

Translate this page into:
Multispectroscopic and molecular modeling strategy to explore the interaction of cholest-5-en-7-one with human serum albumin: DFT and Hirshfeld surface analysis
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The present study accounts for cholest-5-en-7-one (steroid) synthesis using selenium dioxide and silica as well as its identification using high-resolution mass spectrometry (HRMS), FT-IR, NMR, and elemental analysis. Density functional theory (DFT) calculations were used to optimize the steroid's geometry, which was then compared to single-crystal X-ray diffraction data, indicating that the computational and experimental results were in good agreement. UV–vis absorption titration, fluorescence spectral methods, circular dichroism (CD), and dynamic light scattering (DLS) were used to investigate the well-identified steroid's in vitro molecular interaction with human serum albumin (HSA). The results and binding parameter indicate that the steroid has significant binding affinity for HSA. Molecular docking was performed and found that the steroid can interact with the hydrophobic cavity of HSA through hydrogen and hydrophobic contacts. Molecular dynamics simulation was used to confirm the stability of the HSA-steroid complex. Combined quantum mechanics-molecular mechanics (QM-MM) methods were applied to know binding energy of system and individual components for atomic level interactions indicating a significant interaction between the steroid and the biomacromolecules. The findings presented here contribute to a better understanding of steroid pharmacodynamics.
Keywords
MD simulations
Steroid
Spectroscopy
DFT
Molecular docking

1 Introduction
The synthetic modification of steroids derived from various natural sources has resulted in the preparation and development of several diverse pharmacologically active, efficient, and highly specific commercially important therapeutic medicines (Li et al., 2020, Serrano-Vázquez et al., 2020, Borioni et al., 2021, Boutin et al., 2021, Kuzminac et al., 2021). Synthetic steroid aromatase inhibitors have been studied and reported in the literature in recent years, assisting medicinal chemists in the development and synthesis of new powerful aromatase inhibitors for the treatment of breast cancer (Varela et al., 2012). The structural diversity of steroidal substances is thought to be responsible for their pharmacological characteristics, according to research (Saikia et al., 2015, Zhao et al., 2021). As previously described, the structural diversity of steroidal substances is assumed to be responsible for their pharmacological properties. HSA is the most common negatively charged and highly soluble protein found in vertebrate blood plasma and accounts for 60% of the total protein in plasma. Recently, one of the most widely used methods for determining preferential binding sites and binding orientation of small molecules in target proteins such as HSA has been computational approaches (Meng et al., 2011, Ryde and Soderhjelm 2016). In the present study, the binding mechanism of cholest-5-en-7-one (steroid or steroidal ketone) with HSA was explored by various spectroscopic techniques followed by molecular docking, and the structural alterations of HSA derived from docking studies when ligand is linked to HSA binding sites were explored using a 100 ns molecular dynamics (MD) simulation. In addition, the binding energy of the HSA-steroid interaction at the atomic level was determined using a combined method QM/MM approach. In silico analysis which could provide supportive and comprehensive information about the steroid binding mechanism with HSA.
2 Experimental section
2.1 Preparation of steroidal ketone (2)
To a solution of cholest-5-ene (1) (1 mmol), SeO2 (0.5 mmol) and ButOOH (3 mmol) in dichloromethane (20 ml) were taken in round bottom flask equipped with condenser. After that, 1 g of silica was added, and the resulting mixture was stirred for 1 h under reflux. TLC was used to check the progress and purity. The reaction mixtures were cooled and filtered under suction to remove silica and selenium, then washed with dichloromethane after the reaction was completed as revealed by TLC. After removal of solvents and water is added to the residual mass. The organic layer was separated with ether, then washed thoroughly with water, followed by a 5% aqueous sodium bicarbonate solution, water, and finally anhydrous Na2SO4 before drying. The oil was obtained by evaporating the solvent under reduced pressure and chromatographing it over silica gel with light petroleum (60–80 °C). Elution and evaporation produced a solid that was recrystallized from methanol to produce crystals with a 75% yield. m.p.: 130 °C (2, Scheme 1) (published m.p. 125–129 °C, (Dauben and Takemura 1953, Khan et al., 2010, Mushfiq et al., 2010, Sarkar et al., 2017); Anal. calc. for C27H44O: C, 84.31; H, 11.53%. Found: C, 84.71; H, 11.51%; IR (KBr, cm-1): 1685 and 1621 cm-1 (α,β-unsaturated ketone); UV λmax(methanol) 239 nm; HRMS (ESI): calcd. for C27H44O [M + H]+: 384.3393; Found: 384.3399. Materials and Methods including Chemical shift of compound and its UV–Vis (Fig S1) are given in supplementary information.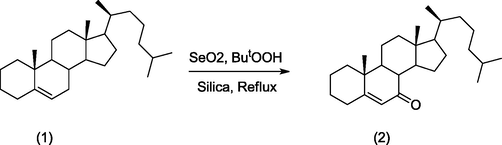
Synthesis of cholest-5-ene-7-one (2) from cholest-5-ene (1) using SeO₂.
2.2 HSA sample preparation
HSA was prepared as a stock solution in 20 mM phosphate buffer (PB) with a pH of 7.4, and its concentration was determined spectrophotometrically with a Perkin–Elmer–Lambda double beam UV–vis spectrophotometer using ε1cm% of 5.30 at 280 nm, and its purity was certified on SDS–PAGE with a single band. A 2 mM stock solution of cholest-5-en-7-one was prepared in small amount of Dimethyl Sulfoxide (in 5% DMSO) and the dilution was made in PB to obtain different concentration of compound. Blanks were 20 mM phosphate buffer (pH 7.4) at 298 K for each experiment. All of the pH readings were taken with an Orion-401-plus pH meter supported with an Orion glass electrode.
2.3 Procedures
UV–Vis absorption, fluorescence, circular dichroism spectrophotometric and Dynamic light scattering (DLS) techniques were applied to study HSA interactions with cholest-5-en-7-one. As interaction parameters, binding constants, binding stoichiometry, and binding modes were all investigated. The Supporting Information contains a detailed procedure.
2.4 Molecular docking
The approach of molecular docking is useful for simulating interactions between molecules and bio-macromolecules. The Lamarckian Genetic Algorithm (LGA) was exercised to undertake an in-silico study on the binding patterns of cholest-5-ene-7-one with HSA utilizing the AutoDock 4.2 program under AutoDockTools (ADT) (Morris et al., 1998, Morris et al., 2009). (Details can be found in the supplementary information).
2.5 Molecular dynamics simulation
Desmond (Schrödinger, LLC, New York, NY, USA) was used to simulate molecular dynamics for 100 ns at 300 K and 1 atm, as previously explained (Rehman et al., 2019). The best docked posed of the HSA-steroid complex with the lowest binding energy was opted as the complex's initial conformation for simulation. (See supplementary information for details)
2.6 Computational details
The density functional quantum chemical calculations on the molecular structure provided in asymmetric unit of crystal structure of cholet-5-ene-7-one (Scheme 1) in vacuuo and solvent phase were carried out by Gaussian 09 program using B3LYP and basis set 6-311G(d,p) (Frisch and Clemente 2009, Alam and Ahmad 2014). (See supplementary information for details)
3 Results and discussion
3.1 Chemistry and molecular geometry of the synthesized cholest-5-ene-7-one
Several methods have been documented for isolating pure steroidal ketone (2) using hazardous chemicals, including lachrymatory procedure, to synthesize steroidal ketone. In this study, steroidal ketone from cholest-5-ene (1) was synthesized using selenium dioxide, small amount of ButOOH, and silica gel to obtain a single product with minimal hazard chemicals, as opposed to the previously published technique (Khan et al., 2010, Mushfiq et al., 2010), which used maximum hazard chemicals and required a lengthy workup for isolation. The reported technique for synthesis of steroidal ketone utilizing tert-butyl alcohol, CrO3, acetic acid, and acetic anhydride is still useful in producing large quantities of the chemical (Dauben and Takemura 1953). The synthesized compound was identified using spectral techniques as given in the experimental section. The IR spectrum of steroidal ketone revealed characterized peaks at 1685 and 1621 cm−1 corresponding to alpha–beta-unsaturated ketone molecule, indicating that the product was successfully formed. Moreover, the melting point of the compound was 130 °C, which is nearly identical to what has been described in the literature (Dauben and Takemura 1953, Khan et al., 2010, Mushfiq et al., 2010, Sarkar et al., 2017). For a single crystal X-ray investigation, the synthesized ketone was effectively crystallized to produce good crystal quality; but, a single crystal X-ray study of steroidal ketone was found to be published in the literature (Khan et al., 2010).
Cholest-5-ene-7-one (2) crystallizes in the monoclinic system that has the space group P21 and 7 stereocenters. The asymmetric unit includes one molecule, whereas the unit cell contains two molecules that are bound together by non-bonding forces and oriented tail-to-tail as noted elsewhere (Khan et al., 2010). The Gaussian-09 software package was used to do DFT calculations to investigate the nature of the molecule via the electronic structure of steroidal ketone (2). Crystallographic Information File (CIF) was retrieved from CC Data Center (CCDC No# 1433642) and used as initial coordinates of the molecular structure for theoretical calculations.
The optimized structure for cholest-5-ene-7-one was computed at the level of DFT/B3LYP/6–311G(d,p) basis det and is given in Fig. 1(a) with the numbering scheme used. The XRD (Fig. 1(b) and DFT molecular structures of the compound were superimposed atom by atom (Fig. 1c; green-experiment; white-calculated), and the root mean square error (RMSE) for the B3LYP/6-311G(d,p) method was noticed to be 0.182. In the isolated gas phase geometry that contributes to the magnitude of the RMSE, intermolecular Coulomb interactions with adjacent molecules are not considered, so the side chain of the compound contributes most of the error. In contrast, the experimental result in the crystal lattice is correlated with interacting molecule (2) (See the Supplementary Table S1 for Comparison of selected optimized structural parameters with corresponding XRD data).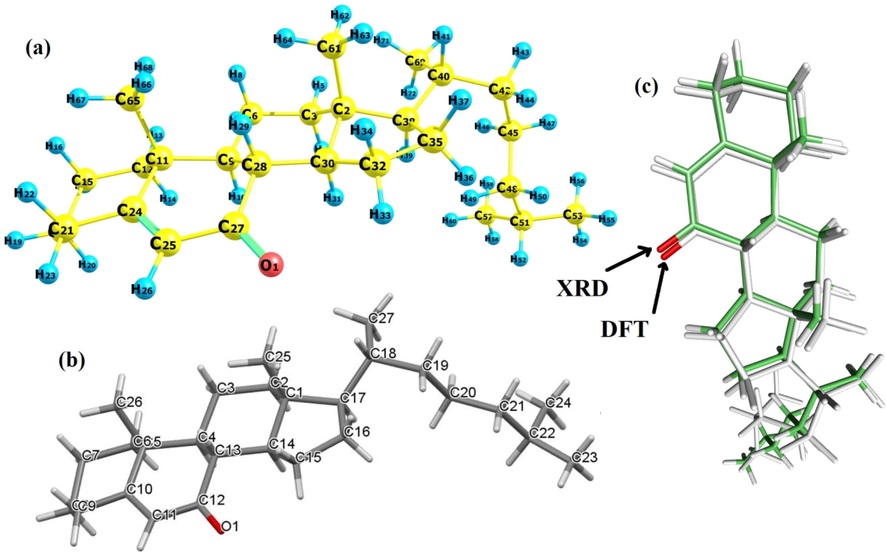
(a) Geometry optimization at B3LYP/6-311G(d,p) level, (b) XRD and (b) (C) Overlay of crystal structure and DFT calculated structure; RMSE 0.182 Å.
3.2 Frontier molecular orbital and molecular electrostatic potential (MEP)Analysis
The properties of molecules generally determined by molecular orbitals (MOs) are known as Frontier Molecular Orbitals. A molecule with a higher HOMO value is more likely to donate electrons to a suitable acceptor molecule with a low energy of the vacant molecular orbitals (Zarrouk et al., 2014). Molecular reactivity decreases as the energy gap between HOMO and LUMO widens, resulting in increased molecular stability and a sign of chemical hardness (Gil et al., 2016). The arrangement of HOMO and LUMO is crucial in determining which portion of the ligand is most suitable for receptor engagement. In Fig. 2a, HOMO and LUMO are predominantly delocalized over carbonyl system and a few steroid rings of compound (2).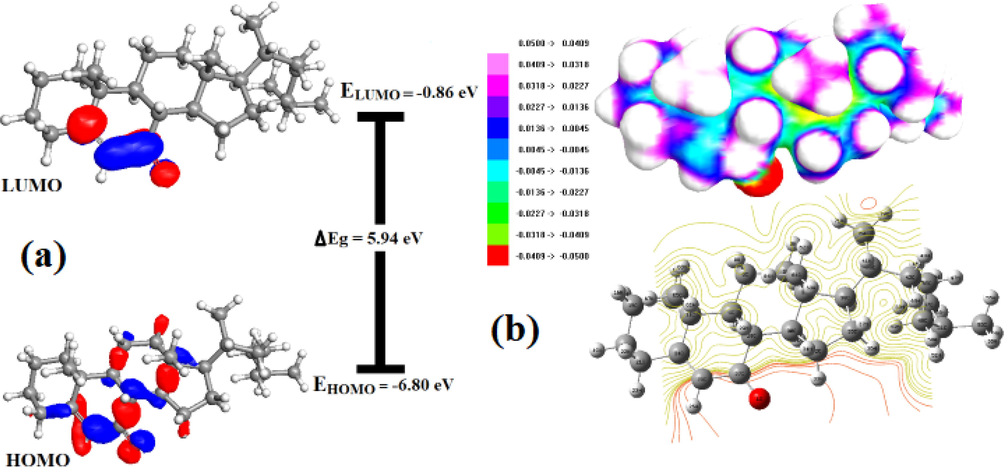
(a) Spatial plots of HOMO and LUMO and (b) MEP and contour plots for cholest-5-ene-7-one.
Fig. 2a depicts HOMO and LUMO plots, with positive and negative phases indicated by red and blue colors, respectively. The energies of EHOMO and ELUMO appear to be −6.80 and −0.86 eV, respectively, and the gap between HOMO and LUMO of 2 is 5.94 eV, which explains the strength and stability of cholest-5-ene-7-one (2) as well as the nature of the skeleton. According to surface inspection of HOMO and LUMO, AOs of the oxygen atoms of the carbonyl group at the 7-position of the steroid skeleton (C27-O1 in Fig. 1) lead to the creation of π – type HOMO and π∗ - LUMO. Steroid appears to be a thermodynamically stable species with little chemical activity due to the significant magnitude of FMOs with energy difference (5.94 eV). The ability of molecule electron donation can be assessed by referring to HOMO energy values, as increasing HOMO energy facilitates the oxidation process. The energies of the Frontier molecular orbital (FMO) and its band gap determines not only the conventional chemical reactions of certain types, but also the thermodynamic and chemical kinetic properties of the system under consideration. Thus, a variety of quantum mechanical reactivity descriptors given in Table S2 such as chemical hardness (η), chemical softness (S), electronegativity (χ), electronic chemical potential (μ), and global electrophilicity index (ω) can be calculated using HOMO energy, LUMO and their energy gap for a steroid (2) using the following equations (1–5) (Masroor et al., 2017, Garg et al., 2021).
≈ −1/2(I + A), where I represent -EHOMO and A for -ELUMO compared to ionization potential and electron affinity, respectively
According to the ionization potential of steroid in Table S2, an energy value of 6.80 eV is required to remove an electron from the HOMO. The lower Electron Affinity (0.86 eV) value shows that the compound (2) easily takes electrons to form bonds, indicating a higher molecular reactivity with nucleophilies. The higher hardness (2.97 eV) and lower softness (0.33 eV) data support the higher molecular hardness of molecule and imply low chemical reactivity with a potential net electrophilicity of 2.46 eV. Because the lower chemical potential values and higher Electrophilicity index are comparable to those of biologically active chemicals, therefore, naturally occurring steroid compounds or synthetic steroidal compounds are physiologically active in the therapeutic sector. The molecular electrostatic potential (MEP) (Fig. 2b) potentials formed in the space around a molecule by its nuclei and electrons were used to determine the behavior and reactivity of the molecules. In order to save space/pages, the MEP is explained in the supporting information, as directed by journal guidelines.
3.3 Molecular Hirshfeld surfaces analysis
The 3D Hirshfeld surface analysis of compound 2 (Fig. S2) is a useful way of evaluating the intermolecular and intramolecular interactions that occur in crystal packing. CrystalExplorer 21.5 was used to construct two-dimensional fingerprint plots based on a Hirshfeld surface study as explained in supporting data.
3.4 HSA binding properties
To the best of our knowledge, studies on steroid chemicals and HSA have rarely been published, and structural interactions have never been rigorously analyzed. The binding interactions of steroid and HSA must be studied in order to evaluate their biological activities at the molecular biological level.
3.5 UV–vis absorption studies
UV–Vis absorption is sensitive and has been used to monitor and examine protein–ligand complex formation as well as to investigate structural changes inside proteins as a result of complex formation (Zhang et al., 2008, Guo et al., 2009). The approach is based mostly on the extension of UV–vis light absorption by the sample and the protein molecule. Light is absorbed by the presence of aromatic moieties largely in tryptophan, tyrosine and phenylalanine. HSA is recorded at a maximum of 280 nm, which is normal for the three aromatic residues. Tryptophan, in particular, is extremely sensitive to changes in the microenvironment and is absorbed mostly at 280 nm. In general, ligand binding is followed by changes in the microenvironment to aromatic residues, which offer information on the phenomena of binding. Fig. 3 shows the absorption spectra of HSA in the absence and presence of cholest-5-ene-7-one. The microenvironment around tyrosine and tryptophan residues of HSA is the focus of the absorbance peak at 278 nm. As the varied steroid concentrations were added to the fixed amount of HSA, the intensity of the peaks decreased. Overall, the peak of intensity indicates that when HSA interacts with steroids, the skeleton loosens and opens up significantly, confirming structural alterations of HSA.![UV absorption spectra of HSA and HSA-steroid (2) complex, fixed [HSA] = 5 µM, increasing concentration of [steroid] = 5, 10, 15, 20, 25, 30 and 50 µM.](/content/185/2021/33/8/img/10.1016_j.jksus.2021.101661-fig4.png)
UV absorption spectra of HSA and HSA-steroid (2) complex, fixed [HSA] = 5 µM, increasing concentration of [steroid] = 5, 10, 15, 20, 25, 30 and 50 µM.
3.6 Fluorescence studies
Protein-drug interaction, mechanism of quenching as well as conformational change can be examined with in protein interaction of drugs by fluorescence spectroscopy (Gowda and Nandibewoor 2014). Tryptophan, tyrosine, and phenylalanine are three main fluorophores that regulate protein fluorescence. The tryptophan residue in subdomain IIA having active cavity contributes significantly to the fluorescence of HSA with a maximum fluorescence intensity at 340 nm, whereas the other fluorophores do not participate notably. The negligible contribution of phenylalanine is due to its poor quantum yield, whereas tyrosine is completely quenched if ionized or next to an amino group, a carboxyl group, or a tryptophan (Yue et al., 2012, Shinde et al., 2021). HSA was titrated against cholest-5-ene-7-one in order to investigate the binding mechanism and complex formation in this work. HSA concentrations were fixed while cholest-5-ene-7-one concentrations were changed. HSA spectra were obtained in the presence and absence of cholest-5-ene-7-one, as shown in the Fig. 4a. The results show that HSA has a fluorescence emission maximum around 338 nm, with quenching as the steroid concentration rises, implying the formation of a complex and binding with HAS(Kabir et al., 2016). Cholest-5-ene-7-one (steroids) triggered progressive fluorescence quenching in HSA without affecting the wavelength (λem) or peak morphology. At the maximum dose of steroid (µM), the fluorescence intensity of HSA decreased by around 43%. This shows that the steroid has a concentration-dependent interaction with HSA, quenching its intrinsic fluorescence. Fluorescence quenching was found to be more prominent in 50 µM based on the calculation of fluorescence intensity as a wide gap can be seen between 30 µM and 50 µM in Fig. 4a. Mechanistic approach for the fluorescence quenching of HSA in presence of drug can occur through either a static or dynamic fluorescence. Static fluorescence quenching is primarily caused by the formation of a stably bound protein–quencher association. As a result, the quenching constant (Kq) increases with decreasing diffusion frequency, indicating an inverse relationship with temperature. Dynamic fluorescence quenching, on the other hand, is largely caused by transitory intermolecular collisions between the protein and the quencher. As a result, as collision frequency increases, the rate constant for quenching (Kq) increases in a temperature-dependent manner. Furthermore, dynamic fluorescence quenching does not need actual ligand interaction, therefore conformational shape or function of HSA are unaffected. Furthermore, dynamic fluorescence quenching does not necessitate actual ligand binding, resulting in no alteration in the conformational shape or function of HAS (Gowda and Nandibewoor 2014). The Stern-Volmer equation was utilized to establish the mechanism of fluorescence quenching by using the mathematical relationship between protein fluorescence quenching and quencher concentration. The Stern-Volmer plot of HSA in the presence of steroid (2) is presented in Fig. 4b. The quenching constant (Ksv) and bimolecular quenching constant (Kq) for the binding of HSA are important factors in knowing the interaction of HSA with drugs. KSV followed by Kq values derived for steroid (2) interaction with HSA at ambient temperature by linear regression plotting F0/F against [Q] and found to be in order 104 showed that quenching mechanism between HSA and cholest-5-ene-7-one was most likely begun by static quenching mechanism (Li et al., 2010). The bimolecular quenching rate constant (Kq) for the binding of HSA with steroid (2) was estimated through Stern Volmer equation and found to be approximately 3.12 × 1012 M−1 s−1. The obtained value of Kq is two orders of magnitude greater than the maximum collision quenching constant 1010 (2 × 7.4 × 1010 L∙mol−1∙s−1 at 298 K) confirming a static quenching mechanism that supports the design of an equilibrium ground state steroid–HSA complex. Furthermore, the binding constant (Kb) and binding sites (n) on HSA can be estimated using a modified Stern-Volmer equation based on a double logarithmic plot of log (F0-F)/F vs log[Q] (or log (F0-F)/F vs log[steroid]) where the obtained slope is the number of binding sites (n) and the intercept (ordinate at the origin, in Fig. 4c) is the logarithm value of Kb (log Kb). In general, a drug with a higher binding constant binds to the receptor more strongly than a drug with a lower binding constant. The slope of the regression equation is close to one, indicating that under defined conditions there is only one binding site with high affinity for steroid to HSA molecule. The binding constant of compound was determined to be 4.5 × 105 M−1. The results clearly show that cholest-5-ene-7-one binds to HSA well. The value of Gibbs free energy (ΔGo) for HSA-steroid binding was determined to be −7.70 Kcal/mol, indicating that the binding occurs spontaneously in nature.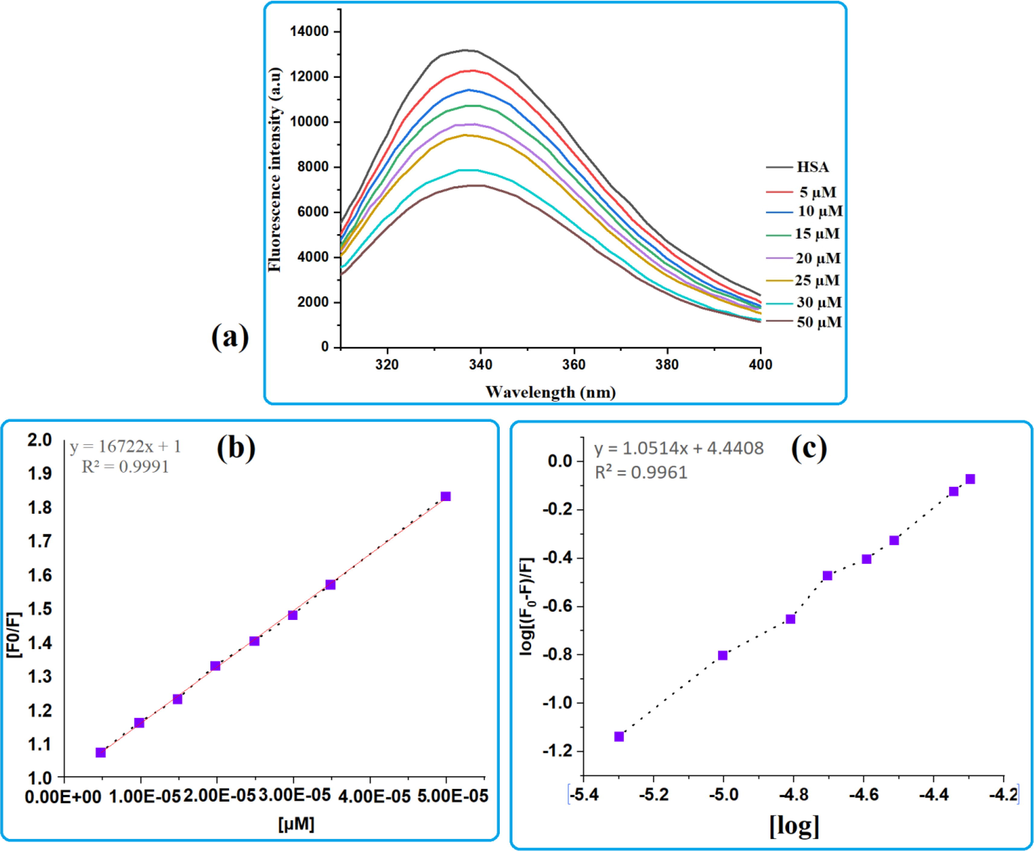
(a) Fluorescence spectra of HSA and HSA-steroid 2 complex in the range of 300–400 nm; fixed concentration of HSA and increasing concentration of steroid 2 (b) Stern-Volmer plot for steroid and (c) modified Stern-Volmer equation based on a double logarithmic plot.
3.7 Synchronous fluorescence spectroscopy
Synchronous fluorescence spectroscopy is a sensitive biophysical technique for examining changes in the conformation and polarity of a binding pocket following ligand attachment to a protein. It frequently demonstrates that ligand-induced protein conformational changes result in binding pocket realignment, which might influence the microenvironment or polarity of neighboring fluorophores in a macromolecule upon ligand or drug engagement. In this study, the excitation and emission monochromators are simultaneously set for synchronous scans at specified wavelength differences (Δλ) between the excitation wavelengths (λex) and the observed emission wavelengths (λem) for the steroid-HSA complex. The wavelength difference (Δλ) between excitation and emission is unique to each fluorophore. After cholest-5-one-7-one binding, the emission and excitation wavelengths are set at 15 nm and 60 nm, after cholest-5-one-7-one binding, referring to alternation between tyrosine (Tyr) and tryptophan (Trp), respectively. The change in emission maxima, i.e. blue shift or red shift, can be used to interpret any change in polarity in adjacent Tyr & Trp residues as shown in Fig. 5 (a-b). The synchronous fluorescence spectra of HSA are shown in Fig. 6 related to the absence and presence of steroid (2). As seen in Fig. 5a, intensity of HSA synchronous fluorescence spectra decreases as increasing steroid concentration, as opposed to when the steroid is absent. This confirms the binding of the steroid to HSA and backs up the fluorescence quenching results (He et al., 2008). Moreover, it was revealed that there is a slight blue shift when subjected to Δλ = 60 nm, implying that Tyr has been exposed to a hydrophobic environment. As a result, the interaction changes the hydrophobic environment, increasing the fluorophore environment's hydrophobicity. However, there is no discernible change in blue shift or redshift in the emission maxima when Δλ was at 15 nm, despite the fact that the fluorescence intensity of HSA dropped with increasing steroid concentration. This observation indicates that steroid binding mostly alters the conformation of the nearby binding pocket to Trp-214 in site I (Fig. 5b) and hence polarity of its microenvironment, with very minor impacts on conformation near the Tyr residues (411, 452) in site II.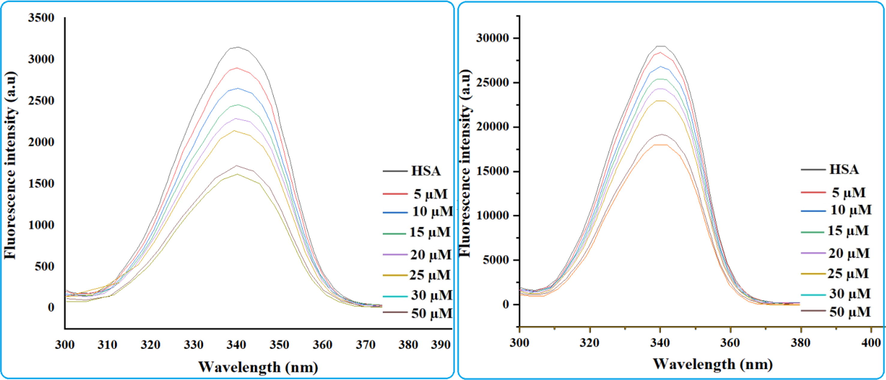
Synchronous fluorescence spectra of human serum albumin and cholest-5-ene-7-one (2) - human serum albumin complex (a) shows synchronous spectra (a) for tyrosine residues, (b) for tryptophan residues of steroid (2).
Circular dichroism spectra of HSA in absence and presence of steroid.
3.8 Circular dichroism measurements
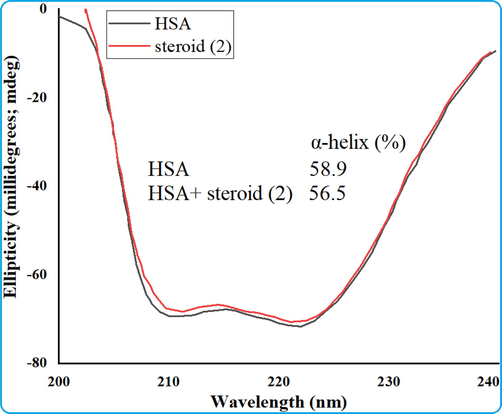
Circular dichroism spectroscopy is a useful and efficient tool for studying changes in the secondary structure of proteins and nucleic acids caused by drug interactions. Thus, CD measurements were performed to track whether cholest-5-ene-7-one binding to HSA caused any secondary structural changes in HAS (Hamed-Akbari Tousi et al., 2010). Due to the involvement of n → π * and π → π * transfer of the peptide bonds of α–helix, the far–UV CD spectra of HSA show two negative bands at 209 and 222 nm, which are diagnostic of the typical α–helical structure of protein. The far–UV CD spectra of HSA were recorded in the absence and presence of steroid to gain a better understanding of the changes in the secondary structure of HSA when it interacts with steroid. The Fig. 6 shows the CD spectra of HSA with and without the addition of steroid, and it exhibits two negative absorption bands at 208 and 222 nm, which are indicative of the protein's α-helical shape. Moreover, in the presence of cholest-5-ene-7-one, the negative peak faded at 208 nm somewhat, while the negative peak at 222 nm shifted somewhat to the short wavelength. The detailed and reduced value of typical α–helical contents of HSA upon interaction with steroid is calculated using the stated equation and determined to be 56.5 from 58.9%. The CD spectrum of HSA after drug exposure demonstrates that the steroid alters the protein conformation, suggesting that the steroid can penetrate into the serum albumin rather than bind to the surface of the protein, disrupting the α-helix conformation of the protein. The general overview shows changes in the secondary structure of HSA, nevertheless, the α-helical content and protein stability are closely linked to its biological roles.
3.9 Dynamic light scattering (DLS) measurements
The size of the proteins and drug-protein complexes was evaluated using the differential light scattering (DLS), which is a very sensitive technology for monitoring changes in protein molecule size by estimating the hydrodynamic radii (Rh) of the complex formed upon ligand or drug engagement. DLS measurements were carried out to obtain DLS parameters named as hydrodynamic radii (Rh) and polydispersity (Pd) and shown in Fig. S3 to determine the hydrodynamic radii of HSA in the presence of cholest-5-ene-7-one in a ratio of 1:10 and compared with hydrodynamic radii of HSA reported in the literature (Alam et al., 2016, Ali et al., 2017, Siddiqi et al., 2018). The Rh and Pd of native HSA have been reported to be 3.6 and 3.4 nm and 11.0 and 13.5% in literature (Alam et al., 2016, Ali et al., 2017), which decreases upon interaction with steroid, Rh was found to be 2.8 ± 0.02 and polydispersity (Pd) was found to be 7.5 %. As interpreted in the literature, this may be due to ligand interaction with albumins disrupting the solvent shell enclosing the protein, causing the molecule to collapse, resulting in a decrease in hydrodynamic radii. The shape and size of the displayed Fig. S3 follow the same pattern as suggested previously in keto-cholestane as well as the explored steroid (2).
3.10 Molecular docking analysis
Molecular docking is a handy computational approach for Molecular modeling in order to investigate the manner of interaction between macromolecules and small molecule/ligand/drug utilizing the Autodock 4.2 application to determine the involvement of HSA amino acids residues. As illustrated in Fig. 7, the cordated molecule HSA has 585 amino acid residues that are split into three structurally similar domains: domain I (residues 1–195), domain II (residues 196–383), and domain III (residues 385–585) (Fig. 7a). The steroidal molecule (Fig. 7b) was docked with the I, II, and III active pockets of HSA individually to study the preference of binding sites of cholest-5-ene-7-one on HSA (Fig. 7c). The lowest-energy docked pose of HSA with steroid in site I, was displayed in Fig. 7d-I (Site II and site III were shown in supplemental material for clarification in the docking structure). The lowest binding energies of cholest-5-ene-7-one were −11.72 kcal mol−1 at site I, −10.44 kcal mol−1 at site II, and −8.34 kcal mol−1 at site III in the corresponding docked pose of steroid into cavities of HSA. The docked pose with the lowest energy as well as presence of hydrophobic cavity at site I was chosen out of three for interpreting the non-bond interaction of steroid to active amino acid residues involved in the formation of the active pocket of HSA. The carbonyl group of cholest-5-ene-7-one formed a hydrogen bond with LYS190 of HSA and occupied the hydrophobic cavity surrounding by ALA191, ALA194, LYS432, VAL433, VAL455 and VAL456 as well as amphipathic amino acids such as TYR452 and hydrophilic amino acids; ASP187, ARG197, ASN429, GLN459 and Glu425 via alky, pi-alkyl, and van der walls interactions to stabilize molecule inside of cavity (8d, f). The pose obtained for ligand interaction is showing one conventional hydrogen bond with Lysine that also takes part in formation alkyl bond formation. The hydrophobic amino acids surrounded ligand and participate in the establishment of non-bonding interactions that lead to the strength of the docked molecule in proper orientation (Fig. 7e). HSA-steroid interactions were also compared to a previously published HAS-ligand interaction profile at site 1 that included amino acids like Asp, Leu, Val, Lys Phe, Tyr Ser, Asn Ala, Glu, Gln, Val, Leu, Lys, Leu, Glu, Arg and Gly, some of which were also found in HSA-steroid interactions(Poór et al., 2015, Shen et al., 2017, Veeralakshmi et al., 2017). Most of the amino acid residues around the reported molecules in literature were hydrophobic in nature, suggesting that hydrophobic effects may also play a role in drug-HSA system stabilization. The majority of the amino acid residues around the steroid were obviously hydrophobic, implying that hydrophobic effects may also contribute to the stability of the steroid–HSA system. The interaction patterns of cholest-5-ene-7-one to HSA at sites II and III are also shown in supplementary Figure S4 (d, e, f & g), which depicts numerous interactions between steroid and active amino acids in the cavity of HSA. Finally, molecular docking analyzes provide a valuable framework for visualizing individual ligand–protein interactions at the atomic level, as well as quantitative analysis of the binding affinity score, which is similar to that of the ligand–protein interactions. correlated well with the previously reported marker competition results(Shen et al., 2017, Veeralakshmi et al., 2017).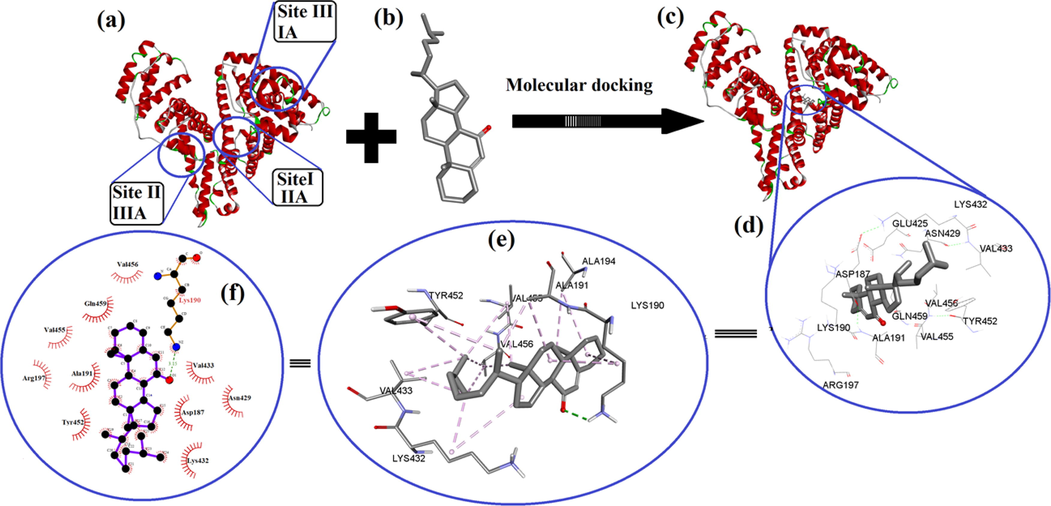
Molecular docking analysis, (a) HSA receptor with different active binding sites, (b) docking ligand (steroid), (c) steroid docked in the binding sites I (e) ligand interaction (steroid) in the cavity of HSA at binding site I surrounding various hydrophobic amino acids and, (f) steroid docked into the cavity of an HSA at binding site 1 showing various interaction and its 2-dimensional interactions generated by LIGPLOT + .
3.11 Analysis of molecular dynamics simulation
The stability of the HSA-steroid complex derived from the best-docked pose was analyzed using a 100 ns molecular dynamics simulation at 300 K and atmospheric pressure (Fig. 8). Root Mean Square Deviation (RMSD) provides information about the structural composition of the protein and indicates whether the simulation is equilibrated. The complex is stable, with minimal RMSD deviation, and the ligand was able to form an H bond with the protein, according to post-simulation analysis of the trajectory (Fig. 8a). However, RMSD plot changes from 0 to 20 ns of MD simulation at the start of the calculation (Fig. 8a), the change is minimal, and no major structural changes in the protein were identified during 20 to 100 ns of MD simulation. It is also found in this study that the RMSD value of HSA converges to a fixed value (∼2.0 Å) towards the end of the simulation, indicating a stable conformation of the protein (Fig. 8a). Furthermore, the ligand RMSD (fit to protein) analysis demonstrates how stable the ligand is in relation to the protein and its binding pocket. In Fig. 8a, it was observed that the RMSD value of the steroid stabilized after the initial fluctuations for 30 ns and was within the upper limit of 2.5 Å. Fig. 8a also revealed that the steroid detached from the protein for a short period of time (20 to 30 ns), after which both the protein and the steroid interacted and stabilized the complex during the course of simulation (9a).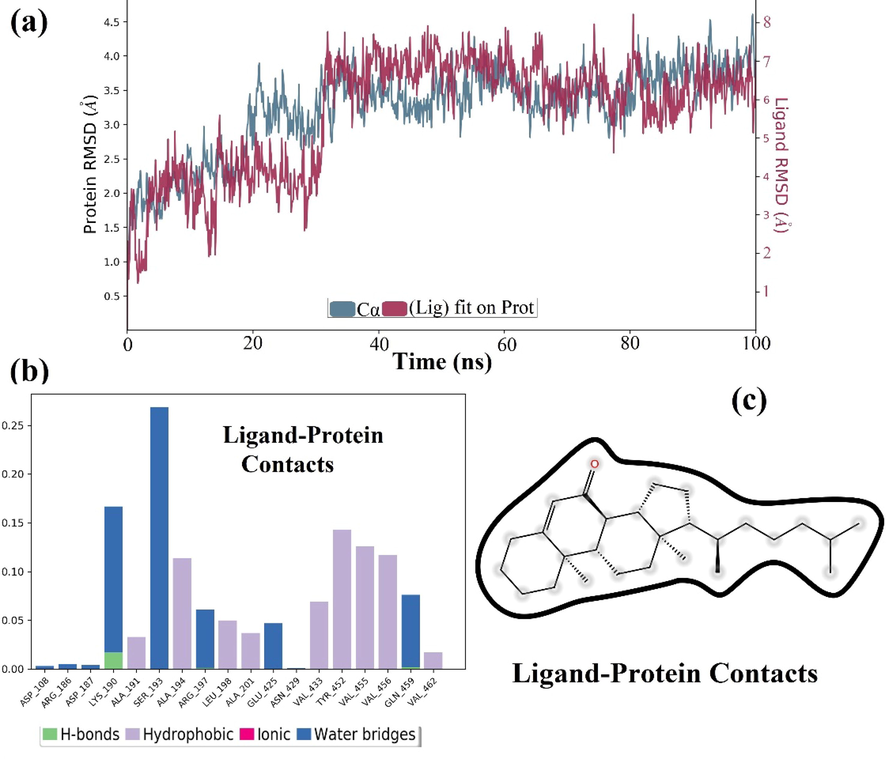
(a) Time dependent RMSD values for HSA and steroid-HSA complex over a 100 ns MD simulation and (b & c) protein-Ligand contact for to show different types of interaction of steroid with amino acid residues of HSA.
The bar diagram in Fig. 8b clearly shows that steroid interactions with amino acid residues present in close proximity to the active site of HAS (Fig. 8c) during the 100 ns trajectory include hydrogen bonding, hydrophobic, ionic, and water bridging. During the MD simulation, it is discovered that Lys exhibits both hydrogen bonding and water bridging interaction. Other amino acid residues appeared to be involved only in hydrophobic interactions.
Torsional angle profiles obtained from MD studies over the course of 100 ns MD simulations analyze the difference in conformational flexibility of each rotatable bond in the steroid structure throughout simulation in terms of specific scaffold dihedrals, i.e. frequency histograms of scaffold dihedral angles for the MD geometries (Fig. 9).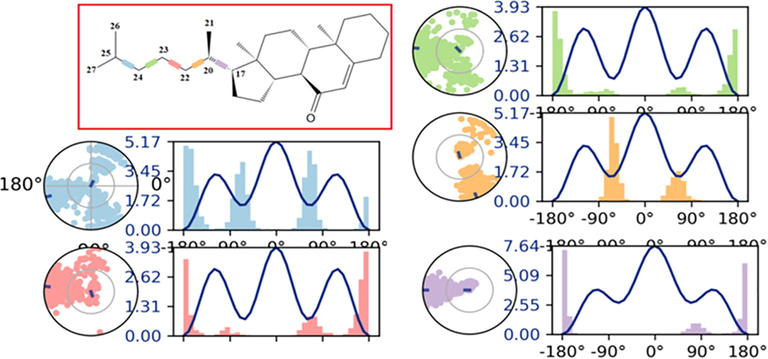
Over the course of the simulation, the ligand torsion profile of cholest-5-en-7-one that represent the distribution of scaffold dihedrals. The graphs summarize the conformational changes during the course of the simulation (0.00–100.00 ns). On 2D structures, the color-coded scaffold dihedrals are highlighted. A dial plot and bar plots of the same color accompany each dihedral. Dial plots depict the torsion's conformation during the course of the simulation. The simulation begins at the center of each dial plot, and time evolution is depicted radially outwards. The frequency distribution of dihedral angle values is represented by the bar charts.
The side chain of the steroidal skeleton is flexible and contributes to the creation of the profile of torsion angles in MD simulation (Fig. 9). The radial plot's center corresponds to the beginning of the simulation, and the simulation's evolution records various conformations of each rotatable bond radially outwards. The probability density of each torsional rotation is represented by the bar charts. When a steroid interacts with a receptor, it occupies a rigid structure that can keep the same binding orientation throughout the simulation, resulting in a narrower band with less variance. The steroid torsion profile analysis gives information for deriving the pharmacophore characteristics essential for interaction with key residues in proteins. The ligand torsion profile in steroids displayed significant conformational flexibility as indicated by five rotatable bonds (RB) such as C17-C20, C20-C22, C22-C23, C23-C24, and C24-C25, which are illustrated in Fig. 9 by blue, pink, green, yellow, and purple, respectively. At the start of the simulation (0°), the ratable bonds (RB) impacted by rigid steroidal rings and methyls (C210 & C26-C27) such C17-C20, C20-C22, and C24-C25 have high potential energies of 7.64, 5.17, and 5.17 kcal/mol, respectively. In addition, the core of the steroid is rigid, and its oxygen atom is involved in a non-binding interaction, it remained stable in complexes during the MD simulation, demonstrating that the oxygen atom of steroidal ring was able to attach HSA tightly. Ligand properties related to Radius of Gyration (rGyr), Intramolecular Hydrogen Bonds (intraHB), Molecular Surface Area (MolSA), Solvent Accessible Surface Area (SASA), and Polar Surface Area (PSA) contributed only by oxygen atom are displayed in graph form in Figure S5. The lowest-energy docked pose of HSA with steroid was also selected for optimization using the hybrid QM/MM approach to calculate the important interaction energy of steroid–HSA system, as given in additional information (Fig S6) under QM/MM approach.
4 Conclusions
DFT analysis was performed on easily synthesized cholest-5-ene-7-one to examine geometry, FMOs, MEP, and chemical reactivity descriptors of steroid using FMOs. Hirshfeld surface analysis and accompanying 2D finger print plots revealed that the predominant interactions controlling solid state packing are H…H, C…H, and O…H interactions. Based on the HSA results, the binding constant for the HSA-steroid complex was calculated to be 4.5 × 105 M−1. The values of ΔGo for the binding of HSA with steroidal ketone was determined to be −7.70 Kcal/mol, indicating that the binding occurs spontaneously in receptor-ligand complex. The interaction of non-bonding and participating hydrophobic amino acids, which are largely involved in the formation of active sites in HSA, was revealed by molecular docking on HSA-steroid. To identify stability of complex and interactions between the synthesized compound and HSA, 100-ns Molecular dynamic (MD) simulation were performed. The binding energy (BE) of −111.0 Kcal/mol calcualted by QM/MM method indicates that HSA accepts the steroid in its cavity by forming an HSA-ligand complex with natural stabilization via multiple non-binding interactions. The features of the HSA-steroid complex derived in this work, although premature, could have a substantial impact on future studies and applications.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Molecular structure, anharmonic vibrational analysis and electronic spectra of o-, m-, p-iodonitrobenzene using dft calculations. J. Mol. Struct. 2014
- [Google Scholar]
- Interplay of multiple interaction forces: Binding of tyrosine kinase inhibitor nintedanib with human serum albumin. J. Photochem. Photobiol., B 2016
- [Google Scholar]
- Dft/b3lyp calculations, in vitro cytotoxicity and antioxidant activities of steroidal pyrimidines and their interaction with hsa using molecular docking and multispectroscopic techniques. Bioorg. Chem. 2017
- [Google Scholar]
- Design, synthesis and evaluation of cholinesterase hybrid inhibitors using a natural steroidal alkaloid as precursor. Bioorg. Chem. 2021
- [Google Scholar]
- Synthesis of 17β-hydroxysteroid dehydrogenase type 10 steroidal inhibitors: Selectivity, metabolic stability and enhanced potency. Eur. J. Med. Chem. 2021
- [Google Scholar]
- A study of the mechanism of conversion of acetate to cholesterol via squalene1. J. Am. Chem. Soc. 1953
- [Google Scholar]
- Frisch, M., Clemente, F., 2009. Gaussian 09, revision a. 01, mj frisch, gw trucks, hb schlegel, ge scuseria, ma robb, jr cheeseman, g. Scalmani, V. Barone, B. Mennucci, GA Petersson, H. Nakatsuji, M. Caricato, X. Li, HP Hratchian, AF Izmaylov, J. Bloino, G. Zhe.
- In acid-aminopyrimidine continuum: Experimental and computational studies of furan tetracarboxylate-2-aminopyrimidinium salt. RSC Adv. 2021
- [Google Scholar]
- Gas-phase structure of 2, 2, 2-trichloroethyl chloroformate studied by electron diffraction and quantum-chemical calculations. PCCP 2016
- [Google Scholar]
- Binding and conformational changes of human serum albumin upon interaction with 4-aminoantipyrine studied by spectroscopic methods and cyclic voltammetry. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014
- [Google Scholar]
- Spectroscopic studies on the interaction between sodium ozagrel and bovine serum albumin. J. Mol. Struct. 2009
- [Google Scholar]
- Comparing the interaction of cyclophosphamide monohydrate to human serum albumin as opposed to holo-transferrin by spectroscopic and molecular modeling methods: Evidence for allocating the binding site. Protein Peptide Lett. 2010
- [Google Scholar]
- Binding of puerarin to human serum albumin: A spectroscopic analysis and molecular docking. J. Fluoresc. 2008
- [Google Scholar]
- Interaction of a tyrosine kinase inhibitor, vandetanib with human serum albumin as studied by fluorescence quenching and molecular docking. J. Biomol. Struct. Dyn. 2016
- [Google Scholar]
- Synthesis and anticancer potential of novel 5, 6-oxygenated and/or halogenated steroidal d-homo lactones. Bioorg. Med. Chem. 2021
- [Google Scholar]
- Design, synthesis and biological evaluation of novel 5α, 8α-endoperoxide steroidal derivatives with hybrid side chain as anticancer agents. Steroids 2020
- [Google Scholar]
- Comparative studies on the interaction of caffeic acid, chlorogenic acid and ferulic acid with bovine serum albumin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010
- [Google Scholar]
- The novel iminium surfactant p-benzylidene benzyldodecyl iminium chloride as a corrosion inhibitor for plain carbon steel in 1 m hcl: Electrochemical and dft evaluation. RSC Adv. 2017
- [Google Scholar]
- Meng, X.-Y., Zhang, H.-X., Mezei, M., Cui, M., 2011. Molecular docking: A powerful approach for structure-based drug discovery. Current computer-aided drug design.
- Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998
- [Google Scholar]
- Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009
- [Google Scholar]
- Poór, M., Lemli, B., Bálint, M., Hetényi, C., Sali, N., Kőszegi, T., Kunsági-Máté, S.J.T., 2015. Interaction of citrinin with human serum albumin.
- Rehman, M.T., AlAjmi, M.F., Hussain, A., Rather, G.M., Khan, M.A.J.I.j.o.m.s., 2019. High-throughput virtual screening, molecular dynamics simulation, and enzyme kinetics identified zinc84525623 as a potential inhibitor of ndm-1.
- Ligand-binding affinity estimates supported by quantum-mechanical methods. Chem. Rev. 2016
- [Google Scholar]
- Marine steroids as potential anticancer drug candidates: In silico investigation in search of inhibitors of bcl-2 and cdk-4/cyclin d1. Steroids 2015
- [Google Scholar]
- P-tsoh-catalyzed one-pot transformation of di-and trihydroxy steroids towards diverse a/b-ring oxo-functionalization. New J. Chem. 2017
- [Google Scholar]
- Synthesis of 23e-ethylidene spirostanes via the bf3· et2o-catalyzed aldol condensation of steroid sapogenins and acetaldehyde. Tetrahedron Lett. 2020
- [Google Scholar]
- Shen, F., Liu, Y.-X., Li, S.-M., Jiang, C.-K., Wang, B.-F., Xiong, Y.-H., Mao, Z.-W., Le, X.-Y.J.N.J.o.C., 2017. Synthesis, crystal structures, molecular docking and in vitro cytotoxicity studies of two new copper (ii) complexes: Special emphasis on their binding to hsa.
- Effect of quercetin on the amiloride–bovine serum albumin interaction using spectroscopic methods, molecular docking and chemometric approaches. Luminescence 2021
- [Google Scholar]
- Siddiqi, M., Nusrat, S., Alam, P., Malik, S., Chaturvedi, S.K., Ajmal, M.R., Abdelhameed, A.S., Khan, R.H., 2018. Investigating the site selective binding of busulfan to human serum albumin: Biophysical and molecular docking approaches. International journal of biological macromolecules.
- New structure–activity relationships of a-and d-ring modified steroidal aromatase inhibitors: Design, synthesis, and biochemical evaluation. J. Med. Chem. 2012
- [Google Scholar]
- Veeralakshmi, S., Sabapathi, G., Nehru, S., Venuvanalingam, P., Arunachalam, S.J.C., Biointerfaces, S.B., 2017. Surfactant–cobalt (iii) complexes: The impact of hydrophobicity on interaction with hsa and DNA–insights from experimental and theoretical approach.
- The investigation of the binding behavior between ethyl maltol and human serum albumin by multi-spectroscopic methods and molecular docking. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012
- [Google Scholar]
- A theoretical study on the inhibition efficiencies of some quinoxalines as corrosion inhibitors of copper in nitric acid. J. Saudi Chem. Soc. 2014
- [Google Scholar]
- Study on the interaction between $${\mathrm {cu}}{\left ({{\mathrm {phen}}}\right)}^{{2+}} _ 3 $$ and bovine serum albumin by spectroscopic methods. Biol. Trace Elem. Res. 2008
- [Google Scholar]
- Zhao, D.-K., Zhao, Y., Chen, S.-Y., Kennelly, E.J., 2021. Solanum steroidal glycoalkaloids: Structural diversity, biological activities, and biosynthesis. Natural product reports.
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2021.101661.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




