

Non covalent interactions analysis and spectroscopic characterization combined with molecular docking study of N′-(4-Methoxybenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide
⁎Corresponding authors. issaoui_noureddine@yahoo.fr (Noureddine Issaoui), omar@ksu.edu.sa (Omar Al-Dossary)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The structure, spectroscopic features, and pharmaceutical effect of N′-(4-Methoxybenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide (MBPPC) has been studied by DFT modeling, X-ray diffraction, FT-IR, 1H and 13C NMR, and molecular docking investigation. Molecular structure analysis was carried out using DFT calculation. Then, the low RMSD value indicates the good agreement between calculated and observed data. In order to understand the electronic charge transition, the electron difference density technical was performed. 1H and 13C NMR spectra were recorded in the region of 0–15 and 5–225 ppm, respectively, and FT-IR spectrum of MBPPC was obtained from 4000 to 500 cm−1. Electronic structure characteristics were achieved at the level of B3LYP/6-311+G(2d,p). Inter and intramolecular interactions are discussed by topological (AIM, RDG) and Hirshfeld surface analyses. TD-DFT calculations were conducted to reveal molecular orbital based reactivity characteristics and nonlinear optical features up to third order. In addition, thanks to the broad biological field of compounds based on pyrazole and hydrazone groups, molecular docking of the title compound was carried out to study their clinical activities. Docking simulation shows the potential of MBPPC against Vibrio cholera, Mycobacterium tuberculosis, and estrogen receptor.
Keywords
FT-IR
NLO
Hydrogen bonding interaction
Anti-microbial
Anti-tubercular

1 Introduction
Heterocyclic compounds have been attracting much attention since their importance in organic and clinical chemistry (Noureddine et al., 2021; Noureddine et al., 2020). Pyrazole ring is one of the most effective group in new drug manufacturing design. It present in the chemical combination of several efficientanticancer drugs such as Crizotinib, Pyrazomycin, Tartrazine (Faria et al., 2017). It can also be used in anti-obesity (Rimonabant) (Hampp et al., 2008) and antidepressant (Fezolamine) (Luttinger and Hlasta, 1987) treatment. Pyrazole derivatives considered as one of the largest active field of heterocyclic compounds. It considerably used in biological activities as anticancer, anti-tubercular, anti-microbial, anti-inflamatory, cardiovascular and analgesic (Koca et al., 2013; Gokhan-Kelekci et al., 2007). In addition, N-acylhydrazone scaffold (-CO-NH-N=CH-) derivatives showed significant clinical activities such as antimicrobial, anti-inflammatory, anticancer…etc. The presence of pyrazole and hydrazone groups in some compounds prove their ability in the treatment of several intractable diseases such as diabetes mellitus (Karrouchi et al., 2020). Thanks to its extraordinary pharmacological application of compounds based on pyrazole and hydrazone groupment, several studies (Karrouchi et al., 2020; Karrouchi et al., 2017) show their special interest in new drug industry. To the best of our knowledge, neither spectroscopic and electronic analyses nor molecular docking investigation of MBPPC have been published as yet. For this reason, quantum computation has been performed using the hybrid functional B3LYP with 6-311+G(2d,p) for monomeric conformation and 6-31G(p) for dimeric structure. B3LYP represents Becke’s three parameters hybrid exchange functional method with Lee-Yang-Parr’s correlation functional (LYP), it considered the most useful functional for aromatic molecules since it shows good agreement between X-ray and estimated parameters (Gatfaoui et al., 2020; Sagaama et al., 2020; Sagaama and Issaoui, 2020; Issa et al., 2020). The RMSD values were calculated demonstrating good correlation among experimental data and theoretical ones. Using a TD-DFT calculation, the electron difference density (EDD) surface was plotted to simulate the electronic charge transfer upon an excitation. However, the topological analysis (AIM), the reduced density gradient (RDG) and Hirshfeld surface (HS) have been adopted to investigate the non covalent interactions in the crystal arrangement. The frontier molecular orbitals analysis (HOMO-LUMO) was conducted in order to determine the chemical reactivity of the studied compound. To complete the molecular orbital description, the density of state (DOS, PDOS, EPDOS) representation was plotted. Besides, the nonlinear optical parameters up to third order (μ, α, β, γ) have been calculated to examine the ability of MBPPC to be a nonlinear optic molecule. Then, the spectroscopic analyses were performed using nuclear magnetic resonance and infrared spectra. The theoretical 13C, 1H NMR and FT-IR spectra of the title compound have been compared with the experimental ones. Finally, owing to multiple clinical benefits of our compound, a biological study was achieved via docking calculation. Molecular docking analysis of MBPPC was performed with three proteins in order to examine the anti-microbial, anti-tubercular and estrogen receptor modulator agents.
2 Theoretical calculations
All calculation was carried out with Gaussian 16 package (Frisch et al., 2016) by using B3LYP hybrid function. The structural parameters of monomer and dimer conformation of the studied molecule were optimized in ground state via DFT computation using 6-11+G(2d,p) level. To evaluate the strength of intermolecular hydrogen interactions, topological analysis (AIM, RDG) were performed via Multiwfn (Lu and Chen, 2012) and VMD (Humphrey et al., 1996) programs. The type of non-covalent contacts was identified by Hirshfeld surfaces using crystal explorer 3.1 program (Wolff et al., 2012). The vibrational frequency assignments were determined with a high degree of accuracy by combination between findings of Gauss view (GaussView, 2020) and Veda4 (Jamróz, 2004) software. The 1H and 13C isotropic chemical shifts were computed using the gauge invariant atomic orbital (GIAO) method. The pharmaceutical effect of MBPPC was examined using molecular docking calculation with the help of iGEMDOCK (Yang and Chen, 2004) and discovery visualizer studio (Discovery Studio) which considered as visualization surface. The protein structures 5UL7, 6VW0, and 5TOA of Vibrio cholera, Mycobacterium tuberculosis, and estrogen receptor, respectively, were obtained from RCSB Protein Data Bank (Nie et al., 2017) database.
3 Results and discussions
3.1 Geometric structure
The optimized structure of MBPPC was simulated by DFT level using B3LYP/6-311+G(2d,p) method. The experimental geometry of the studied molecule was obtained from Cambridge Crystallographic Data Center (CCDC 1437241) (https://www.ccdc.cam.ac.uk). X-ray diffraction evince that the title compound belongs to monoclinic system with a = 10.9734(4) Å, b = 11.8835(5) Å, c = 13.0996 Å cell dimensions. Then, MBPPC crystallizes in space group P21/c with four atoms for each unit cell (Z = 4). The stabilization energy and dipole moment value in monomer and dimer geometries are calculated. The SCF energies are-1065.2778 Hartree (monomer) and −2129.9412 Hartree (dimer). The dipole moment values are3.87 and7.35 Debye for monomer and dimer, respectively. The bond lengths and bond angles parameters along with experimental ones are tabulated in Table 1 while the atom labeling scheme is given in Fig. 1. The comparison between computed and experimental bond lengths and angles was established based on RMSD (Root Mean Square Deviation) value. The lower RMSD values calculated for bond lengths 0.0774 (monomer)/0.0784 (dimer) indicate the reasonable correlation between theoretical and experimental parameters. The angle RMSD values of monomeric and dimeric conformation demonstrate a slight difference between experimental and theoretical findings. The deviation between X-ray diffraction and calculated parameters is probably owing to establishment of intermolecular interactions in crystalline structure which resulted by the formation of contacts between proton acceptor and proton donor. The H-bond interactions (N-H…O and C–H…O) formed between the two unit are represented with dashed lines in Fig. S1. The bond lengths and bond angles of these non-covalent contacts range experimentally between 1.91–2.86 Å and 152-157°, respectively. According to the finding results, the hydrogen bonding interactions are considered medium.
| Geometricalparameters | Monomer | Exp. | Dimer | Geometricalparameters | Monomer | Exp. | Dimer | Geometricalparameters | Monomer | Exp. | Dimer |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bond lengths (Å) | |||||||||||
| O1-C22 | 1.214 | 1.232(2) | 1.221 | C9-H10 | 1.083 | 0.950 | 1.086 | C23-C25 | 1.458 | 1.463(3) | 1.463 |
| O2-C30 | 1.361 | 1.365(2) | 1.366 | C9-C11 | 1.392 | 1.387(3) | 1.395 | C25-C26 | 1.398 | 1.397(2) | 1.411 |
| O2-C35 | 1.421 | 1.428(2) | 1.419 | C11-H12 | 1.083 | 0.950 | 1.086 | C25-C33 | 1.402 | 1.397(3) | 1.401 |
| N3-N4 | 1.340 | 1.347(2) | 1.339 | C11-C13 | 1.390 | 1.388 | 1.397 | C26-H27 | 1.082 | 0.950 | 1.085 |
| N3-C18 | 1.366 | 1.356(2) | 1.370 | C13-H14 | 1.083 | 0.950 | 1.067 | C26-C28 | 1.386 | 1.385(3) | 1.384 |
| N3-H39 | 1.007 | 0.89(2) | 1.012 | C13-C15 | 1.389 | 1.383(3) | 1.392 | C28-H29 | 1.080 | 0.950 | 1.085 |
| N4-C21 | 1.334 | 1.339(2) | 1.338 | C15-H16 | 1.084 | 0.950 | 1.086 | C28-C30 | 1.399 | 1.393(3) | 1.407 |
| N5-N6 | 1.355 | 1.380(2) | 1.358 | C15-C17 | 1.400 | 1.396(3) | 1.405 | C30-C31 | 1.397 | 1.393(3) | 1.398 |
| N5-C22 | 1.375 | 1.349(2) | 1.381 | C17-C18 | 1.464 | 1.468(2) | 1.466 | C31-H32 | 1.082 | 0.950 | 1.083 |
| N5-C40 | 1.017 | 0.98(2) | 1.022 | C18-C19 | 1.382 | 1.382(3) | 1.388 | C31-C33 | 1.382 | 1.379(3) | 1.397 |
| N6-C23 | 1.279 | 1.277(2) | 1.286 | C19-H20 | 1.075 | 0.950 | 1.079 | C33-H34 | 1.084 | 0.950 | 1.088 |
| C7-H8 | 1.083 | 0.950 | 1.086 | C19-C21 | 1.407 | 1.393(2) | 1.411 | C35-H36 | 1.087 | 0.979 | 1.091 |
| C7-C9 | 1.388 | 1.385(3) | 1.393 | C21-C22 | 1.488 | 1.480(3) | 1.488 | C35-H37 | 1.093 | 0.980 | 1.098 |
| C7-C17 | 1.400 | 1.393(3) | 1.406 | C23-H24 | 1.097 | 0.951 | 1.094 | C35-H38 | 1.093 | 0.980 | 1.097 |
| RMSD | 0.0774 | – | 0.0784 | ||||||||
| Bond Angles (°) | |||||||||||
| C30-O2-C35) | 118.548 | 117.3(2) | 118.143 | C13-C15-C17 | 120.686 | 120.2(2) | 120.676 | C26-C25-C33 | 118.169 | 118.1(2) | 118.065 |
| N4-N3-C18 | 113.433 | 112.8(1) | 113.701 | H16-C15-C17 | 120.096 | 119.8 | 119.346 | C25-C26-H27 | 118.909 | 119.5 | 118.616 |
| N4-N3-H39 | 119.155 | 115(1) | 118.565 | C7-C17-C15 | 118.564 | 119.1(2) | 118.537 | C25-C26-C28 | 121.135 | 120.9(2) | 120.795 |
| C18-N3-H39 | 127.192 | 132(1) | 127.680 | C7-C17-C18 | 120.074 | 119.4(2) | 121.464 | H27-C26-C28 | 119.955 | 119.5 | 120.584 |
| N3N-4-C21 | 104.465 | 104.3(1) | 104.366 | C15-C17-18 | 121.360 | 121.5(2) | 119.998 | C26-C28-H29 | 119.185 | 120.0 | 121.197 |
| N6-N5-C22 | 121.729 | 119.0(2) | 120.140 | N3-C18-17 | 122.896 | 122.7(2) | 122.839 | C26-C28-C30 | 119.905 | 120.0(2) | 120.431 |
| N6-N5-40 | 120.588 | 122(1) | 120.200 | N3-C18-C19 | 105.323 | 106.1(2) | 105.145 | H29-28-C30 | 120.909 | 120.0 | 118.370 |
| C22-N5-40 | 117.682 | 119(1) | 119.180 | C17-C18-C19 | 131.777 | 131.2(2) | 132.014 | O2-C30-C28 | 124.493 | 125.1(2) | 115.593 |
| N5-N6-C23 | 117.036 | 115.3(2) | 116.855 | C18-C19-H20 | 128.099 | 127.5 | 128.264 | O2-C30-C31 | 115.918 | 115.3(2) | 124.773 |
| H8-C7-C9 | 119.906 | 119.7 | 119.137 | C18-C19-C21 | 105.266 | 105.2(2) | 105.164 | C28-C30-C31 | 119.588 | 119.6(2) | 119.630 |
| H8-C7-C17 | 119.466 | 119.6 | 120.221 | H20-C19-C21 | 126.601 | 127.5 | 126.539 | C30-C31-H32 | 118.762 | 120.1 | 121.177 |
| C9-C7-C17 | 120.620 | 120.7(2) | 120.620 | N4-C21-C19 | 111.506 | 111.8(2) | 111.618 | C30-C31-C33 | 119.950 | 119.9(2) | 119.391 |
| C7-C9-H10 | 119.611 | 120.2 | 119.425 | N4-C21-C22 | 122.247 | 119.2(2) | 122.269 | H32-C31-C33 | 121.287 | 120.1 | 119.428 |
| C7-C9-C11 | 120.329 | 119.7(2) | 120.331 | C19-C21-C22 | 126.246 | 128.9(2) | 126.102 | C25-C33-C31 | 121.250 | 121.4(2) | 121.674 |
| H10-C9-C11 | 120.057 | 120.1 | 120.240 | O1-C22-N5 | 125.074 | 124.1(2) | 124.798 | C25-C33-H34 | 119.513 | 119.3 | 119.333 |
| C9-C11-H12 | 120.225 | 119.9 | 120.253 | O1-C22-C21 | 121.922 | 121.4(2) | 121.780 | C31-C33-H34 | 119.236 | 119.3 | 118.991 |
| C9-C11-C13 | 119.574 | 120.2(2) | 119.507 | N5-C22-C21 | 113.003 | 114.4(2) | 113.420 | O2-C35-H36 | 105.815 | 109.5 | 105.926 |
| H12-C11-C13 | 120.199 | 119.9 | 120.239 | N6-C23-H25 | 121.076 | 119.0 | 121.719 | O2-C35-H37 | 111.336 | 109.4 | 11.543 |
| C11-C13-H14 | 120.114 | 120.0 | 120.106 | N6-C23-C25 | 122.375 | 121.9(2) | 121.090 | O2-C35-H38 | 111.322 | 109.5 | 111.553 |
| C11-C13-C15 | 120.221 | 120.1(2) | 120.321 | H24-C23-C25 | 116.548 | 119.1 | 117.121 | H36-C35-H37 | 109.373 | 109.5 | 109.319 |
| H14-C13-C15 | 119.659 | 120.0 | 119.570 | C23-C25-C26 | 122.225 | 122.9(2) | 122.143 | H36-C35-H38 | 109.386 | 109.5 | 109.276 |
| C13-C15-H16 | 119.187 | 119.9 | 119.969 | C23-C25-C33 | 119.605 | 118.9(2) | 119.757 | H37-C35-H38 | 109.519 | 109.4 | 109.142 |
| RMSD | 1.3498 | – | 12.2267 |
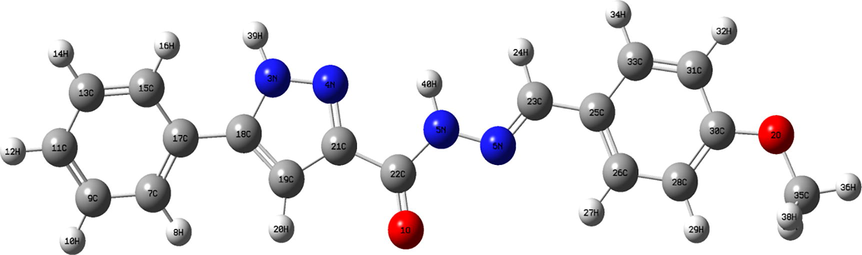
- Optimized structure of N'-(4-Methoxybenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide compound.
3.2 Electron difference density (EDD) analysis
In order to estimate the bonding character between atoms, the EDD technical was conducted. The electron distribution overlapping and electronic density accumulation between atoms display the covalent type of bonding. Whereas, the transfer of electron and the detachment of electronic density distribution show the ionic bonding (Zhang et al., 2018). The EDD surface is also used to understand the electronic charge transition in crystal structure of compound under investigation. The direction of electronic charge transfer is from electron-surplus (nucleophilic sites) areas to electron-deficient (electrophilic sites) areas. To think about the electron reorganization due to the formation of chemical bonds, the EDD map between ground and excited state was drawn in Fig. 2. In intramolecular charge transfer, the nucleophilic region represented on orange correspond to increased electron density. While, the electrophilic regions with increased electron density are shown by blue color. The blue and orange areas contribute to the accumulation and reduction of charge density. As clearly seen from EDD plot, the orange surfaces around oxygen and azote atoms demonstrate the strong electronegativity of these atoms. Therefore, they participate as electron donor in bonding formation. Then, the presence of blue areas at hydrogen atoms of pyrazole and phenyl rings indicate electron density depletion. For benzene ring and carbohydrazide chain, the H atoms with orange surfaces show the accretion of charge density. This fact proves the ability of H-bonding interactions formation in MBPPC crystal arrangement.
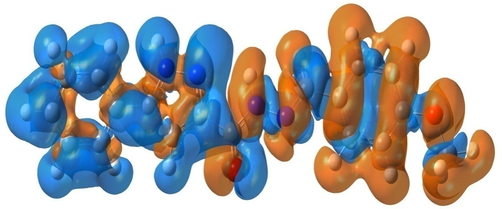
- Electron difference density map of N'-(4-Methoxybenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide compound.
3.3 AIM study
In order to discover the existence of the various interactions in crystalline structure via toplogical analysis, Bader has used the atom in molecule (AIM) theory (Sagaama et al., 2020). Thus, the topological analysis of MBPPC was performed, the AIM graphs generated by Multiwfn program are provided in Fig. 3. The topological parameters at Bond critical point (BCP): electron density ρ(r), Laplacian value Δρ(r), the eigenvalue (λ1,2,3), Hessian matrix H(r), electrostatic potential V(r) and interaction energy Eint are illustrated in Table 2. In this section we focused on non-covalent interactions present in dimeric structure. Table 2 reveals the existence of five hydrogen bond types. For MBPPC, the density ρ and Laplacian charge density values of these hydrogen bonding interactions ranges respectively between 0.0017 and 0.01 and 0.0075–0.0356 a.u. The positive sign of Laplacian Δρ(r) disclose the electronic load depletion at the level of interatomic path. In addition, the C23-H24…O41 andN5-H39…O41 H-bonds considered the powerful interactions compared to the other ones with 18.11 and 18.90 kJ/mol values, respectively. According to interaction energy value, the increasing order of these H-bonds is C53-H54…O2 < C59-H60…C25 < N3-H40…C66 < C23-H24…O41 < N5-H39…O41. As clearly seen from dimer conformation, the oxygen atom O41 is related to two hydrogen atom (H24 and H39) forming two type of H-bond (N–H…O, C–H…O). From AIM plot, we notice the presence of 3 ring critical points (RCP) in the monomer while in dimer there are 6 RCPs as well as the training of 9 new ring critical points (NRCP). In addition, Fig. 3 show the presence of intra-molecular interaction such as N45-H80…N44 and intermolecular interaction different from H-bond of C-N…N nature.
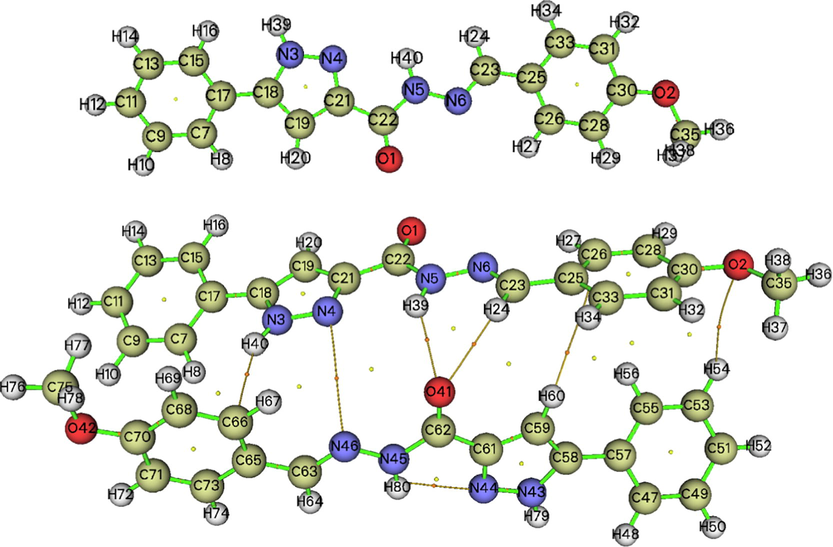
- AIM graphs of monomeric and dimeric structures mapped using Multiwfn package.
| Interactions | ρ(r) (a.u) | Δρ(r) (a.u) | λ1 | λ2 | λ3 | G(r) (a.u) | H(r) (a.u) | V(r) (a.u) | Eint (kJ.mol−1) |
|---|---|---|---|---|---|---|---|---|---|
| N3-H40…C66 | 0.0087 | 0.0287 | −0.0076 | 0.0434 | −0.0071 | 0.0059 | 0.0012 | −0.0047 | 12.33 |
| N5-H39…O41 | 0.0095 | 0.0356 | −0.0091 | 0.0540 | −0.0099 | 0.0080 | 0.0008 | −0.0072 | 18.90 |
| C23-H24…O41 | 0.0100 | 0.0354 | −0.0095 | 0.0542 | −0.0092 | 0.0079 | 0.0009 | −0.0069 | 18.11 |
| C59-H60…C25 | 0.0047 | 0.0149 | −0.0032 | 0.0193 | −0.0011 | 0.0029 | 0.0008 | −0.0020 | 5.25 |
| C53-H54…O2 | 0.0017 | 0.0075 | −0.0005 | −0.0012 | 0.0093 | 0.0013 | 0.0005 | −0.0008 | 2.10 |
3.4 Reduced density gradient analysis
Molecular stability is ensured by inter and intramolecular interactions, reduced density gradient (RDG) analysis was carried out to visualize these non-covalent interactions (NCI). This method can identify the NCI by using the
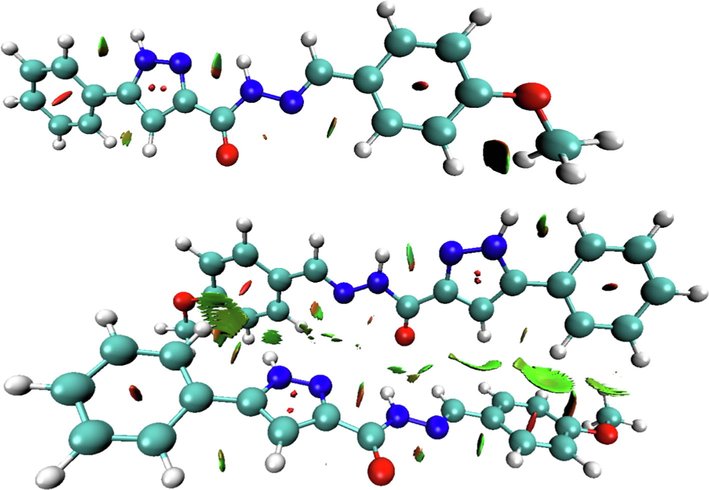
- VMD plots of non covalent interactions existing in the monomer and dimer structures of our molecule.
3.5 Hirshfeld surface
Hirshfeld surfaces analysis is a practical technical to explore the construction of supramolecular assembly (Ramalingam et al., 2020). The hydrogen bonding interactions and neighboring contacts are discovered by making Hirshfeld surface studies. The non-covalent interactions were estimated using 3D Hirshfeld surfaces and 2D fingerprint plot which mapped by crystal explorer 3. 1 software. These surfaces were plotted starting from the input file CIF (Crystallographic Information File). The 2D dimensional surfaces that have been plotted over dnorm, de, di, shape index, curvedness are provided in Fig. S3. The dnorm surface was generated over distance range from −0.6182 to 1.4971 Å. The di and de surface with rescale surface property varies between 0.7313 and 2.7380 Å. The shape index ranging from −1 to 1 Å, whereas the curvedness ranging between −4 and 4 Å. Red-white-blue is the color scale of dnorm map. The red is an indicator of shorter contacts, white color denotes van der Waals interactions while the blue shows the longer intermolecular contacts. The di and de image make out the hydrogen donors and hydrogen acceptors via red circles. The red triangle illustrated in shape index map as well as the green flat surface limited by blue contour in curvedness surface point to π-π stacking interactions. For clear visualization of intermolecular interactions types, dnorm surface of crystalline molecular arrangement was plotted in Fig. 5. This figure show the presence of six categories of hydrogen bond which are N-H…O, N-H…C, N-H…N, C-H…N, C-H…C and C-H…O. The red spots color intensity gives an idea on intermolecular interaction strength. In this context, the most powerful hydrogen contacts are N-H…O, N-H…C and N-H…N.
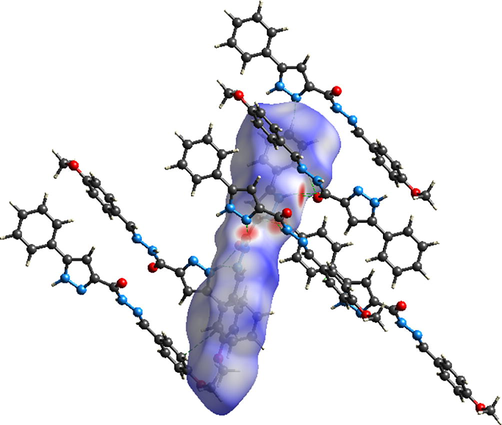
- The intermolecular interactions in crystalline molecular arrangement.
To identify the directional interaction, the two-dimensional fingerprint plots for reciprocal contacts and their percentage from the total surface. In 2D fingerprint plots, the contours of total print and by element are illustrated respectively by grey and blue regions. As clearly seen from Fig. S4, the O…H/H…O (1.8 Å) interaction appear as symmetrical wings. The H…H (35.6%) and C…H/H…C (33.5%) are the highest interactions due to the presence of abundance hydrogen and carbon atoms on the surface. The weaker contributions are associated to O…N/N…O (0.2%), O…C/C…O (0.8%), N…N (0.5%), N…C/C…N (2%) and finally C…C (0.3%). The other contacts: O…H/H…O and N…H/H…N have percentages equal to 15.2% and 11.7%, respectively.
3.6 HOMO-LUMO and DOS analysis
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular (LUMO) considered the most popular frontier orbitals. HOMO represent the nucleophile that gives electron while LUMO is considered as electrophile which accept electron. Energy difference between HOMO-LUMO orbitals determine the energy gap, as illustrated in Fig. S5. The gap energy gives information about chemical reactivity of a molecule (Bhavani et al., 2015). The weaker gap energy value characterizes the strong reactivity of the title compound. The red color represents the positive phase and the negative phase is colored by the green. The ELUMO = −1.86642 eV; EHOMO = −6.01916 eV and EHOMO-LUMO is equal to −4.15274 eV. Starting from HOMO and LUMO energies, the ionization potential, electron affinity, electronegativity, chemical hardness, chemical potential are calculated and listed in Table S1. The gap energy value of dimer (−3.6245 eV) was lower than that of monomer (−4.1527 eV), the dimeric conformation was comparatively more reactive.
To full molecular orbital description, the density of state (DOS) approach was carried out. It gives the orbital population contribution of each entity to the total compound. TDOS, PDOS, OPDOS are respectively the total density of state, partial density of state and overlap density of state. TDOS provide the molecular orbital population of compound under investigation. Whereas, PDOS and OPDOS consists in dividing the compounds into fragments to know the contribution of each group and to predict the nature of interactions between each other. In addition, OPDOS is a helpful tool to discovering the type of contacts between orbitals, atomic groups or atoms. To plot PDOS and OPDOS per fragment, our compound is divided to four groups; methoxybenzylidene (fragment 1), phenyl (fragment2), pyrazole (fragment 3) and carbohydrazide (fragment 4). The Fig. S6 demonstrate the variation of OPDOS (right side) and TDOS as well PDOS (left side) in term of energy (a.u). The vertical dashed line in this figure corresponds to the highest occupied molecular orbital (HOMO) with energy equal to 0.22 a.u. Ranging from −0.35 to −0.50 a.u, the TDOS spectrum show the presence of two intense peaks. According to band intensities of PDOS diagrams, the increasing order of contribution is; pyrazole < carbohydrazide < phenyl < methoxybenzylidene. The methoxybenzylidene groupment predominantly provide to the molecular orbitals. Based on orbital overlap sign, the OPDOS technical classified the interactions on bonding (positive value), anti-bonding (negative value) and non-bonding (zero value) (Muthu et al., 2013). From OPDOS diagram, we can say that the interaction between the four fragments had non-bonding nature. Thus, this spectrum illustrates the non-bonding character of HOMO and LUMO orbitals.
3.7 First and second nonlinear optical properties
Nowadays, the non-linear optical (NLO) compounds are extremely used in optoelectronic and photonic field manufacturing (Govindharajan et al., 2020). They are applied in optical switching and communication as well as information storage. The purpose of this part is to discover the non-linear optical properties of the studied compound. The dipole moments μ, the average polarizability α, the anisotropy of the polarizability Δα, and the first hyperpolarizability β, and the second hyperpolarizability γ of MBPPC are tabulated in Table S2 in comparison to those of urea. The diagram in Fig. S7 shows the nonlinear optical parameters (μ, α, β, and γ) of both systems. The dipole moment of the urea is higher than MBPPC by 0.51 Debye. The component dipole moment μy = 1.2 Debye possess the largest contribution to the total electric moment. Then, the isotropic alpha polarisability of MBPPC (44.34 10−24esu) was more than eight times larger than urea (5.09 10−24esu). The anisotropy of linear polarizability Δα(0;0) of MBPPC and Urea were equal to 50.89, 2.03 10−24esu, respectively. Regardless of direction, the α(−w;w) with 1064.6 nm laser pulsation value are 45.71(MBPPC)/ 5.13 10−24esu (Urea). The linear polarisability anisotropy of Δα(−w;w) is 54.11 10−24esu for compound under investigation and 2.05 10−24esu for the compared molecule. Take into account the laser pulsation, the total polarisability and the anisotropy of lineair polarisability values show a little increase compared to parameters without consideration of pulsation. Therefore, the first hyperpolarisability of title molecule is almost 45 times higher than that of urea. The highest first hyperpolarisability value is directly related to intramolecular electronic charge transfer ability emerged from donor to acceptor groups throughout π conjugated contacts (Romani et al., 2020). Under electric field application effect, the first dipole hyperpolarizability β (−w;w,0)|| parallel component to dipole orientation is 3.6 10−30esu for MBPPC and 0.02 10−30esu for urea. This parameter is a measure of a molecule to act as an electrooptical Pockels effect environment, and the title molecule is a good candidate in this sense. In addition, the second harmonic generation SGH impact allow to rise the first dipole hyperpolarisability β (−2w;w,w)|| which range at 31.99 10−30esu (MBPPC) and 0.41 10−30esu (urea). The electric field induced SHG augment the second hyperpolarisability value with 40.5 10−30esu (MBPPC)/0.20 10−30esu (urea) compared to first order β NLO. The second-order NLO γ value of the studied molecule is 52 times higher than that of reference system (urea). Second dipole hyperpolarisability γ(−2w;w,w,0) value with electro-optical Kerr effect (EOKE) are equal to 362.87 and 4.11 10−30esu for MBPPC and urea, respectively.
From this analysis, we can deduce that the first and second order NLO parameters are significantly influenced by the application of electric field. Based to calculation results of polarisability also first and second-order hyperpolarisability, the studied compound could be a good candidate for non-linear optical properties.
3.8 Vibrational studies
The MBPPC molecule is formed from 40 atoms, so it possess 114 (3N-6) normal modes. The vibrational assignments of studied compound has been determined using Veda4 program. Table S3 displays the recorded and computed wavenumbers obtained by the hybrid functional B3LYP and 6-311+G(2d,p) basis set. In vibrational theoretical spectrum, there are no imaginary frequencies so the optimized structure of MBPPC possess the lowest potential energy. In Fig. S8 (a), the experimental infrared spectrum of the title compound in solid state was provided ranging from 500 to 4000 cm−1. The theoretical IR and Raman spectra were illustrated in Fig. S8 (b) and S9, respectively. The FT-IR spectra can be divided into two parts: 500–1750 cm−1 and 1750–4000 cm−1. As shown in Fig. S8, a sensible agreement between the experimental and theoretical spectra in the first region (500–1750 cm−1). Whereas, in the second one (1750–4000 cm−1) there are a difference between them. This deviation can be explained by the fact that the calculated and observed spectra were obtained respectively in gas phase and solid state. In the present work, the calculated wavenumbers are scaled, the scaling factors is 0.958 for frequencies higher than 1700 cm-1 while frequencies weaker than 1700 cm−1 are multiplied by 0.983.
3.8.1 N–H and N-N vibrations
The peaks assigned to N–H stretching vibration range normally between 3500 and 3300 cm-1 (Karrouchi et al., 2020). The absorption bands at 3496 and 3339 cm−1 are consistent with the N–H stretching modes of pyrazole (N3-H39) and hydrazonoic (N5-H40) group, respectively. As clearly seen from Fig. S8 and Table S3, the IR intensity of νN3-H39 is five times more intense than that of νN5-H40. The νN-H mode for our compound is observed at 3014.77 cm−1 is probably associated to N3-H39 interaction. In addition, the N-N stretching mode matching to pyrazole and carbohydrazide groups were expected and recorded respectively in the ranges 1215–1080 cm−1 and 1056–1032 cm−1. The finding values are in good agreement with those reported for pyrazole derivatives (Özkınalı et al., 2018). The most intensive HNN bending mode in IR spectra are predicted at 1543 cm−1 and recorded at 1564 cm−1 with intensity higher than 335 Å4 (a.m.u)−1. The other δHNN vibrations are expected at 1449 and 1419 cm−1 with weak intensity in FT-IR spectrum unlike in Raman.
3.8.2 C-N vibrations
C-N stretching vibration of the title compound are assigned at 1394 and 1419 cm−1 and observed in the range 1406 cm−1. The C=N stretching vibration are experimentally assigned at 1313 cm−1 and predicted at 1639, 1363 cm−1 in FT-IR spectrum. As clearly seen in Raman spectrum (Fig. S6), the υC=N vibration appointed at 1639 cm−1 correspond with intense absorption peak. The C=N stretching modes associated to pyrazole and hydrazonoic groups were observed at 1593 and 1547 cm−1 for (E)-N′-(2,4-dichlorobenzylidene)-5-phenyl-1Hpyrazole-3-carbohydrazide (E-PPC) (Karrouchi et al., 2020). For MBPPC, the in plane CCN bending vibration are presented in 600–73 cm−1 range of frequency. While δCNN deformation are expected between 1394 and 968 cm−1. The torsion modes which involve nitrogen atoms of carbohydrazide chain such as τCCNN and τNCCN are obtained in a low frequency range, between 267 and 14 cm−1.
3.8.3 C–H vibrations
The aromatic molecules generally give rise to several weak peaks in the area 3200–3000 cm−1 (Noureddine et al., 2020), it correspond to C–H stretching modes. These bands are visualized in Raman spectra better than in FT-IR spectra. For MBPPC, four bands located at 3071, 3057, 3055 and 3025 cm−1 are assigned to C–H stretching vibration of benzene ring. The C–H stretching modes occur in the region 3058–3026 cm−1 are associated to phenyl ring. The νC23-H24 vibration of carbohydrazide group is assigned to the band exhibited at 2284 cm−1. Concerning the methoxy group which has three powerful C–H stretching vibration in Raman spectra with value almost equal to 3000 cm−1. Then, the Raman absorption peak at 3274 cm−1 correspond to the vibrational νC-H mode of pyrazole ring, which is in correlation with E)-N′-(2,4-dichlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide (Karrouchi et al., 2020). The second forceful peak located at 1215 cm−1in theoretical spectra is related to the in plane HCC deformation with high intensity value in IR and Raman spectra. This peak was recorded at 1169 cm−1 in experimental FT-IR. Another powerful absorption band observed at 1512 cm−1 assigned to δHCC with 1517 cm−1 calculated value. However, the τHCCC torsion and γOCCC out of plane deformation modes are coupled at the calculated frequency 829 cm−1 with 827 cm−1 experimental values.
3.8.4 C–C vibrations
Generally, the aromatic C=C and C–C stretching modes range between 1650 and 1200 cm−1 (Karrouchi et al., 2020; Iramain et al., 2020). In our case, the region ranging from 1616 to 1566 and 1309–1232 cm−1 show a coupling between C–C and C=C stretching modes of benzene and phenyl as well as pyrazole rings. The most intensive band in Raman spectrum with wavenumber equal to 1613 cm−1 is assigned as υC=C of benzene ring with contribution 62%. Experimentally, the νC-C vibration is observed at 1603, 900 and 848 cm−1. In the region 1467–1394 cm−1, some νCC modes are coupled with other in plane bending vibrations like δHCC and δHNN. For the MBPPC molecule, the δCCC deformations modes appear with medium frequencies and low intensity value. The C–C out of put vibrations appears as three weak absorption bands at 320, 309 and 93 cm−1. Then, the C–C torsion modes occur in the region 724–395 cm−1 and observed 767 cm−1, almost coupled with other vibrations.
3.8.5 C-O vibration
The C=O stretching mode of pyrazole derivatives appear generally in the range 1800–1600 cm-1 (Özkınalı et al., 2018; Karrouchi et al., 2020). The frequencies 1674 and 1259 cm−1 coincide respectively with two intensive bands in FT-IR and Raman spectra, they are assigned to νOC of carbohydrazide chain (C22=O1) and methoxy groupment (C35-O2). The experimental values of these vibrations are respectively 1656 and 1250 cm−1. The peak identified at 526 cm−1 are assigned to C-O in bending vibration with percentage equal to 51%. The following modes; τCCOC and τCCCO are predicted in lowest range of frequency. The OCCC out of plane bending deformations of the compound under investigation occur at 529 and 724 cm−1.
3.9 NMR spectral analysis
In order to identify the reactive organic and ionic species, the chemical shifts are habitually employed. NMR spectroscopy has considered as a convenient tool to clarify the molecular structure of compound under investigation. Therefore, structural optimization of the title compound were carried out by B3LYP/6-311G+(2d,p) level. The 1H and 13C isotropic chemical shifts were calculated by using Gauge-independent atomic orbital (GIAO) method with respect to TMS. The recorded and calculated 1H and 13C NMR spectra of MBPPC are illustrated in Fig. S10. The generated 1H and 13C NMR spectra of monomer and dimer conformation were depicted in Fig. S11. The 1H and 13C observed and computed chemical shifts of monomer and dimer are tabulated in Table 3. As plainly seen from this table that the two hydrogen atoms H39 and H40 bonded to the nitrogen atoms N3 and N5 which forms the N–H bond have the largest isotropic shift. For phenyl protons (H16, H14, H12, H10, H8), the chemical shifts observed in the range 7.458–7.830 ppm. While the calculated value are ranging from 7.76 to 8 ppm. The chemical shifts of H20 and H39 of pyrazole ring appear at 7.216 and 13.803 ppm, and computed at 7.31 and 10.02 ppm, respectively. Concerning the carbohydrazide chain, the shifts values of H24,40 are experimentally founded to be 11.741 and 8.097 ppm and theoretically are equal to 9.95 and 8.19 ppm. The methyl proton (H36,37,38) possess the weaker chemical shifts, the theoretical values were predicted at 4.06 and 3.84 ppm. Experimentally they were observed in the range 2.482–3.783 ppm. Besides, the 13C chemical shifts are recorded in the range −5–225 ppm. The major deviation between experimental and theoretical 13C shifts is noticed for C31 with −80.37 ppm. The signal predicted and recorded in the interval 168.72–113.83 ppm and 40.203–161.263 ppm, respectively, are assigned to aromatic carbon atoms of phenyl and benzene rings. The peaks occurred at 148.026, 147.272, 39.927 are associated respectively to C21, C18, C19 of pyrazole ring. While, the signals seen at 158.496 and 144.158 ppm correspond to C22 and C23 carbon atoms. Comparing the isotropic chemical sifts of monomer and dimer conformation, there is a lowering of the chemical shifts values especially for the carbon atoms that the protons.
| δ theoretical (ppm) | |||
|---|---|---|---|
| Monomer | Dimer | Exp. | |
| Atom | TMS B3LYP/6–311 + G (2d,p) GIAO | TMS B3LYP/6–311 + G (2d,p) GIAO | |
| C30 | 168.72 | 11.75 | 161.263 |
| C22 | 161.15 | 19.20 | 158.496 |
| C21 | 154.28 | 27.93 | 148.026 |
| C 18 | 151.84 | 31.02 | 147.272 |
| C 23 | 148.92 | 29.97 | 144.158 |
| C 33 | 136.68 | 43.31 | 127.447 |
| C 17 | 134.58 | 47.49 | 125.818 |
| C 9 | 133.85 | 47.83 | 127.447 |
| C 11, C 13 | 133.73 | 48.29, 47.88 | 125.818 |
| C 25, C 7 | 131.81 | 46.82, 53.78 | 114.799 |
| C 26 | 130.80 | 50.59 | 103.871 |
| C 15 | 128.95 | 50.41 | 55.755 |
| C 31 | 120.86 | 67.85 | 40.482 |
| C 28 | 113.83 | 58.33 | 40.203 |
| C 19 | 107.21 | 74.85 | 39.925 |
| C 35 | 55.79 | 126.20 | 39.370 |
| H39 | 10.02 | 10.38 | 13.803 |
| H 40 | 9.95 | 9.29 | 11.741 |
| H 27 | 8.60 | 8.72 | 8.445 |
| H 24 | 8.19 | 9.40 | 8.097 |
| H 8 | 8.00 | 6.27 | 7.830 |
| H 16 | 7.87 | 7.90 | 7.805 |
| H 14, H 10 | 7.81 | 7.80, 7.58 | 7.663, 7.636 |
| H 12 | 7.76 | 7.71 | 7.458 |
| H 34 | 7.58 | 8.13 | 7.434 |
| H 20 | 7.31 | 7.29 | 7.216 |
| H29, H32 | 7.21 | 7.50, 7.38 | 7.015, 6.986 |
| H36 | 4.06 | 4.31 | 3.783 |
| H37, H38 | 3.84 | 3.91, 4.02 | 2.488, 2.482 |
3.10 Molecular docking calculation
3.10.1 MBPPC as anti-microbial agent
Cholera is a contagious epidemic enteric infection caused by a strictly human bacterium “Vibrio cholerae”. This latter is an incurved mobile bacterium inhabited in water has a great resistance to survive in the environment. The intestinal toxi-infection transmitted by ingestion of infected food and water or by direct faecal-oral way. The principal drug used in the treatment of this microbial bacterium is Bactrim. Looking at the sensitivity of certain patients against this type of drug and also in order to have an alternative of Bactrim, molecular docking analysis was carried out. In the current section, we perform a comparative analysis between the useful drug “Bactrim” and the candidate compound “MBPPC” based on binding affinities values. Docking results of both ligands with 5UL7 (Vibrio cholerae) protein were collected in Table S4. This Table display the small difference in binding score between the two ligands which equal to −0.77 kcal/mol. For MBPPC, the interaction energy is equal to −114.11 kcal /mol. The VDW and H-bond interactions are found to be −101.27 and −12.84 kcal/mol, respectively. Concerning Bactrim, the total energy, H-bond and VDW interactions values are found to be respectively −114.88, −81.54 and −33.34 kcal/mol. However, the most stable poses of MBPPC and Bactrim in protein sites are pictured in Fig. 6a. According to this representation, the applicant molecule is located in the protein center while the Bactrim is placed at the edge. Then, Fig. 6b show the interactions established between MBPPC atoms and pocket atoms of residues. There are many categories of interactions indicated with discontinuous lines such as conventional hydrogen bond (green), π-alkyl (purple) and π-anion (mustard yellow). The following residues: Leu 250, Ala 125, Arg 252 forms three π-alkyl interactions with benzene ring. The pyrazole ring works together with Ala 429, Phe 428 and Val 255 to form respectively π-alkyl, conventional hydrogen bond and π-lone pair interaction. Similarly, the π-anion interaction was created among Asp 121 residue and benzene ring. The obtained close binding value between MBPPC and Bactrim along with the numerous weak interactions established among our compound and amino acid residues demonstrate the significant capacity of our compound to be an anti-microbial drug against Vibrio cholerae.
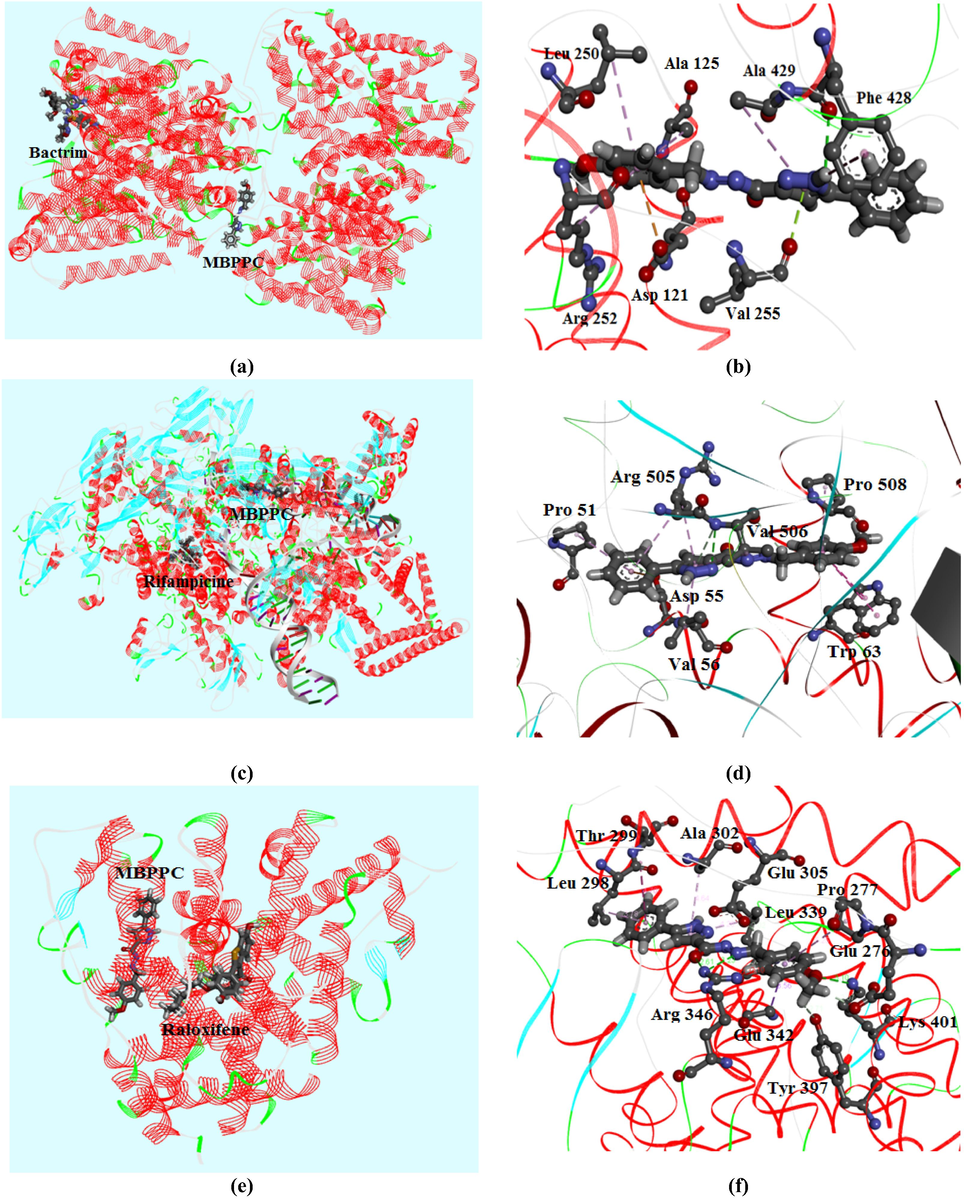
- Best poses of MBPPC, Bactrim, Rifampicine and Raloxifene in each protein along with the different interactions between ligand atoms and amino residues.
3.10.2 MBPPC as Anti-tubercular compound
The tuberculosis is a contagious illness generated by human pathogenic bacterium Mycobacterium tuberculosis which belong to Mycobacteria family, generally attacks the lungs. At times it can affect other members like the bones kidneys and lymph nodes. This disease touch both children and adults and spread through the air. A rigorously followed treatment using antibiotics can be effective way to treat this sickness. Despite the effectiveness of antibiotics such as Rifampicine in treating most cases of this malady, thousands of people die of tuberculosis annually. Therefore, several investigations were carried out in order to obtain more reliable treatment than antibiotics, cures the patient at any stage of the disease. In our case, docking study of MBPPC with Mycobacterium tuberculosis (6VW0) was conducted to examine the anti-tubercular agent of the title compound. Docking results are tabulated in Table S4, illustrating the comparison between 6VW0-MBPPC and 6VW0-Rifampicine complexes in terms of interaction energy. In addition, Table S4 demonstrate the close value of binding scores of the compared complexes. The energetically difference between the two protein–ligand complexes is found to be −9 kcal/mol. The van der Waals interaction of MBPPC (−117.43 kcal/mol) is higher than that of Rifampicine (−103.67 kcal/mol). Whereas, the hydrogen bonding interaction of the title molecule (−10.45 kcal/mol) is weaker than the energy of the standard antibiotic (−33.40 kcal/mol). Then, the docked poses of MBPPC and Rifampicine in amino acids assembly are mapped in Fig. 6c. The non-covalent interactions in protein–ligand systems are plotted in Fig. 6d. The interaction Val 506-N4 is of conventional hydrogen bond nature. Likewise, the amino acids possess four π-alkyl bonds with benzene (Pro 508), pyrazole (Val 56, Arg 505) and phenyl (Pro 51) rings. Furthermore, the finding results prove the anti-tubercular effect of the studied molecule on front of Mycobacterium tuberculosis bacterium.
3.10.3 MBPPC as estrogen receptor modulator compound
The female sex hormone “estrogen” perform an significant function in female reproduction. It derives from steroid group synthesized by the ovaries. This ovarian hormone allows the progress of cardiovascular system and central nervous but it causes dangerous diseases for women such as breast and endometrial cancer. Thus, the moderation of the level of this hormone by using the modulator of estrogen receptors reduces the percentage of exposure to these diseases. The main estrogen receptors are estrogen alpha (ERα) and beta (ERβ) receptors which are intracellular proteins. The modulators of these receptors are compounds able to get linked to estrogen receptors demonstrating the inhibition of estrogen-receptor interaction. Arzoxifene, raloxifene are commonly used as estrogen receptor modulator, also it a useful drug for breast cancer chemoprevention. The goal of the present section is to research a new estrogen receptor modulator. For this reason, molecular docking calculation of MBPPC with estrogen receptor (5TAO) was performed to simulate the binding score and discovering the interactions into 5TAO-MBPPC complex. The finding results are compared then with those of estrogen receptor-raloxifene. The best positions of MBPPC and raloxifene in 5TAO protein site are depicted in Fig. 6e demonstrating the close poses of the two ligands. As clearly seen from Table S4, MBPPC and raloxifene have comparable binding score values which found to be −102.01 and −116.21 kcal/mol, respectively. From Table S4, the raloxifene interaction energy is totally van der Waals type. While the binding energy of the studied compound was divided to VDW interaction with 73.45 kcal/mol and hydrogen bonding with −28.56 kcal/mol. The electrostatic interaction energies of both complexes are equal to zero value due to the absence of electronic charge transfer from protein to ligand and vice versa. In addition, Fig. 6f show the different non-covalent contacts established between MBPPC atoms and amino acid residues of 5TAO protein. The Arg 346 residue form with N6 and O1 of carbohydrazide chain two conventional H-bond which drawn by green discontinuous lines. A carbon hydrogen bonding interaction (light green color) was founded between carbon atom (C35) of methyl group and Glu 276 amino acid. Also, the Ala 302, Leu 339 and Pro 277 present three π-alkyl (pink dashed lines) interactions with pyrazole and benzene ring, respectively. However, molecular docking results confirms the ability of our compound to be an effective estrogen receptor modulator minimizing breast cancer injury.
4 Conclusion
In the present investigation, the optimized molecular geometric, electronic and spectroscopic, molecular docking analyses of our molecule have been computed via DFT calculation using B3LYP functional. The structural bond lengths and angles of stable conformer were calculated. These parameters were compared with experimental data in term of RMSD indicating good agreement. The chemical reactivity of the title compound was obtained by plotting electron difference density (EDD) map. The π-π stacking interactions and various hydrogen bond types (N-H…O, N-H…C, N-H…N) which responsible for crystal stability were identified using Hirshfeld surface and topological analyses. The weak HOMO-LUMO gap energy also prove the electronic transition within the molecule. The non-linear optical parameters (first and second order) were calculated indicating that our system was much better than urea. The vibrational IR spectrum of MBPPC was recorded. The harmonic modes and frequency assignements were compared with experimental ones indicating good results. Besides, the isotropic chemical shifts of 1H and 13C were computed with respect of TMS and were compared to NMR experimental spectrum. The calculated isotropic chemical sifts demonstrate good results with recorded ones especially for hydrogen atoms. However, the high reactivity of MBPPC anticipate the strong interactions of ligand–protein complex. For this reason, molecular docking study was performed to discover the anti-microbial, anti-tubercular and receptor estrogen modulator on front of Vibrio cholera, Mycobacterium tuberculosis and estrogen, respectively. Compared to standard drugs, docking study verify the performance of our compound in the treatment of these tractable diseases.
Acknowledgements
Researchers supporting project number (RSP-2021/61), King Saud University, Riyadh, Saudi Arabia
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- (a) K. Bhavani, S. Renuga, S. Muthu. Quantum mechanical study and spectroscopic (FT-IR, FT-Raman, 13C, 1H) study, first order hyperpolarizability, NBO analysis, HOMO and LUMO analysis of 2-acetoxybenzoic acid by density functional methods. Spectrochim. Acta A Mol. Biomol. Spectrosc. 136 (2015) 1260-1268. (b) T.K. Kuruvilla, S. Muthu, J.C. Prasana, J. George, S. Sevvanthi, Spectroscopic (FT-IR, FT-Raman), quantum mechanical and docking studies on methyl[(3S)-3- (naphthalen-1-yloxy)-3-(thiophen-2-yl)propyl]amine, J. Mol. Struct. (2018).
- Discovery Studio 4.5 Guide. Accelrys Inc., San Diego.http://www.accelrys.com.
- Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem.. 2017;25:5891-5903.
- [Google Scholar]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratch, W.C., Gaussian16 Revision A.03, (2016).
- Synthesis, experimental, theoretical study and molecular docking of 1-ethylpiperazine-1,4-diium bis(nitrate) Solid State Sci.. 2020;106:106326
- [Google Scholar]
- GaussView, Guassian, Inc. (Carnergie Office Parck-Building6 Pittsburgh PA 151064 USA), Copyright © 2000-2003 Semichem. Inc.
- A new therapeutic approach in Alzheimer disease: some novel pyrazole derivatives as dual MAO-B inhibitors and anti-inflammatory analgesics. Bioorg. Med. Chem.. 2007;15:5775-5786.
- [Google Scholar]
- Spectroscopic characterization and DFT investigation of L-isoleucine D-Norvaline: a potential NLO crystal. Mater. Today: Proc. 2020
- [Google Scholar]
- Cost-utility analysis of rimonabant in the treatment of obesity. Value Health.. 2008;11:389-399.
- [Google Scholar]
- https://www.ccdc.cam.ac.uk/(CCDC-N° 1437241).
- J. Molec. Graphics. 1996;14:33-38.
- Spe1ctroscopic studies on the potassium 1-fluorobenzoyltrifluoroborate salt by using the FT-IR, Raman and UV–Visible spectra and DFT calculations. J. Mol. Struct.. 2020;1204:127534
- [Google Scholar]
- Computational study of 3-thiophene acetic acid: molecular docking, electronic and intermolecular interactions investigations. Comput. Biol. Chem.. 2020;2020:107268
- [Google Scholar]
- M.H. Jamróz, Vibrational Energy Distribution Analysis, VEDA 4, computer program, Poland, 2004.
- New pyrazole-hydrazone derivatives: x-ray analysis, molecular structure investigation via density functional theory (DFT) and their high in-situ catecholase activity. Int. J. Mol. Sci.. 2017;18:2215.
- [Google Scholar]
- Synthesis, crystal structure, hirshfeld surface analysis, DFT calculations, anti-diabetic activity and molecular docking studies of (E)-N’-(5-bromo-2-hydroxybenzylidene) isonicotinohydrazide. J. Mol. Struct.. 2020;1221:128800
- [Google Scholar]
- Synthesis, structural, molecular docking and spectroscopic studies of (E)-N'-(4-methoxybenzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct.. 2020;1225:129072
- [Google Scholar]
- Synthesis, X-ray structure, vibrational spectroscopy, DFTinvestigation and biological evaluation studies of (E)-N'-(4-(dimethylamino)benzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct.. 2020;1219:128541
- [Google Scholar]
- Synthesis, X-ray, spectroscopy, molecular docking and DFT calculations of (E)-N’-(2,4-dichlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct 2020
- [Google Scholar]
- Synthesis and anticancer activity of acyl thioureas bearing pyrazole moiety. Bioorg. Med. Chem.. 2013;21:3859-3865.
- [Google Scholar]
- Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem.. 2012;33:580-592.
- [Google Scholar]
- Quantum mechanical, spectroscopic studies (FT-IR, FT-Raman, NMR, UV) and normal coordinates analysis on 3-([2-(diaminomethyleneamino) thiazol-4-yl] methylthio)-N′-sulfamoylpropanimidamide. Spectrochim. Acta A Mol. Biomol. Spectrosc.. 2013;108:307-318.
- [Google Scholar]
- (a) R. Nie, S. Stark, J. Symersky, R. S. Kaplan, M. Lu (2017) Structure and function of the divalent anion/Na+ symporter from Vibrio cholerae and a humanized variant Nat. Commun. 8: 1-10, (b) M. Lilic, J. Chen, H. Boyaci, N. Braffman, E. A. Hubin, J. Herrmann, E. A. Campbell (2020) The antibiotic sorangicin A inhibits promoter DNA unwinding in a Mycobacterium tuberculosis rifampicin-resistant RNA polymerase Proc. Natl. Acad. Sci. 117: 30423-30432, (c) P. C. Souza, L. C. Textor, D. C. Melo, A. S. Nascimento, M. S. Skaf, I. Polikarpov (2017) An alternative conformation of ERβ bound to estradiol reveals H12 in a stable antagonist position Sci. Rep. 7: 1-11.
- Structural, docking and spectroscopic studies of a new piperazine derivative, 1-Phenylpiperazine-1, 4-diium bis (hydrogen sulfate) J. Mol. Struct.. 2020;1202:127351
- [Google Scholar]
- Experimental and DFT studies on the molecular structure, spectroscopic properties, and molecular docking of 4-phenylpiperazine-1-ium dihydrogen phosphate. J Mol. Struct.. 2020;1207:127762
- [Google Scholar]
- DFT and molecular docking study of chloroquine derivatives as antiviral to coronavirus COVID-19. J. King Saud Univ. Sci.. 2021;33:101248.
- [Google Scholar]
- Synthesis of new azo schiff bases of pyrazole derivatives and their spectroscopic and theoretical investigations. J. Mol. Struct.. 2018;1174:74-83.
- [Google Scholar]
- (a) A. Ramalingam, S. Sivakumar, K. Rajkumar, J. P. Jasinski, M. Kaur, A. Thiruvalluvar,. Synthesis, Crystal Structure, DFT Calculations and Hirshfeld Surface Analysis of 3-Chloro-3-methyl-r (2), c (6)-bis (p-methoxyphenyl) piperidin-4-one. J. Chem. Crystallogr. 50 (2020) 41-51. (b) A. Ramalingam, Hirshfeld surface analysis, Interaction energy calculation and spectroscopical study of 3-chloro-3-methyl-r(2),c(6)-bis(p-tolyl)piperidin-4-one using DFT approaches J. Mol. Struct. (2021).
- Properties and reactivities of niclosamide in different media, a potential antiviral to treatment of COVID-19 by using DFT calculations and molecular docking. Biointerface Res. Appl. Chem.. 2020;10:7295-7328.
- [Google Scholar]
- Design, molecular docking analysis of an anti-inflammatory drug, computational analysis and intermolecular interactions energy studies of 1-benzothiophene-2-carboxylic acid. Comput. Biol. Chem.. 2020;88:107348
- [Google Scholar]
- Searching potential antiviral candidates for the treatment of the 2019 novel coronavirus based on DFT calculations and molecular docking. Heliyon. 2020;6:e04640.
- [Google Scholar]
- Molecular docking studies, structural and spectroscopic properties of monomeric and dimeric species of benzofuran-carboxylic acids derivatives: DFT calculations and biological activities. Com. Biol. Chem.. 2020;87:107311
- [Google Scholar]
- S.K. Wolff, D.J. Grimwood, J.J. McKinnon, M.J. Turner, D. Jayatilaka and M.A. Spackman, University of Western Australia (2012).
- GEMDOCK: a generic evolutionary method for molecular docking. Proteins Struct. Funct. Bioinforma.. 2004;55:288-304.
- [Google Scholar]
- Research on inhibitors and hindered groups in ultra-deep hydrodesulfurization based on density functional theory. Catal. Today. 2018;314:170-178.
- [Google Scholar]
Further reading
- Cation-anion interactions in 1- buthyl-3-methyl imidazolium nitrate ionic liquid and their effect on their structural and vibrational properties. J. Mol. Struct.. 2018;1164:1-14.
- [Google Scholar]
- Synthesis, vibrational spectra, DFT calculations, Hirshfeld surface analysis and molecular docking study of 3-chloro-3-methyl-2, 6-diphenylpiperidin-4-one. Spectrochim. Acta A Mol. Biomol. Spectrosc.. 2020;232:118166
- [Google Scholar]
- Synthesis, crystal structure, DFT calculations and Hirshfeld surface analysis of 3-chloro-2, 6-bis (4-chlorophenyl)-3-methylpiperidin-4-One. J. Chem. Crystallogr.. 2021;51:273-287.
- [Google Scholar]
- S. Seshadri, S. Gunasekaran, S. Muthu, S. Kumaresan, R. Arunbalaji. Vibrational spectroscopy investigation using ab initio and density functional theory on flucytosine. J. Raman Spectrosc.: An International Journal for Original Work in all Aspects of Raman Spectroscopy, Including Higher Order Processes, and also Brillouin and Rayleigh Scattering, 38 (2007) 1523-1531.
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2021.101778.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




