cif), the HSs were analyzed in the Crystal Explorer 3.1program (Wolff et al., 2012). The molecular docking computation of the title molecule was made using the iGEMDOCK (Yang and Chen, 2004) and PyRx 0.8 programs (Dallakyan and Olson, 2015). The protein codes were taken from the RCSB protein data bank (RCSB). The docking results were visualized with the discovery studio visualizer (Discovery Studio) and PyMOL 2.4.1(pymol) package.
3 Results and discussion
3.1 Geometric study
The investigated C17H19NO3 compound crystallizes in orthorhombic sp. gr. P212121. Each cell of the morphine crystal structure contains four atoms. The crystallographic cell lengths are a = 7.6989(10) Å, b = 12.737(4) Å, and c = 13.740(4) Å and the cell angles are α = β = γ = 90°. The geometrical parameters (bond lengths and angles) were calculated using the B3LYP/6−311++G** and HF/6−311++G** method. The SCF energies are −939.8799 and −934.4044 Hartree for the DFT and HF calculations, respectively. The dipole moment is 4.02 Debye (B3LYP) and 4.24 Debye (Hartree–Fock). The optimized geometry and the corresponding atoms are shown in Fig. 1.The crystal packing mapped in Fig. S1 demonstrates the existence of the O—H…O intermolecular interactions. In addition, the results obtained are compared with the experimental parameters in terms of the RMSD value. The B3LYP and H-Fock RMSD values were found to be 0.2434 and 0.2825, respectively. As clearly seen in Table 1, a discrepancy between the predicted geometrical parameters and the experimental data is insignificant and apparently caused by the fact that, in the theoretical prediction, the molecule is considered to be isolated in the gaseous phase, while the X-ray diffraction (XRD) data are recorded in the solid state. It should be noted that, basically, there is a difference between the optimized structure in the gaseous and solid phase outstanding from weak interactions (hydrogen and stacking bonds). According to the SCF energy and RMSD values, the DFT calculation using the B3LYP hybrid functional with the 6−311++G** basis set yields better results than the HF method. Therefore, the quantum calculation of the molecule under investigation was made with the B3LYP/6−311++G** level of the theory.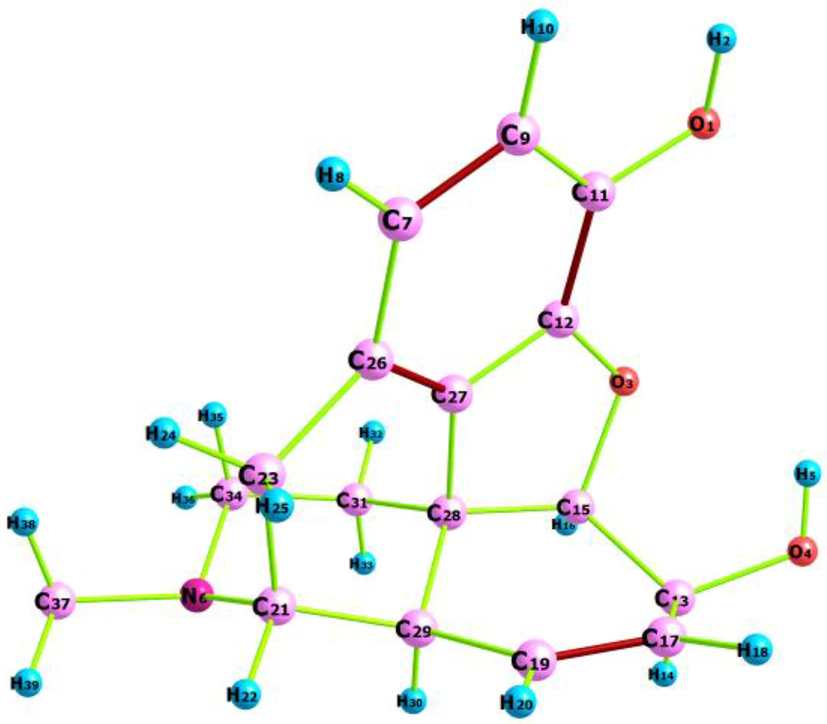
Optimized structure of morphine compound using Chemcraft program.
Geometrical parameters
DFT
HF
Exp.
Geometrical parameters
DFT
HF
Exp.
O1-H2
0.962
0.940
0.840
C19-H20
1.333
1.077
1.33(1)
O1-C11
1.369
1.351
1.380(7)
C19-C29
1.086
1.510
0.950
O3-C12
1.372
1.354
1.384(7)
C21-H22
1.508
1.085
1.512(9)
O3-C15
1.471
1.437
1.483(7)
C21-C23
1.094
1.558
1.000
O4-H5
0.967
0.943
0.840
C21-C29
1.563
1.545
1.567(9)
O4-C13
1.419
1.395
1.442(8)
C23-H24
1.552
1.085
1.537(9)
N6-C21
1.475
1.462
1.474(8)
C23-H25
1.095
1.087
0.991
N6-C34
1.463
1.452
1.456(9)
C23-C26
1.096
1.515
0.990
C7-H8
1.085
0.940
1.460(8)
C26-C27
1.515
1.372
1.515(9)
C7-C9
1.397
1.351
0.950
C27-C28
1.383
1.504
1.371(8)
C7-C26
1.401
1.354
1.388(9)
C28-C29
1.508
1.537
1.506(9)
C9-H10
1.086
1.437
1.397(9)
C31-C28
1.545
1.538
1.510(9)
C9-C11
1.401
0.943
0.950
C29-H30
1.097
1.087
1.535(8)
C11-C12
1.390
1.395
1.39(1)
C31-H32
1.094
1.086
1.000
C12-C27
1.380
1.462
1.374(8)
C31-H33
1.096
1.087
0.990
C13-H14
1.098
1.452
1.387(8)
C31-C34
1.529
1.525
0.990
C13-C15
1.551
0.940
1.000
C34-H35
1.104
1.093
1.512(9)
C13-C17
1.513
1.351
1.541(9)
C34-H36
1.094
1.085
0.990
C15-H16
1.090
1.354
1.49(1)
C37-H40
1.104
1.093
0.979
C15-C28
1.552
1.437
0.999
C37-H39
1.093
1.084
0.981
C17-H18
1.085
0.943
1.565(9)
C37-H38
1.093
1.085
0.980
C17-C19
1.085
1.395
0.950
RMSD
0.2434
0.2825
Angles (°)
H2-O1-C11
109.4
110.8
109.5
C21-C23-H24
108.9
109.3
108.7
C12-O3-C15
107.3
108.5
106.3(4)
C21-C23-H25
108.4
108.3
108.7
C5-O4-C13
106.9
109.0
109.5
C21-C23-C26
114.5
114.2
114.3(5)
C21-N6-C34
113.4
113.9
112.3(5)
H24-C23-H25
105.3
105.8
107.6
C21-N6-C37
114.2
114.6
111.9(5)
H24-C23-C26
111.2
111.1
108.7
C34-N6-C37
112.3
112.5
112.7(5)
H25-C23-C26
107.8
107.5
108.7
H8-C7-C9
118.9
118.8
120.1
C7-C26-C23
124.7
124.8
124.2(5)
H8-C7-C26
120.3
120.4
120.2
C7-C26-C27
116.3
116.2
117.4(5)
C9-C7-C26
120.6
120.5
119.7(6)
C23-C26-C27
118.3
118.3
117.8(5)
C7-C9-H10
119.4
119.2
119.3
C12-C27-C26
123.1
123.1
121.6(5)
C7-C9-C11
121.8
121.8
121.2(6)
C12-C27-C28
109.6
109.3
110.7(5)
H10-C9-C11
118.6
118.7
119.4
C26-C27-C28
126.7
126.8
126.9(5)
O1-C11-C9
124.0
123.7
124.1(6)
C15-C28-C27
100.5
100.0
99.5(5)
O1-C11-C12
119.2
119.5
119.3(5)
C15-C28-C29
116.4
116.7
116.4(5)
C9-C11-12
116.6
116.6
116.4(6)
C15-C28-C31
111.6
111.6
112.0(5)
O3-C12-C11
126.5
126.7
125.7(5)
C27-C28-C29
106.6
106.7
106.5(5)
O3-C12-C27
112.5
112.1
112.3(5)
C27-C28-C31
112.4
112.3
111.7(5)
C11-C12-C27
120.8
120.8
121.8 (5)
C29-C28-C31
108.8
108.9
111.7(5)
O4-C13-H14
105.4
105.6
107.0
C19-C29-C21
115.7
115.7
114.2(5)
O4-C13-C15
110.8
111.1
108.9(5)
C19-C29-28
110.2
110.1
109.8(5)
H2-O1-C11
109.4
110.8
109.5
C19-C29-30
109.2
108.7
108.6
O4-C13-C17
112.9
112.5
113.0(5)
C21-C29-C28
106.5
106.4
106.7(5)
14-C13-C15
106.2
106.3
107.1
C21-C29-H30
105.5
105.7
108.7
H14-C13-C17
107.4
107.0
107.0
C28-C29-H30
109.2
109.7
108.7
C15-C13-C17
113.3
113.4
113.5(5)
C28-C31-H32
109.9
110.0
109.2
O3-C15-C13
109.0
110.0
108.5(5)
C28-C31-H33
108.1
108.2
109.3
O3-C15-H16
104.9
105.5
109.5
C28-C31-C34
112.1
112.16
111.8(5)
O3-C15-C28
106.2
106.0
107.0(5)
H32-C31-H33
107.6
107.5
107.9
C13-C15-H16
109.2
108.7
109.5
H32-C31-C34
109.4
109.4
109.2
C13-C15-C28
114.1
113.8
112.8(5)
H33-C31-C34
109.2
109.1
109.3
H16-C15-C28
112.6
112.3
109.5
N6-C34-C31
111.2
111.1
110.7(5)
C13-C17-H18
116.0
116.2
119.4
N6-C34-H35
111.8
111.8
109.5
C13-C17-C19
121.7
121.7
121.2(6)
N6-C34-H36
107.9
108.1
109.6
H18-C17-C19
122.0
121.8
119.3
C31-C34-H35
109.7
109.8
109.5
C17-C19-H20
120.5
120.4
120.1
C31-C34-H36
109.3
109.1
109.5
C17-C19-C29
120.3
120.2
119.7(6)
H35-C34-H36
106.4
106.5
108.1
H20-C19-C29
119.0
119.2
120.2
N6-C37-H38
114.1
114.0
109.5
N6-C21-H22
105.8
106.0
107.0
N6-C37-H39
109.7
109.9
109.5
N6-C21-C23
114.8
114.9
115.3(5)
N6-C37-H40
109.2
109.3
109.5
N6-C21-C29
107.1
107.0
107.9
H38-C37-H39
108.0
108.1
109.5
H22-C21-23
107.3
107.1
106.9
H38-C37-H40
107.4
107.4
109.5
H22-C21-C29
107.9
107.8
107.0
H39-C37-H40
107.9
107.7
109.5
C23-C21-C29
113.2
113.4
112.4(5)
RMSD
1.4152
1.3858
–
4 Molecular topological analysis
4.1 QTAIM
The Quantum Theory of Atoms in Molecules (QTAIM) (Becke, 2007) was employed to identify the nature of non-bonded interactions in the crystal structure (Tahenti et al., 2020). In this context, the topological analysis was performed for the title compound. Electron density ρ(r), Laplacian Δρ(r), kinetic energy density G(r), potential energy density V(r), ratio |V(r)|/G(r), and the hydrogen bonding interaction Eint = V(r)/2 for the morphine structure were calculated. The results obtained are summarized in Table 2 and an AIM map is shown in Fig. 2. The Laplacian at the bond critical points (BCPs) determines the accumulation or depletion electron density and can be exploited to distinguish the shared- and closed-shell bonding interactions (Bazargan et al., 2020; Malaganvi et al., 2019). The positive Δρ(r) sign corresponds to the ionic interaction and the negative one stands for the covalent interaction. In addition, the parameter |V(r)|/G(r) at the BCP (3, −1) allows one to distinguish the nature of an interaction. The ionic and hydrogen bond and van der Waals (vdW) interactions are characterized by a ratio of smaller than unity. The covalent interactions have the |V(r)|/G(r) value>2. The mixed interaction corresponds to 1<|V(r)|/G(r) < 2 (Rad et al.,2021). The AIM representation proved the existence of nine weak H-bond interactions. These intermolecular contacts were classified as O—H…O, C—H…O, C—H…C, and O—H…H categories.
Name
ρ(r)
Δρ(r)
G(r)
V(r)
|V(r)|/G(r)
Eint(kcal/mol)
O1-H2…O44
0.0263
0.0924
0.0236
−0.0241
1.0211
−7.56
C9-H10…O44
0.0050
0.0210
0.0041
−0.0030
0.7317
−0.94
O121-H122…O4
0.0263
0.0923
0.0207
−0.0241
1.0211
−7.56
C129-H130…O4
0.0058
0.0210
0.0041
−0.0030
0.7317
−0.94
C34-H36…C91
0.0068
0.0267
0.0051
−0.0036
0.7058
−1.12
C37-H40…C97
0.0053
0.0170
0.0032
−0.0022
0.6875
−0.69
C37-H40…C87
0.0030
0.0095
0.0018
−0.0013
0.7222
−0.37
O84-H85…H36
0.0020
0.0084
0.0015
−0.0009
0.6000
−0.28
C93-O84…H33
0.0030
0.0116
0.0021
−0.0014
0.6666
−0.43
O44…O43
0.0192
0.0929
0.0207
−0.0182
0.8792
−5.71
O3…O4
0.0192
0.0927
0.0207
−0.0182
0.8792
−5.71
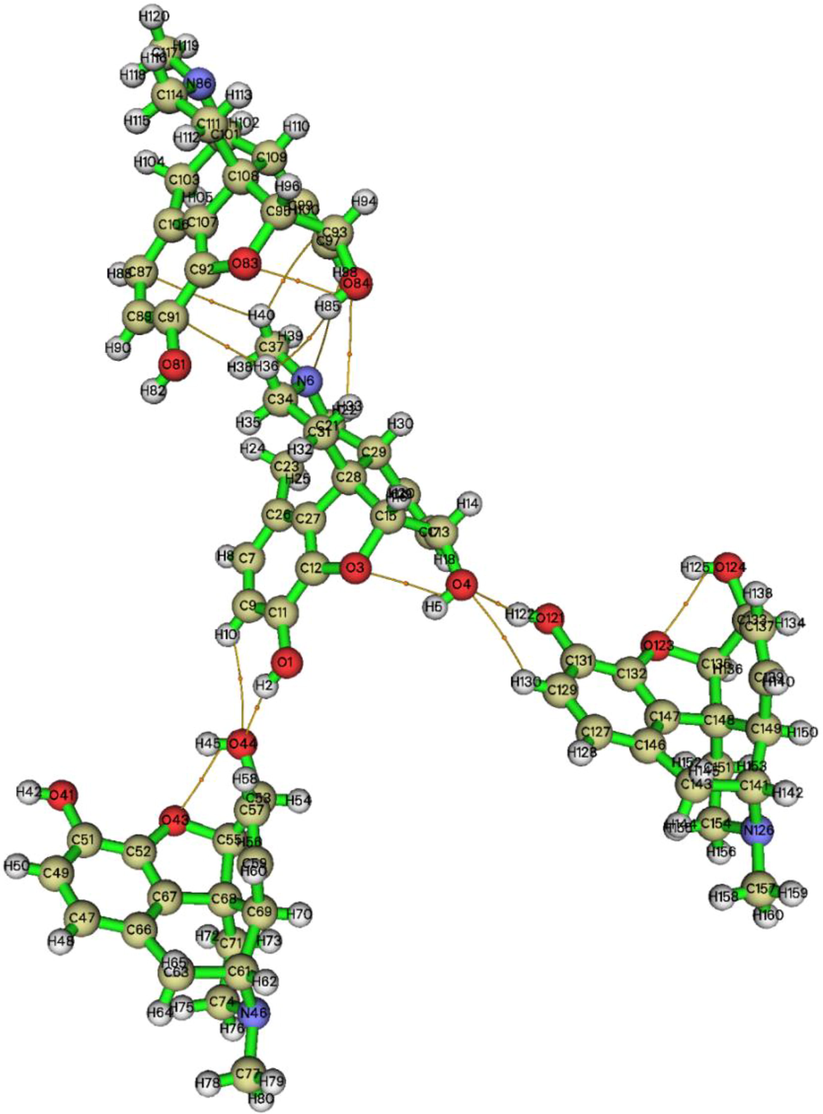
The non covalent interactions of morphine crystal conformation.
The electron density and Laplacian values for the H-bonds range between 0.0020 and 0.0263 and 0.0084–0.0929 a.u., respectively. The value of the interaction energy suggests the high chemical reactivity of oxygen atoms, since they are involved in the four H-bond interactions. According to the Eint value, the O1—H2…O44 and O121—H122…O4 (−7.56 kcal/mol) bonds can be considered to be the stronger ones. In addition, as shown in Fig. 2, the interaction between oxygen atoms O44…O43 and O3…O4 probably belongs to the ionic interactions. The binding energy of these intramolecular contacts was found to be −5.71 kcal/mol. It can be clearly seen in Table 2 that the other non covalent interactions C—H…O, C—H…C, and O—H…H are very weak as compared with the O—H…O ones. Their binding energies range from −0.28 to −1.12 kcal/mol.
4.2 The ELF and LOL analysis
The atomic or molecular localization of compounds is estimated using the ELF (Becke and Edgecombe, 1990a). The ELF and LOL topological analysis has been widely used in the classification of chemical bonds and description of the electronic structure (Noureddine et al., 2021a; Noureddine et al., 2021b). The excess kinetic energy density due to the Pauli repulsion was determined by the ELF analysis. The latter identifies the localization of electrons pairs in covalent bonds, including bonding and lone pairs (Becke and Edgecombe, 1990b). Thus, basing on the electron localization and Pauli repulsion, the electron localization descriptor (ELF and LOL) studies were carried out using the Multiwfn 3.8 program. The ELF and LOL representations are shown in Fig. 3a, b. The surface map with the ELF projection effect is presented in Fig. S2. The color scale of the ELF and LOL maps varies from blue to red in the ranges of 0–1 and 0–0.8, respectively. The area colored in red (Fig. 3a) corresponds to the maximum Pauli repulsion with the unity value and the blue-colored regions refer to the minimum Pauli repulsion with zero value. However, the highest repulsive interaction, which is related to the most localized electron state, is shown in red. The strong localization (ELF > 0.5) of electrons corresponds to the covalent bond, lone pair, or inner shells (Noureddine et al., 2021b). As can be seen in Fig. 3a, almost all hydrogen atoms (H14, 22, 30, 38, 39, and 40) are surrounded by the red-colored regions, which indicate the greatest electric charge accumulation areas. In addition, the ELF values for two oxygen atoms (O1 and O3) are in the range of 0.5–0.8 a.u. According to their red-light color, the region around O1 and O2 is characterized by the weak electron localization. The blue spots (ELF < 0.5) around the oxygen (O4) and carbon (C11, C12, C27, C28, and C13) atoms show charge delocalization regions. This is indicative of the existence of non covalent interactions in the molecular structure. In our case, these weak interactions are probably of the O–H…O and O—H…C types, which were previously observed during the AIM investigation. In addition, the green regions overlapping the morphine atom on the ELF surface and LOL color filled maps point out the VDW interactions. The white spots concentrated at hydrogen atoms in the LOL scheme show that the electron density exceeded the maximum color scale (0.8). The white color indicates that the bonds are dominated by a single localized orbital (Noureddine et al., 2021b).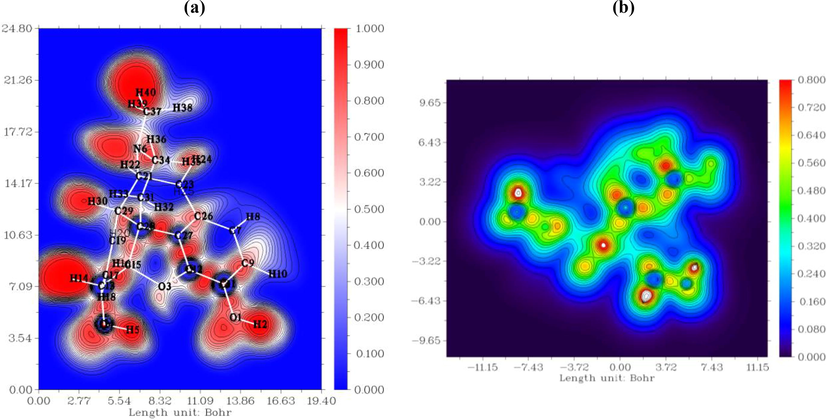
The ELF (a) and LOL (b) pictorial representations generated via Multiwfn software.
5 HS study
To find the weak non covalent interactions in the morphine crystal structure, the HS study with two-dimensional (2D) fingerprint plots were carried out using the crystal Explorer 17.5 program (Wolff et al., 2012). The HSs were generated by computing the electron distribution as a sum of electron densities of spherical atoms (Spackman and Jayatilaka, 2009). Thanks to it importance in revealing weak interactions, the HSs analysis was the objectives of several papers (Arulraj et al., 2020a; Arulraj et al., 2020b; Anitha et al., 2020; Arulraj et al., 2019). The HSs of the morphine structure were plotted with the dnorm value from −0.6089 to 1.1436 Å, the di value from 0.7363 to 2.5007 Å, the devalue from 0.7349 to 2.3999 Å, the shape index from −1 to 1 Å, and a curvedness of −4,4 Å (Fig. S3). For the dnorm surface, the shorter contacts with the negative dnorm value are indicated by red spots and the longer contacts with the positive dnorm value are colored in blue. The white color corresponds to dnorm = 0 and characterizes the contact with a distance close to di + de. The large deep-red circles on the dnorm surface correspond to the strongest hydrogen bonding interactions; the weaker H-bond contacts are indicated by light-red areas. Using the di and de surfaces, we can determine whether the molecule acts as an acceptor or as a donor. In particular, the hydroxyl group (O—H) acts as an electron acceptor on the di surface. The oxygen atom of the other hydroxyl group acts as an electron donor on the de surface. In addition, the shape index and curvedness were used to establish the existence of the C—H…π and π… π stacking interactions. The red concave triangles on the shape index surface characterize the acceptor atoms, while the blue convex area indicates the donor hydrogen atoms. The existence of a blue region around hydrogen atoms of the methyl group and a red region near the phenyl rings confirm the establishment of the C—H…π (C—H…C) interactions in the crystal. Concerning the curvedness map, the existence of the π…π interactions is justified by the appearance of a huge green region bounded by blue lines. In Fig. 4, different types of the hydrogen bond interactions are identified. The figure shows the presence of two O—H…O, two O—H…C, and three C—H…C bonding interactions between morphine units. Therefore, the O—H…O and O—H…C interactions are stronger than the C—H…C contacts. The intermolecular contacts responsible for the crystalline stability can also be seen in the 2D fingerprint representation. However, the molecular interaction contribution of each element was individually analyzed. The diverse contributions of the close reciprocal contacts are mapped in Fig. 4.The main contribution to the HSs is made by the H…H, O…H/H…O, H…C/C…H, and H…N/N…H contacts. The fingerprint plots of these interactions, along with each percentage, are mapped in Fig. S4. The most significant contribution (62.6%) to the total HSs is made by the H…H contact. This is owing to plenty of hydrogen atoms in the morphine geometry. The O…H/H…O contacts constitute 20.4% of the total HS, resulting from the presence of three oxygen atoms. These contacts, which involve the O—H group, are attributed to the O—H…O and O—H…C hydrogen bonding interactions. The H-bond interactions ensure the molecular packing stability; in the fingerprint plot, they appear as two symmetrical spikes. The left spike related to the donor atom corresponds to de > di (H…O), while the right one is related to the acceptor atom with de < di (O…H). In addition, the H…C/C…H and H…N/N…H contacts constitute15.6% and 1.7% of the total HS, respectively. These contacts appear as two short spikes in the fingerprint plots.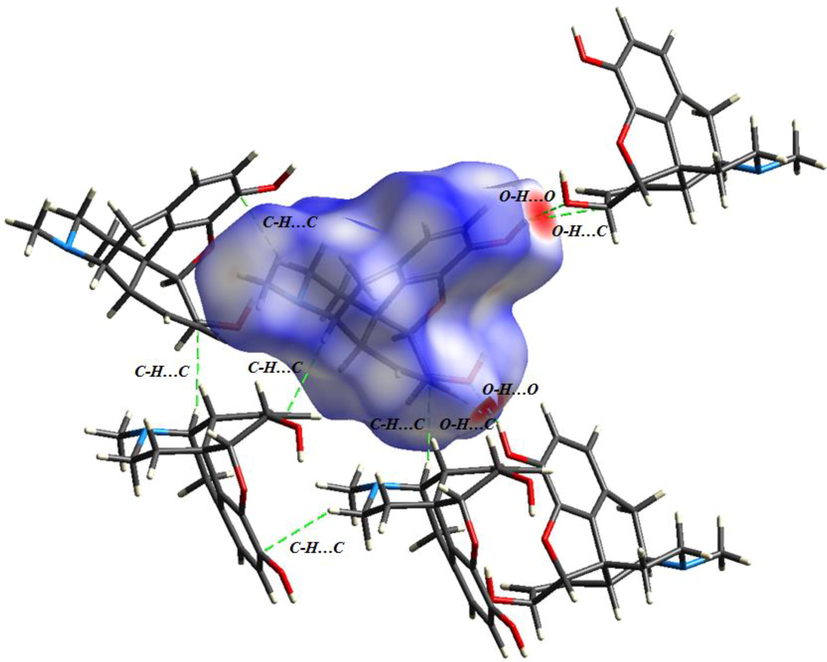
The non covalent interactions existing in morphine packing structure.
6 Study of the non covalent interactions
6.1 RDG
Generally, the molecular stability was ensured by weak inter- and intramolecular interactions. In this study, such interactions were predicted using the RDG analysis based on the non covalent interaction (NCI) method. The λ2 sign was exploited to differentiate between the bonded (λ2 < 0) and non-bonded (λ2 > 0) interactions. In our case, the λ2 sign ρ function ranges from −0.05 to 0.05 a.u. in the RDG scatter graph (Fig. 5). The RDG spectra are coded by three colors: red, green, and blue. The red peaks in the range of λ2 > 0 correspond to the effect of steric repulsion in the ring (Jia et al., 2019). The spikes appearing in the region of λ2 = 0 represent the dipole–dipole or London dispersions forces (Khan et al., 2020). The blue-colored spikes in the regions ρ > 0 and λ2 < 0 represent the electrostatic interactions (hydrogen and halogen bonds). The 2D reduced density graph of the monomeric and dimeric structures together with the 3D isosurface are shown in Fig. 5.The strong attractive interaction (H-bond) appear between −0.01 and −0.05 a.u. The isosurface plot reveals the presence of three blue flaky patches between the two units corresponding to the O—H…O hydrogen bonding interactions. The strong repulsion interactions (steric effect) occur in the range of 0.05–0.01 a.u. These interactions look like an elliptical slab at the center of the rings. The vdW interactions range from 0.01 to −0.01 a.u. The obtained results of the NCI/RDG study of the title compound agree well with the AIM and XRD data.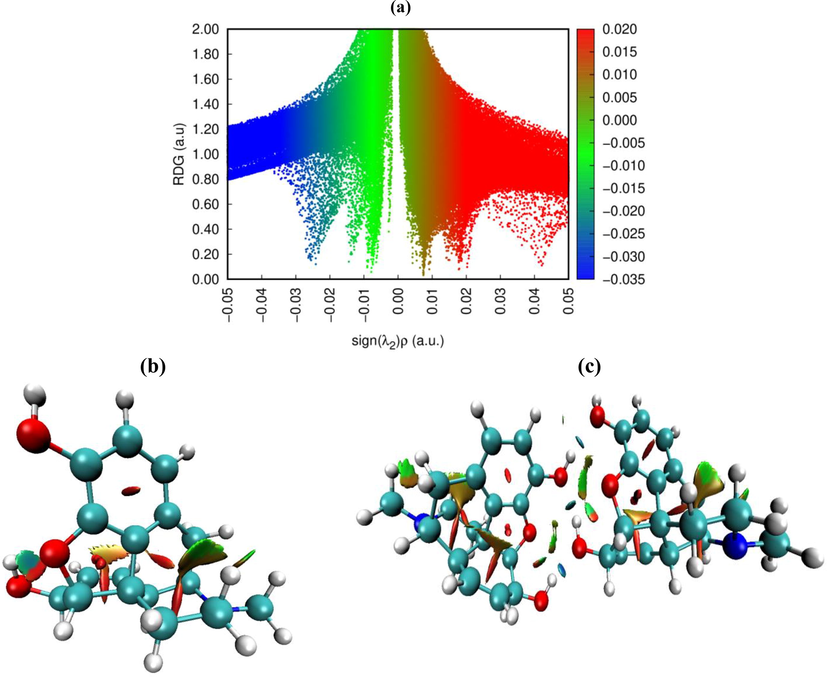
. Reduced density graph and isosurface plots of monomeric and dimeric structures.
6.2 IRI and DORI analysis
In this Section, the new scalar fields, interaction region indicator (IRI), and density overlap regions indicator (DORI) are introduced. These indicators depend on the electron density and its derivatives (Mebs, 2016). The IRI and DORI techniques can simultaneously reveal the covalent and non covalent interactions. They have analogous goals, but the IRI is simpler than the DORI, since it was only defined with the electron density and its gradient. As in the RDG analysis, the (λ2)ρ function sign was plotted on the IRI and DORI isosurfaces through a color scale in order to disclose the nature of the interactions. The largest λ2 value of the Hessian matrix ensures a clear distinction between the attractive and repulsive interactions (Johnson et al., 2010). The region with λ2 < 0 corresponds to the bonding interactions, whereas the non-bonding region is identified with λ2 > 0. Fig. 6 shows the strong similarity, but the rough extremities of the interaction regions presented on the DORI decreases its graphic resolution. The IRI method demonstrates the lower calculation cost as compared with the other technique. In addition, both methods offer a coherent description of the chemical interactions in molecular structures. The covalent bonds are characterized by a positive electron density (ρ ≫ 0) and a weak negative second eigenvalue (λ2 ≪ −0.04). In addition, the regions of weak intermolecular interactions are shown satisfactorily, as in the RDG study. The different types of interactions, including the steric effect, vdW, H-bond, and covalent bonds, are presented in the IRI map. The covalent bond regions are shown by blue surfaces, which demonstrate the huge electron density and the strong bonding effect in these regions. It can be seen in Fig. 6 that the carbon–carbon and carbon–oxygen covalent contacts were established.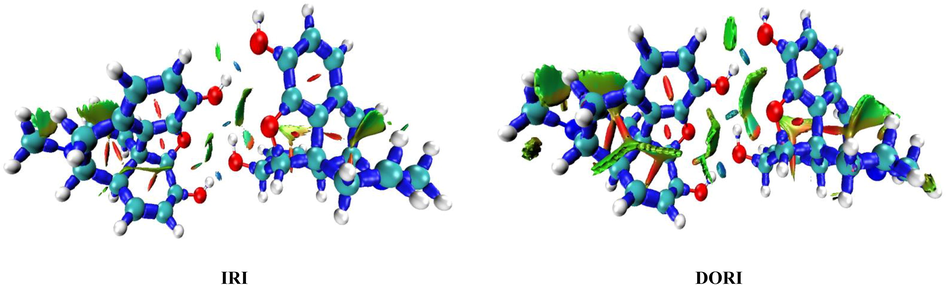
IRI and DORI representations mapped for morphine dimer conformation.
7 Molecular docking study
The molecular docking simulation is highly important for new drug design. It can help identify the most stable position of a docked compound within a target enzyme. This part of the study is aimed at investigations of more pharmacological activities of morphine. For this purpose, the docking analysis of the molecule under study into proteins active sites was performed using the iGEMDOCK and PyRx programs. The five proteins used were Humane immunodeficiency, Monoamine oxidase B (MOAB), COX-2, Candida albicans, and Phosphodiesterase taken from our previous works (Sagaama and Issaoui, 2020; Sagaama et al., 2020a). The target protein structures were chosen to test the anticancer (1DLO), anti-parkinson (2BK3), anti-inflammatory (3LN1), antimicrobial (4HOE), and anti-schizophrenia (5K95) activities. These enzymes were initially downloaded from the RCSB protein data bank website. Using the discovery studio visualizer, the five proteins were cleaned by eliminating previous ligands and water molecules. The binding score and interaction energy were estimated with the iGEMDOCK and PyRx packages, respectively. The obtained energy data are given in Table 3 and the docking poses between the morphine conformation and different structures generated using the PyMOL software (pymol) are presented in Fig. 7.The total energy computed using the iGEMDOCK graphical environment is a sum of the hydrogen bonding, vdW, and electronic interactions. In our case, the electronic contribution was found to be zero for different complexes. According to Table 3, the interaction forces are mainly the hydrogen bonding and vdW interactions. The interaction score decreases in the order of 2BK3 > 3LN1 > 4HOE > 1DLO > 5K9R. As can be seen in Table 3, the highest interaction score corresponds to the MOAB enzyme with energy of −105.04 kcal/mol. The 3LN1 protein is also characterized by a high binding energy, which was found to be −101.85 kcal/mol. The morphine–MOAB and morphine-3LN1 complexes have the similar vdW and H-bond energies. For the other enzymes, the interaction score ranges between −86 and −93 kcal/mol. The higher vdW and H-bond energies are related to 3LN1 (−87.64 kcal/mol) and 4HOE (−30.95 kcal/mol), respectively. Table 3 compares the obtained docking results with the previously published data (Sagaama and Issaoui, 2020; Sagaama et al., 2020a). It can be seen that the five morphine–protein complexes exhibit better energy results as compared with the previous scores.
Protein name
Protein code
Ligand
Binding affinities (kcal/mol)
Interaction score (kcal/mol)
H-bond
VDW
Humane immunodeficiency
1DLO
Morphine
−7.9
−87.13
−15.61
−71.51
2BT
–
−79.33
−69.65
−7.75
Monoamine oxidase B (MOAB)
2BK3
Morphine
−7.2
−105.04
−25.91
−79.13
Farnesol
–
−92.49
−5.95
−86.53
COX-2
3LN1
Morphine
−8.7
−101.85
−14.20
−87.64
2BT
–
−81.44
−66.34
−9.98
Candida albicans
4HOE
Morphine
−7.3
−92.90
−30.95
−61.95
2BF
–
−88.47
−22.18
−66.60
Phosphodiesterase
5K9r
Morphine
−8.4
−85.57
−21.21
−64.36
Imadazopyrazine
–
−69.56
−25.74
−43.82
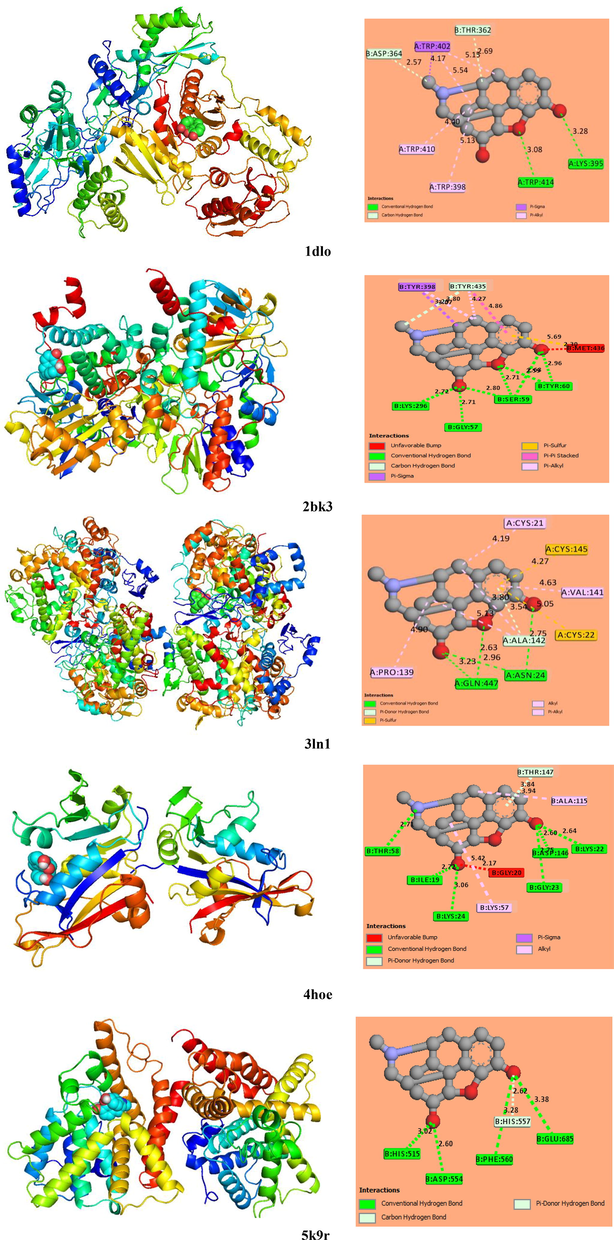
The best localization of morphine in the five target protein along with 2D interactions map.
The 2D representations of the protein–ligand interaction profile were plotted using the discovery studio visualizer (Fig. 7). As can be clearly seen in Fig. 7, the ligand protein interactions are principally a hydrogen bonding type. The latter was divided into three categories: the conventional, carbon, and π-donor hydrogen bond. The ligand docked in the active site of pocket atom 2BK3 has greater hydrogen bonding interaction number, justifying the highest corresponding binding score. Ser 59 interacts with three oxygen atoms of the morphine structure and Tyr 60 forms two H-bonds with O4 and O3. In addition, two H-bonds are introduced between Lys 296, Gly 57, and O1. The interaction bond lengths range between 2.7 and 3 Å. The shorter distances demonstrate the strength of the established H-bonds. The formed carbon hydrogen bond links carbon atom C37 and residue Tyr 435. For the morphine-3LN1 interaction, four conventional hydrogen bonds are formed between oxygen atoms O1, 3, and 4 and amino acids Asn 24 and Gln 447. In addition, a π-donor hydrogen bond (3.54 Å) was found between Ala 142 and the phenyl ring. In addition, residues Cys21, Val 141, Ala 142, and Pro 139 were implicated in five alkyl interactions with the morphine conformer. Concerning the 4HOE protein, six conventional hydrogen bonds (Thr58, Ile19, Lys 24, Gly 23, Asp 146, and Lys 22) and one π-donor hydrogen bond (Thr 147) were established between the Candida albicans residues and the candidate ligand. Thus, amino acids Lys 57 and Ala 115 interact with atoms 4HOE with the formation of the π–σ and alkyl interactions, respectively. For the Humane immunodeficiency protein, two conventional hydrogen bonds between Lys 395-O1 and Trp 414-O3 with respective bond lengths of 3.28 and 3.08 Å were observed. In addition, two carbon hydrogen bonds between C34 and The 362, the carbon atom of the methyl group, and Asp 364 were established. The π–sigma and π–alkyl interactions were also generated, which connect ligand atoms and amino acid residues Thr 402, Thr 410, and Trp 398. The 2D morphine-5K9R plot shows the formation of five H-bonds with oxygen atoms O1 and O4 via intervention of amino acid residues His 515, Asp 554, Phe 560, His 557, and Glu 685.
8 Conclusions
In this study, the molecular optimization of the compound under investigation was performed using the B3LYP/6−311++G** level of the theory. The calculated and experimental bond lengths and angles were found to be in good agreement. The Hirshfeld surfaces investigation disclosed various intermolecular interactions in the crystal structure and identified the electrophilic and nucleophilic sites. The AIM, ELF, and LOL studies were carried out to elucidate the potential of the inter- and intermolecular interactions based on the electron localization regions. It was shown that these interactions can be classified as weak contacts. In addition, the van der Waals and hydrogen bonding interactions were determined via the RDG analysis using a color code. However, the IRI and DORI yielded a detailed representation of the inter- and intramolecular contacts simultaneously. Finally, the docking simulation of morphine with the selected proteins was made. The results obtained proved the analgesic effect and treatment ability of our compound for the cancerous, Parkinson, inflammatory, microbial, and schizophrenia diseases.
Acknowledgement
Researcher supporting project number (RSP-2021/61), King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis, Crystal Structure, DFT Calculations and Hirshfeld Surface Analysis of 3-butyl-2,6-bis(4-fluorophenyl)piperidin-4-one. Acta Cryst E.. 2020;76(5):651-655.
- [Google Scholar]
- Synthesis, crystal structure, DFT calculations and Hirshfeld surface analysis of 3-chloro-3-methyl-r(2), c(6)-bis(p-methoxyphenyl)piperidin-4-one. J. Chem. Crystallogr.. 2019;50(1):41-51.
- [Google Scholar]
- Synthesis, Crystal Structure, DFT Calculations and Hirshfeld Surface Analysis of 3-Chloro-2,6-Bis(4-Chlorophenyl)-3-Methylpiperidin-4-One. J. Chem. Crystallogr.. 2020;51:273-287.
- [Google Scholar]
- Synthesis, vibrational spectra, DFT calculations, Hirshfeld surface analysis and Molecular docking study of 3-chloro-3-methyl-2,6-diphenylpiperidin-4-one. Spectrochim. Acta A.. 2020;232:118166
- [Google Scholar]
- Supramolecular assembly of a 2D coordination polymer bearing pyridine-N-oxide-2,5-dicarboxylic acid and copper ion: X-ray crystallography and DFT calculations. J. Mol. Struct.. 2020;1202:127243
- [Google Scholar]
- The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design. John Wiley & Sons; 2007.
- A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys.. 1990;92:5397-5403.
- [Google Scholar]
- A simple measure of electron localization inatomic and molecularsystems. J. Chem. Phys.. 1990;92:5307-5403.
- [Google Scholar]
- <https://www.ccdc.cam.ac.uk/> (CCDC-N° 920204).
- Small-molecule library screening by docking with PyRx. Methods Mol Biol.. 2015;1263:243-250.
- [Google Scholar]
- Discovery Studio 4.5 Guide. Accelrys Inc., San Diego. <http://www.accelrys.com>.
- Pharmacological significance of synthetic heterocycles Scaffold. A Review. Adv. Biol. Res.. 2011;5:120-144.
- [Google Scholar]
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Jr., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, N.J., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, Ö., Foresman J.B., Ortiz, J.V., Cioslowski, J. Fox, D.J., 2009. Gaussian, Inc., Wallingford C.T.
- Synthesis, experimental, theoretical study and molecular docking of 1-ethylpiperazine-1,4-diium bis(nitrate) Solid State Sci.. 2020;106:106326
- [Google Scholar]
- GaussView, Guassian, Inc. (Carnergie Office Parck-Building6 Pittsburgh PA 151064 USA), Copyright © 2000-2003 Semichem. Inc.
- Computational study of 3-thiophene acetic acid: Molecular docking, electronic and intermolecular interactions investigations. Comput. Boil. Chem.. 2020;86:107268
- [Google Scholar]
- A density functional theory study on complexation processes and intermolecular interactions of triptycene-derived oxacalixarenes. Theor. Chem. Acc.. 2019;138:113.
- [Google Scholar]
- Synthesis, crystal structure, Hirschfeld surface analysis, DFT calculations, anti-diabetic activity and molecular docking studies of (E)-N’-(5-bromo-2-hydroxybenzylidene) isonicotinohydrazide. J. Mol. Struct.. 2020;1221:128800
- [Google Scholar]
- Adsorption behaviour of chronic blistering agents on graphdiyne; excellent correlation among SAPT, reduced density gradient (RDG) and QTAIM analyses. J. Mol. Liq.. 2020;316:113860
- [Google Scholar]
- Bioactive Heterocyclic Compound Classes: Agrochemicals. Weinheim: Wiley-VCH; 2012.
- J. Comput. Chem.. 2012;33:580-592.
- Experimental, DFT dimeric modeling and AIM study of H-bond-mediated composite vibrational structure of Chelidonic acid. Heliyon. 2019;5:1586.
- [Google Scholar]
- Complex modes of bonding: NCI/ELI-D vs. DORI surface analyses of hapticities and hydrogen–hydrogen contacts in zincocene related compounds. Chem. Phys. Lett.. 2016;651:172-177.
- [Google Scholar]
- Quantum chemical studies on molecular structure, AIM, ELF, RDG and antiviral activities of hybrid hydroxychloroquine in the treatment of COVID-19: Molecular docking and DFT calculations. J. King Saud Univ. Sci.. 2021;33:101334
- [Google Scholar]
- Quantum chemical calculations, spectroscopic properties and molecular docking studies of a novel piperazine derivative. J. King Saud Univ. Sci.. 2021;33:101283
- [Google Scholar]
- Are nickel-and titanium-doped fullerenes suitable adsorbents for dopamine in an aqueous solution? Detailed DFT and AIM studies. J. Mol. Liq.. 2021;322:114942
- [Google Scholar]
- Molecular docking studies, structural and spectroscopic properties of monomeric and dimeric species of benzofuran-carboxylic acids derivatives: DFT calculations and biological activities. Com. Biol. Chem.. 2020;87:107311
- [Google Scholar]
- Design, molecular docking analysis of an anti-inflammatory drug, computational analysis and intermolecular interactions energy studies of 1-benzothiophene-2-carboxylic acid. Comput. Biol. Chem.. 2020;88:107348
- [Google Scholar]
- Searching potential antiviral candidates for the treatment of the 2019 novel coronavirus based on DFT calculations and molecular docking. Heliyon. 2020;6:04640
- [Google Scholar]
- Biological activity study for some heterocyclic compounds and their impact on the gram positive and negative bacteria. Energy Procedia. 2019;157:296-306.
- [Google Scholar]
- Cryst. Eng. Comm.. 2009;11:19-32.
- A tetrachlorocobaltate(II) salt with 2-amino-5-picolinium: synthesis, theoretical and experimental characterization. J. Mol. Struct.. 2020;1207:127781
- [Google Scholar]
- Wolff, S.K., Grimwood, D.J., McKinnon, J.J., Turner, M.J., Jayatilaka, D., Spackman, M.A., 2012. University of Western Australia.
- Effects of nalbuphine on the cardioprotective effect of morphine in rats. Int. J. Cardiol.. 2021;322:207-210.
- [Google Scholar]
- GEMDOCK: a generic evolutionary method for molecular docking. Proteins Struct. Funct. Bioinforma. 2004;55(2):288-304.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2021.101606.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




