Translate this page into:
Molecular modeling and biological activity analysis of new organic-inorganic hybrid: 2-(3,4-dihydroxyphenyl) ethanaminium nitrate
⁎Corresponding authors. issaoui_noureddine@yahoo.fr (Noureddine Issaoui), omar@ksu.edu.sa (Omar Al-Dossary)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
In this paper, experimental and theoretical results of 2-(3, 4-dihydroxyphenyl) ethanaminium nitrate (2DOPN) have been investigated. From DFT calculations, molecular geometry and optimized parameters of 2-(3, 4-dihydroxyphenyl) ethanaminium nitrate have been obtained at B3LYP/ 6–311 ++ G(d, p) level of theory and compared with the available X-ray data. The non covalent interactions of the crystal structure were investigated by QTAIM analysis, ELF, LOL and Hirsfeld surfaces. In addition to study the weak interaction, a visualized approach known as RDG has been carried out. Mulliken atomic charge has been used to describe the process of electronegativity equalization and charge transfer in chemical reactions. NBO analysis was also performed to find the charge transfer within the molecule and their stabilization energy. Electronic proprieties such as MEP, ESP and the frontier molecular orbital analysis HOMO-LUMO of molecule were studied. The dipole moment (µ) and the first hyperpolarisability (β0) have been calculated and found that our compound is a potential NLO material. Thermal behavior (TGA and DTA) of C8H12NO2 (NO3) have also been undertaken and reported. The biological activity of 2DOPN through ligand and proteins interactions has been confirmed theoretically for the treatment of Parkinson disease with respect to chosen proteins. The activities of our molecule with divers proteins were studied in accordance with literature survey and the results were presented here.
Keywords
DFT
HOMO-LUMO
Electronic propriety
RDG
AIM
Non-covalent interactions
Molecular docking

1 1) introduction
Hybrid materials have been a subject of intense research in recent years due to its combination between the two organic–inorganic groups. The applications of these hybrid materials open up a wide field of application in various fields, namely in biology, optics, electricity, photo-catalysis, medicine. Herein, the new hybrid compound 2- (3, 4-dihydroxyphenyl) ethanaminium nitrate is known as dopaminium nitrate exhibits interesting biological and pharmacological properties (Gatfaoui et al., 2014). The molecule studied 2DOPN is a powerful and selective dopamine receptor agonist and has been proved to be effective in the treatment of Parkinson's disease. Parkinson's disease is a degenerative disease manifested by the progressive degeneration of dopaminergic neurons in the substantia nigra of the brain (Adler et al., 1997). In the broad sense, dopamine plays an important role in the regulation of many psychiatric disorders such as Schizophrenia, bipolar disorder, Parkinson and major depression (Campayo et al., 2005). The aim of this paper is to study the structural, vibrational and electronic propriety of our molecule 2DOPN. In this context, the most stable structure in the ground state was obtained by employing the DFT calculation method under the B3LYP/6–311 ++G(d, p) level of theory. The optimized molecular structure and the geometrical parameters have been compared with experimental results ones. The method used to prove that the calculated results can well reproduce the data parameters. Hence, in the atomic arrangement, the structure study show that the stability of 2DOPN crystalline structure arising from N-H···O, C-H···O as well as O-H···O hydrogen bonds. Recently, great deals of non covalent interactions are very frequently used by chemists to build supramolecular assemblies, such as hydrogen-bonding (Mirzaei et al., 201420142014). Taking advantage of DFT calculations and theoretical models, we studied several methods to describe the non covalent interactions and their influence in crystal packing (Mirzaei et al., 201420142014). Thus, computational tools have found to understand the nature and extent of non-covalent interactions. The quantum theory of atoms in molecules (QTAIM) approach has been applied to classify the hydrogen bonding interaction within 2DOPN. In addition, the topological studies ELF and LOL were performed to quantify non covalent interactions. The Hirsfeld surface mapped with dnorm, di, Shap index and Curvedness are a beneficial tool to identify the nature of interactions and their quantitative contributions inside the crystal packing. The RDG approach has been widely applied to classify and understand the hydrogen bonding interactions in our molecule. Further, the NBO charges were calculated to reveal the charge transfer and the stabilization energy of molecules. The Mulliken atomic charges have been performed which were helped to display the distribution of electrons between the atoms forming molecule. MEP and ESP were computed to explore the reactivity of this title compound by predicting their nucleophilic as well as their elcetrophilic sites. Also a TD-DFT calculation has been realized in order to determine the electronic propriety, stability and reactivity of diverse compounds in terms of frontier molecular orbital. The first hyperpolarizabilty (β0), polarizability (α) and the dipole moment (μ) showed that the 2DOPN is a right candidate for NLO applications. Further, the thermal behavior of dopaminuim nitrate has been investigated. For the purpose to discover a new drug for the treatment against Parkinson disease, a molecular docking was simulated with different proteins. For this reason a literature study shows that MAO-B, COMT, AAAD and L-DOPA inhibitors are currently being used as a potential drug for the clinical treatment of Parkinson's disease.
2 Experimental details
2.1 Samples preparation
2-(3,4-dihydroxyphenyl) ethanaminium nitrate crystals were synthesized by mixing 1 mmol of HNO3 in 10 ml of water with 1 mmol of dopamine hydrochloride in 10 ml of water. This solution was stirred for 15 min and then left to stand at room temperature. After a few days colorless single crystals of the title compound were obtained. The reaction mechanism for the synthesis is shown in the following Scheme 1: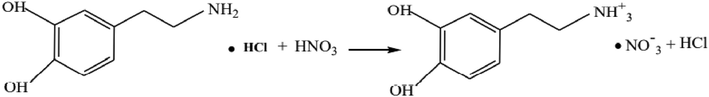
The reaction mechanism of 2DOPN.
2.2 Materials and physics measurement
Measurement of single crystal X-ray diffractions was registered at the room temperature (T = 150 K) using a Bruker APEXII diffractometer with Mo KWα radiation source (λ = 0.71073 Å). Absorption corrections were performed by the multi scan technique by the means of SADABS software (Ross et al., 2006). The total number of measured reflections was 10,787 which 4339 were independent reflections and 3583 reflections had an intensity I > 2σ (I). SIR97 (Altomare et al., 1999) was used to solve the structure and then refined with full-matrix least- square methods based on F2 (SHELXL97) (Sheldrick, 2008) included in WinGX program (Farrugia, 2012). All non hydrogen atoms were refined anisotropically. A final refinement on F2 converged at R [F2 > 2 σ (F2)] = 0.043 and wR (F2) = 0.113. The Table S1 summarizes all the crystallographic details and results of the structure refinement.
2.3 Theoretical calculations
All the quantum computational calculations were performed for 2DOPN by the DFT method with B3LYP/6-311G++(d,p) basis set using Gaussian 09 (Gaussian 09 et al., 2009) program. Molecular structure was visualized with GaussView 6.0 package (GaussView, 0000). The molecular formula of the target compound is C8 H12 N2 O5. It contains 27 atoms and 114 electrons. It is a neutral molecule with a singlet system. The optimized parameters like bond angles and bond lengths were compared with experimental values reported from crystallographic data. To determine the types of intermolecular interactions, many analyses have been used such as QTAIM, ELF, and LOL. These topological analyses were carried out using Multiwfn (Lu and Chen, 2012) software. The Crystal Explorer package (Wolff et al., 2012) was used to perform the Hirsfeld surface in order to quantify the interactions in the crystal structure. Moreover, the non-covalent interactions were investigated in terms of reduced density gradient (RDG). NBO analysis was studied to establish the energy of intermolecular interactions by computing donor acceptor interaction energy. In addition, the Mullikan charges have been used to process the elctronegativity and charge transfer in molecule. The Molecular electrostatic potential analysis (MEP) and Molecular electrostatic potential (ESP) on the molecular VDW surface were visualized to predict the reactive sites of the molecule title. The energy gap between HOMO-LUMO and the global reactivity descriptors were calculated in order to determine the electronic propriety, stability and reactivity of 2DOPN. Gauss-sum program (O’Boyle et al., 2008) has been used to prepare the plots of density of state (DOS). Furthermore, the non linear optical propriety (NLO) has been carried out on our compound. Biological activities of 2-(3, 4-dihydroxyphenyl) ethanaminium nitrate has been investigated using iGemdock (Visualizer, 2005). Discovery studio (Yang and Chen, 2004) software is used to view and design the docking patterns from standard protein data bank
3 Results and discussion
3.1 Description of the crystal structure
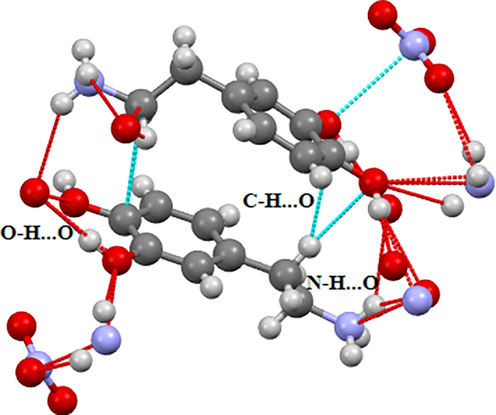
The molecular graphics of 2DOPN single crystals were done using an ORTEP drawing as shown in Figure S1. Dopaminuim nitrate (C8H12NO2·NO3) consists with two units: an organic unit which is the dopaminuim [C8H12NO2]+ and the inorganic unit which is the nitrate [NO3]- .
2DOPN has been crystallized to the triclinic system with space group P1 and the network parameters obtained are a = 8.3066 (4) Å, b = 10.4856 (5) Å, c = 11.2303 (7) Å, V = 953.37 (9) Å3 and Z = 4. The dopaminuim cation are linked to the nitrate anion by means of an assortment of N-H···O, C-H···O and O-H···O hydrogen bonding (Figure S2). As shown in this figure, in the crystal structure the compound are connected through three H-bonds (N-H···O, C-H···O and O-H···O) involve oxygen atoms of the nitrate anion as acceptors, and the nitrogen atoms, carbon and oxygen atoms of dopaminuim cation as donors. These non-covalent interactions help to increase the stability of the structure.
3.2 Molecular geometry and optimized parameters
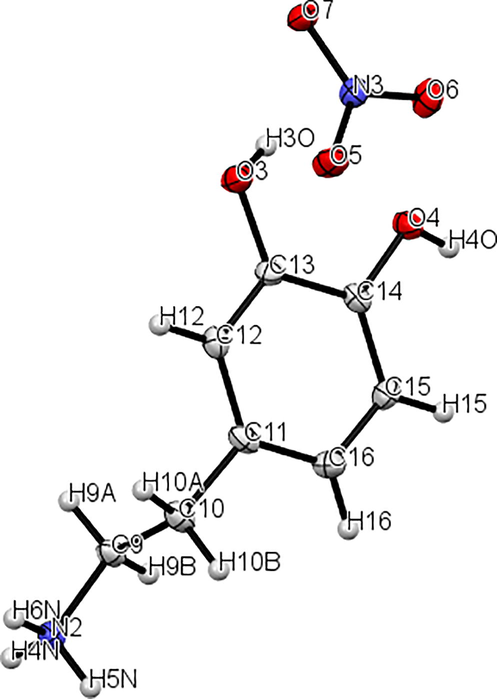
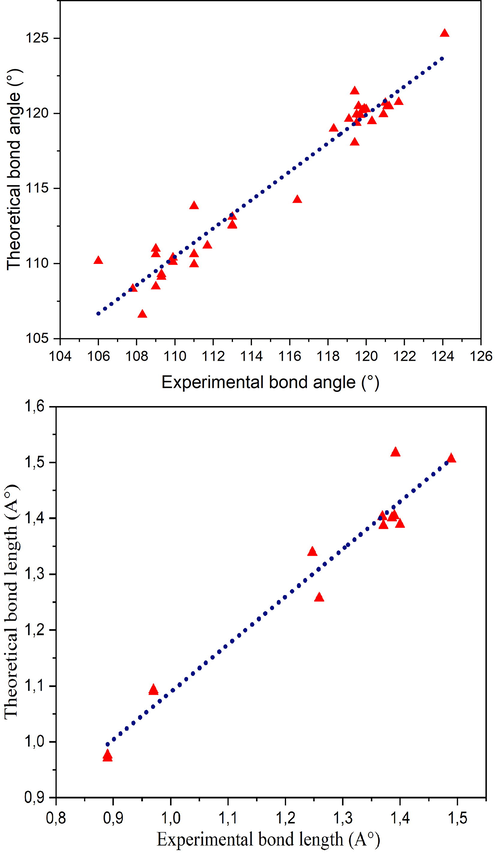
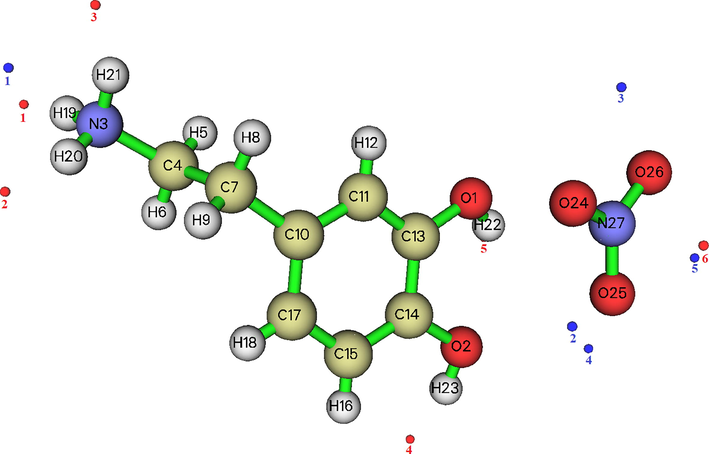
In this research, geometry of 2-(3, 4-dihydroxyphenyl) ethanaminium nitrate were optimized using the DFT calculation of a hybrid functional B3LYP and dispersion correction method WB97XD with the level of 6–311++G (d, p) basis set. Imaginary frequency confirms the absence of negative frequencies and that the molecular system was fully optimized and converged. The optimized structure along with the numbering of atoms of C8H12N2O5 is depicted in Fig. 1. Both the geometrical parameters (bond lengths and bond angles) and the experimental data are selected in Table 1 and compared by using the RMSD values. During the DFT calculation, we find that the optimized structure corresponding to the minimum energy (E B3LYP = -797.568 a.u, E WB97XD = -797.561 a.u) is obtained with B3LYP / 6–311++G(d, p) method. On the other hand, the RMSD is calculated in order to rigorously compare the most adequate method. The lowest RMSD of the bond angles (RMSD B3LYP = 0.99°, RMSD WB97XD = 1.02°) and lengths (RMSD B3LYP = 0.186 A°, RMSD WB97XD = 0.194 A°) is obtained with B3LYP / 6–311++G (d, p). As can seen from the table 1, that the calculated bond lengths were greatly then the experimental ones. Usually, the theoretical bond lengths are stronger than the experimental results because in majority of system the molecules are ensured by inter and intramolecular H bonds (Shukla et al., 2020). The molecule title has eight C–C bond lengths, seven C–H bond lengths, four O-N bond lengths, three N–H bond lengths, two O–H bond lengths and one O-C bond lengths. We have six C–C bond lengths present in the phenyl ring lies in the interval 1.517–1.389 A° in DFT and 0.930–1.38 A° in the experiment. The other two bond lengths C4-C7 (1.539 Å) and C7-C10 (1.517 Å) show stronger values than other ring C–C bond lengths. This is explained by the difference in electronegativity between the atoms, the types of bonds (single, double, …) and the chemical environment. The C–H bond lengths are elongated in the interval 1.082–1.093 A° (DFT) and in 0.970–1.520 A° (Exp.). Also, the bond lengths calculated formed between N3-H19 (1.017 A°), N3-H20 (1.086 A°) and N3-H21 (1.033 A°) atoms are approximately similar values. The two bond lengths O1-H22 and O2-H23 reported in DFT calculations are equal to 0.976 A° and 0.971 A° respectively. The corresponding bond angles C13-O1-H22 (110.63°), C14-O2-H23 (113.82°) demonstrated that the hydrogen bonds which link the crystal stability of our compound are weak types. We note also that all the bond angles calculated are in concord with the experimental bond angles. Moreover, it is observed that the simulated and observed values for the bond angles of the phenyl ring C7-C10-C11 (120.27°, 120°), C7-C10-C17 (120.75°, 121.7°), C11-C10-C17 (118.98°, 118.3°), C13-C14-C15(120.48°, 119.6°), C14-C15-C17 (119.48°, 120.3°) and C10-C17-C15 (120.70°, 121°) are very close to the experimental results. In Figure S3 we shows the calculated correlation coefficients (R2) between the experimental and theoretical bond length and angles. We found that the correlation coefficient is very close to 1 for the bond length (R2 = 0.958) and bond angle (R2 = 0.95). This coefficient showed that the result greatly correlated. From all these results has demonstrated that this method were reliable to reproduce the experimental values. Thus, we continue our calculation by using B3LYP functional at 6–311++G (d, p) level of theory.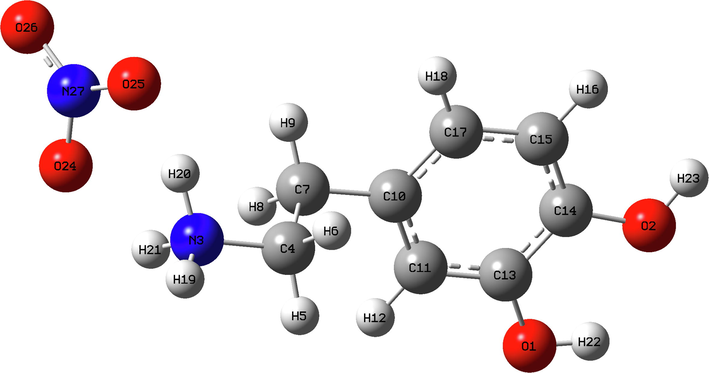
The optimized geometric structure of C8H12N2O5 compound.
Atom position
B3LYP/6–311++G(d.p)
WB97XD/6–311++F(d.p)
Experimental
Bond lengths (Å)
O1-C13
1.387
1.354
1.371(2)
O1-H22
0.976
0.962
0.89(2)
O1-N27
8.758
7.905
–
O2-C14
1.403
1.370
1.369(2)
O2-H23
0.971
0.958
0.89(2)
N3-C4
1.506
1.473
1.489(2)
N3-H19
1.017
1.014
0.92(2)
N3-H20
1.086
1.016
0.93(2)
N3-H21
1.033
1.594
0.970
C4-H5
1.09
1.093
0.970
C4-H6
1.090
1.097
1.520(2)
C4-C7
1.539
1.529
0.970
C7-H8
1.093
1.094
0.970
C7-H9
1.091
1.095
1.515(2)
C7-C10
1.517
1.508
1.392(2)
C10-C11
1.405
1.396
1.390(2)
C10-C17
1.402
1.390
0.930
C11-H12
1.082
1.085
1.386(2)
C11-C13
1.389
1.384
1.400(2)
C13-C14
1.401
1.399
1.386(2)
C14-C15
1.389
1.383
0.930
C15-H16
1.083
1.086
1.394(2)
C15-C17
1.401
1.393
0.930
C17-H18
1.082
1.085
1.247(2)
O24-N27
1.311
1.348
0.930
O25-N27
1.339
1.217
1.247(2)
O26-N27
1.257
1.198
1.259(2)
RMSD
0.186
0.194
–
Bond angles (°)
C13-O1-H22
110.63
108.51
111(1)
C13-O1-N27
38.26
50.64
–
H22-O1-N27
147.79
155.46
–
C14-O2-H23
113.82
110.68
111(1)
C4-N3-H19
112.58
110.72
113(1)
C4-N3-H20
112.53
110.24
113(1)
C4-N3-H21
113.11
113.04
113(1)
H19-N3-H20
110.17
107.24
106(2)
H19-N3-H21
109.95
113.99
111(2)
H20-N3-H21
97.56
101.01
102(2)
N3-C4-H5
108.48
107.90
109
N3-C4-H6
107.54
111.75
109
N3-C4-C7
110.78
110.76
113(1)
H5-C4-H6
108.32
107.35
107.8
H5-C4-C7
110.99
109.52
109
H6-C4-C7
110.62
109.47
109
C4-C7-H8
109.30
109.11
109.3
C4-C7-H9
109.12
109.36
109.3
C4-C7-C10
111.20
111.42
111.7(1)
H8-C7-H9
106.60
106.74
108.3
H8-C7-C10
110.40
110.03
109.9
H9-C7-C10
110.11
110.06
109.9
C7-C10-C11
120.27
119.85
120(1)
C7-C10-C17
120.75
121.20
121.7(1)
C11-C10-C17
118.98
118.92
118.3(1)
C10-C11-H12
121.46
120.91
119.4
C10-C11-C13
120.47
120.92
121.2(1)
H12-C11-C13
118.05
118.16
119.4
O1-C13-C11
119.63
119.89
119.1(1)
O1-C13-C14
120.48
120.60
121.1(1)
C11-C13-C14
119.89
119.51
119.7(1)
O2-C14-C13
114.23
115.15
116.4(1)
O2-C14-C15
125.29
124.73
124.1(1)
C13-C14-C15
120.48
120.12
119.6(1)
C14-C15-H16
120.31
119.84
119.9
C14-C15-C17
119.48
119.94
120.3(1)
H16-C15-C17
120.21
120.23
119.8
C10-C17-C15
120.70
120.60
121(1)
C10-C17-H18
119.93
120.03
119.5
C15-C17-H18
119.37
119.38
119.5
O1-N27-O24
59.91
59.51
–
O1-N27-O25
75.85
92.72
–
O1-N27-O26
136.41
114.06
–
O24-N27-O25
117.94
117.30
120.9(1)
O24-N27-O26
122.12
115.65
119.6(1)
O25-N27-O26
119.94
127.06
120.9(1)
RMSD
0.99
1.02
–
4 Topology analysis
4.1 Quantum theory atoms in molecule (QTAIM) analysis
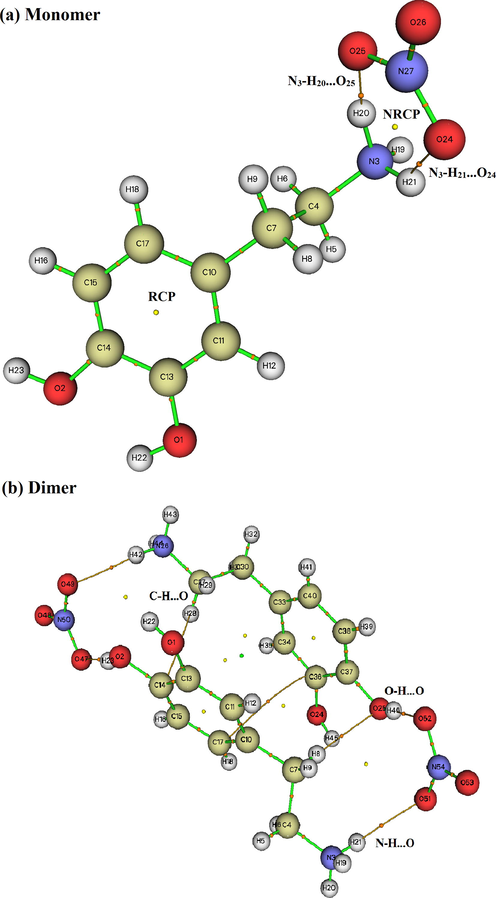
The quantum theory of atoms in molecule (QTAM) is one of the appropriate technique to analyze different types of interaction (inter and intramolecular interaction). This approach provides as more effective information about the presence of non covalent interaction in term electron density of a system analyzed (Issa et al., 2020). We have applied this tool using the Multiwfn program to characterize and determine the proprieties of non covalent interaction. The bond critical points (BCPs) of the intra and intermolecular bonds, ring critical points (RCP) and the new ring critical points (NRCP) of C8H12N2O5 are shown in Figure S4. Also, we are presented in Table 2 the calculated electron density (ρ), Laplacien of electron density (∇2ρ(r)), kinetic energy (G(r)), potential energy (V(r)), total energy (H(r)) and interaction energy (Einteraction). These topology parameters are calculated to identify the natures of the bonds that exist in our molecule. The Fig. 4Sa of monomer shows the presence of hydrogen bonds N-H···O types. In addition to this like, we noted in the dimer (Figure S4) the presence of two other types of hydrogen bonds C-H···O and O-H···O. By analyzing the results obtained in table 2, the ∇2ρ(r) > 0 and G(r) + V(r) < 0 indicating that the hydrogen bond is a medium hydrogen bond. In addition, the value of the rapport –G (rBCP)/V(rBCP) is between 0.5 < -G(rBCP)/V(rBCP) < 1, it indicates that the nature of the H-bond is covalent. Consequently, AIM analysis confirms that these results are in good agreement with the experimental results.
Interactions
∇2ρ(r) (a.u)
ρ (r)(a.u)
G(r) (a.u)
V(r)(a.u)
H(r) (a.u)
ε
Einteractions kJ/mol
Monomer
RCP
0.153
0.021
0.031
−0.023
0.007
−1.187
–
NRCP
0.073
0.016
0.016
−0.014
0.002
−1.580
–
N3-H21…O24
0.096
0.028
0.022
−0.021
0.002
0.045
− 27.57
N3-H20…O25
0.136
0.070
0.052
−0.071
−0.018
0.048
− 93.21
Dimer
RCP1
0.159
0.022
0.032
−0.025
0.007
−1.195
–
RCP2
0.158
0.022
0.032
−0.025
0.007
−1.194
–
NRCP1
0.013
0.003
0.003
−0.002
0.001
−2.087
–
NRCP2
0.011
0.003
0.002
−0.002
0.001
−1.530
–
NRCP3
0.009
0.003
0.002
−0.002
0.001
−3.685
–
NRCP4
0.005
0.002
0.001
−0.001
0.001
−1.493
–
N3-H21…O51
0.048
0.013
0.001
−0.008
0.002
0.104
−10.50
N26-H42…O49
0.045
0.013
0.001
−0.008
0.002
0.147
−10.50
C7-H8…O25
0.031
0.001
0.007
−0.006
0.001
0.250
−7.88
O25-H46…O52
0.118
0.031
0.029
−0.028
0.001
0.069
−36.76
O2-H23…O47
0.125
0.036
0.032
−0.032
−0.001
0.066
−42.01
4.2 ELF and LOL
Electron localization descriptors, such as the electron localization function ELF and the localized orbital locator LOL are calculated by using Multiwfn software. These methods are performed by Selvi et al.(Savin et al., 1997) and provide a visual tool for interpreting the results of electronic structure calculations (Clements et al., 2020). ELF and LOL have the same chemical properties, they make it possible to differentiate covalent, ionic and van der Waals bonds, in terms of local kinetic energy. However, ELF allows a partition of the molecular space in basins of electronic localization within which the excess of kinetic energy due to the repulsion of Pauli is minimum. LOL is usually observed during the overlap of localized orbitals, resulting in high gradients of localized orbitals. The pictures of the ELF and LOL for the title compound are presented in color shade map and contour map which illustrated in Fig. 2.a and Fig. 2.b, respectively. These maps show the areas where the probability of finding an electron pair is high (Silvi and Savin, 1994). From Fig. 2.a, the elevated ELF regions (red regions) indicate the highly localized bond and non-bond electrons around the hydrogen atoms (H19). Blue regions around few carbon atoms indicate the delocalized electron cloud. In Fig. 2. b, it can be seen that the LOL map shows a white region around hydrogen atoms. This region designating that the electron density surpasses the high limit of color scale (0.80). In addition, high LOL values are represented by red colors located between carbon (C10, C7 and C4) indicates the covalent nature of the bonds between them. In contrast, the blue regions between organic and inorganic unit are small (0.12~ 0.24 a.u) gave evidence of existence of non covalent interactions leading to hydrogen bonds. The ELF and LOL results correlate very well with those obtained from QTAIM analyses.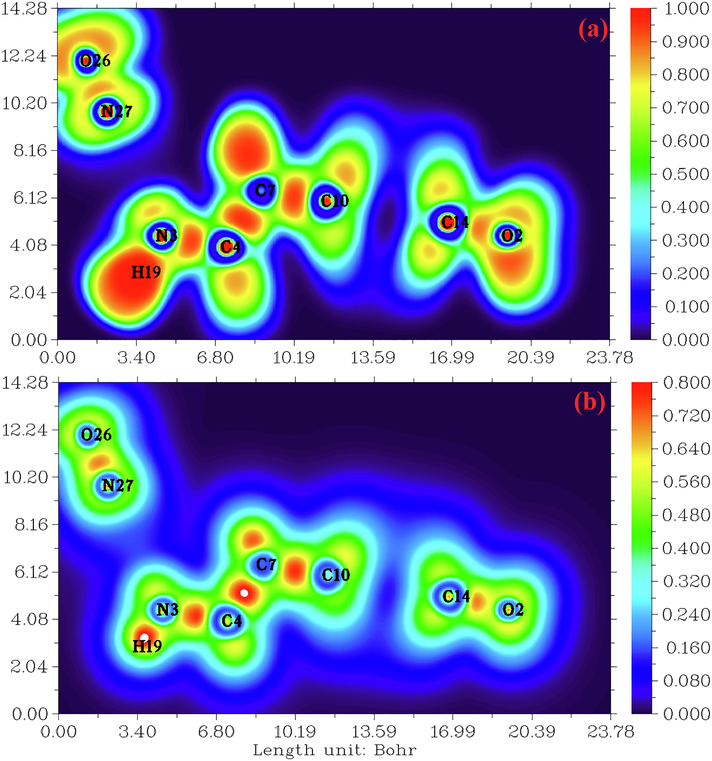
(a) and (b) electron localization function (ELF) and localized orbital locator (LOL) map for the title compound.
4.3 Hirshfeld analysis
Hirshfeld surface analysis is a good method for displaying and comprehending of intermolecular interactions present within the crystal structure of the studied compounds (Gatfaoui et al., 2020). The Hirshfeld surfaces of our 2DOPN molecule were generated with different properties: d norm, d i, Shap index and Curvedness obtained by the software Crystal explorer (Fig. 3). The normalized contact distance dnorm surface displayed three colors: red (negative d norm values), white (zero d norm values) and blue (positive d norm values) used to recognize the shorter, close and longer intermolecular interaction than the Van der Waals radii (Sagaama et al., 2020). Red spots on the d norm surface have been localized around oxygen and hydrogen atoms. This situation shows the presence of O…H/ H…O interactions, which are due to hydrogen bonds N-H···O, O-H···O and C-H···O within the crystal packing. The white color representing intermolecular distances close to Van der Waals contact and indicates the H…H interactions. The blue surfaces illustrate the domains where the neighboring atoms are too moved away to interact with each other. Furthermore, dnorm mapping makes it possible to display the different types of intermolecular interactions in our compound (Fig. 4). The circular deep red color displayed on this surface highlight the shortest intermolecular contacts which are attributed to the hydrogen bonds (due to N-H···O, C-H···O and O-H···O). Fig. 3.d and Fig. 3.b illustrate how the shape index and curvedness mapping can be also be used to identify characteristic packing mode existent in the crystal. The absence of red and blue triangles in the Shap index and Curvedness maps excluded the presence of π-π interactions and even if in C-H···π type interactions in our crystal structure. In addition, the fingerprints of the main contact involved in the studied compound are illustrated in Fig. 5. These plots resulting from the distribution of the pairs (di, de) and allow to highlight the mutual contacts of the atoms participating in close contacts in the crystalline structure. We can notice that the maximum contribution is from O…H/H…O (59%) (represents half of the total contribution) followed by H…H and C…H/H…C with 27.2% and 10.1%, respectively. The participation of the contact observed between carbon and hydrogen (C…H/H…C) is equal to 10.1%. The O…H/H…O contacts (Fig. 5.b) between the oxygen atoms located inside the HS and the hydrogen atoms situated outside and vice versa are characterized by two narrow symmetrical pointed wings with de + di = 1.7 A°. These types of contact provide evidence of N-H···O hydrogen bonds. The graphic associated with hydrogen atoms (Fig. 5.c) is described by an end which points towards the origin on the diagonal and that corresponds to the de + di = 1.1 A°. This result reveals the presence of close H…H contacts within the title compound 2DOPN. The composition of the 2D fingerprint (Fig. 5.e) also shows other contacts such as C…H/H…C (10.1%), O…O (1.8%). This analysis makes it possible to show the different intermolecular interactions obtained by x-ray diffractions, which help to stabilize the molecular structures.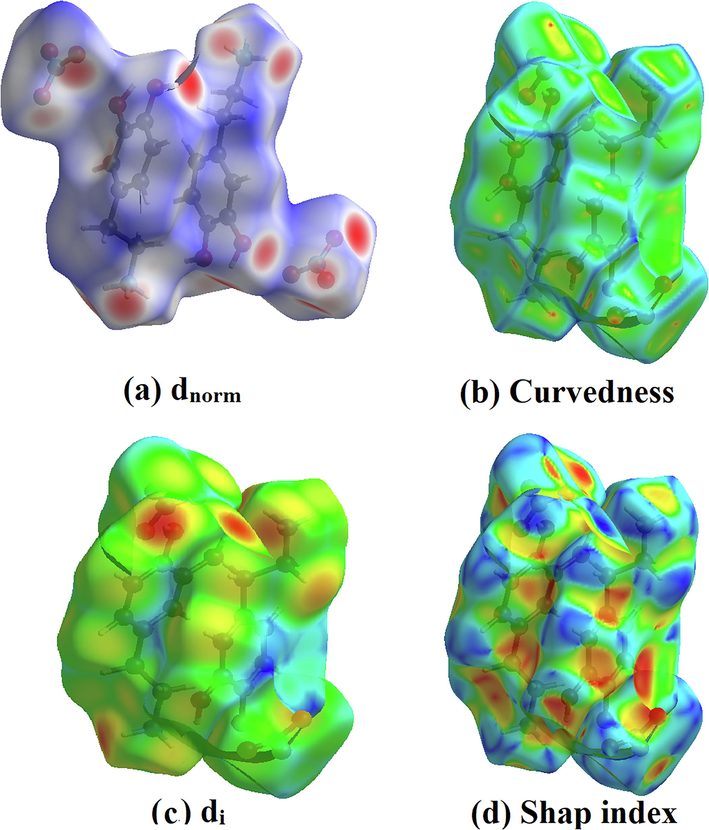
Hirshfeld surfaces: (a) dnorm, (b) Curvedness, (c) di and (d) Shape index of the title molecule.
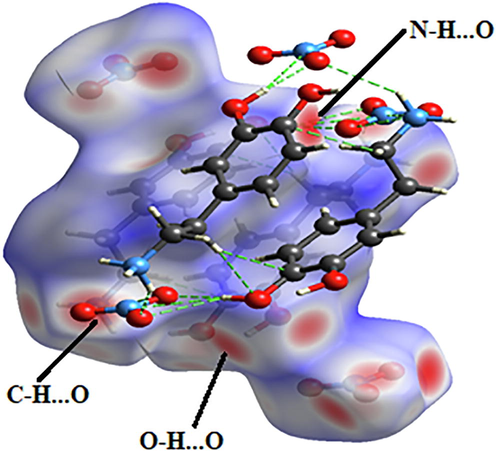
Different hydrogen bonds N–H…O, C–H…o and O–H…O observed by d norm mapping.
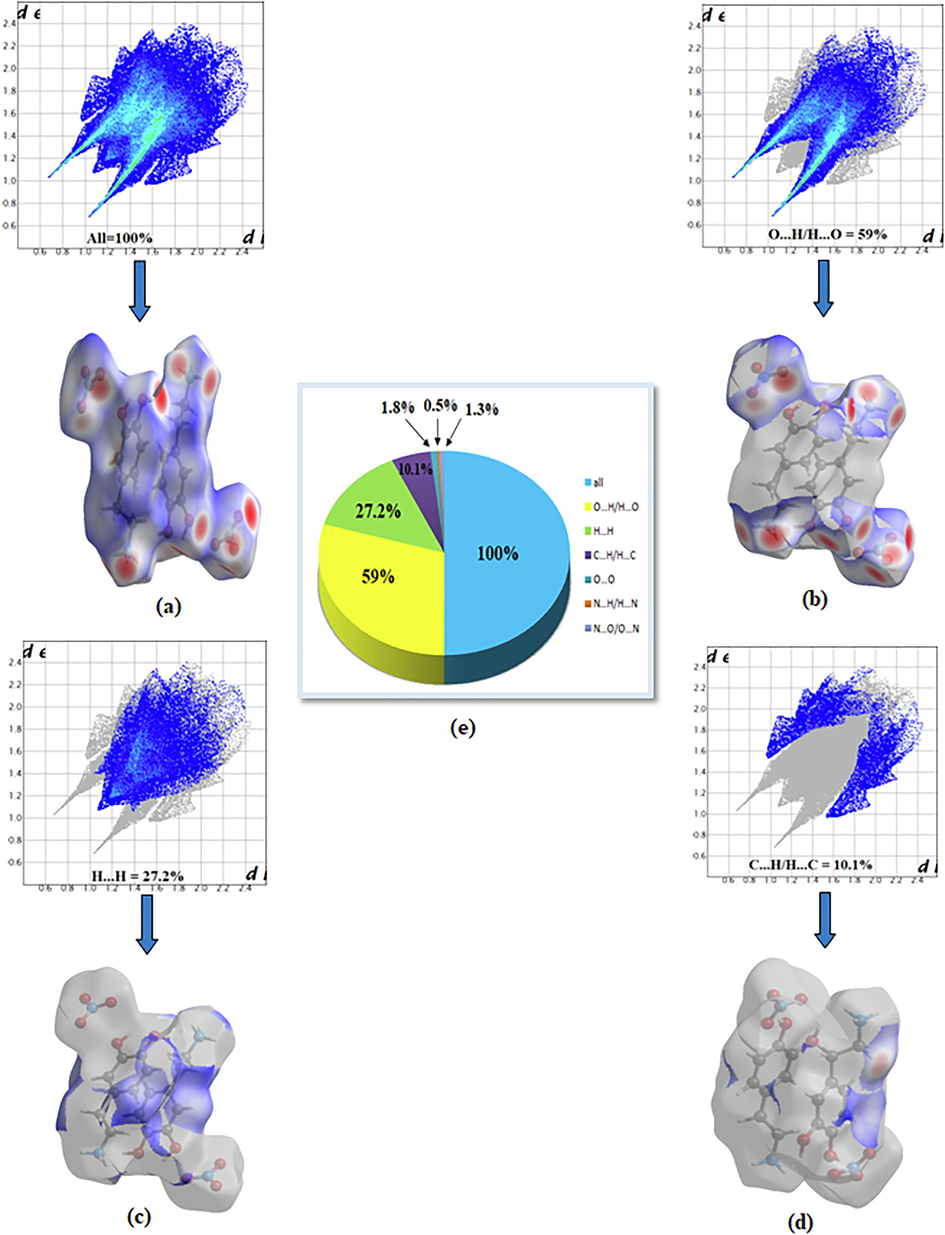
Bidimensional fingerprints of 2DOPN: (a) All intermolecular contact, (b) contact O…H/H…O, (c) H…H, (d) C…H/H…C and (e) percentage contribution to the Hirshfeld surfaces.
4.4 Reduced density gradient (RDG) visualization of non covalent interaction
In addition to the topology analysis, a visual approach known as Reduced Density Gradient analysis (RDG) has been added to study the non-covalent interactions. This method could be used to visualize different interactions such as hydrogen bonding, Van der Waals (VDW) interaction and steric effects (Tahenti et al., 2020). To probe the existence of weak interactions, it was shown that RDG analysis was able to recognize non-covalent interactions such as the hydrogen bonds, Van der Waals and steric effect. We represented on Fig. 6.a and Fig. 6.b, the RDG plots of 2-(3, 4-dihydroxyphenyl) ethanaminium nitrate in two and three dimensional spaces. There are three different interaction regions shown within our molecule located in the low density and have low RDG spaces, were indicated as blue, green and red spots. The red color indicating the steric repulsion (strong repulsion) between the atoms of molecule, which sign (λ2) ρ > 0. Green color appears in the area of the sign (λ2) ρ = 0 correspond to the Van der Waals interaction. The spikes in the region sign (λ2) ρ < 0 are blue in color, representing the strong electrostatic interaction like hydrogen bonding (Gatfaoui et al., 2020). As shown in Fig. 6.a, clear blue spots are presented between the hydrogen atoms of the NH3 group and oxygen atoms of nitrate group results from the weak hydrogen bond like N–H…O (strong attractive interaction). Red color was observed inside the center of aromatic rings, which indicate the steric repulsion (repulsive interaction). Furthermore, the C–H…π interaction is shown by the mixture of the red-green color. Green plates between the hydrogen atoms are attributed to Van der Waals interactions. In addition, to determine the nature and strength of the interactions analyzed using RDG surfaces, we have plotted the graphic of the RDG versus sign (λ2) ρ (Fig. 6.b). This graph shows a color code allows to visualize the interaction zones in 3D space. We noted large negative values of sign (λ2) ρ are indicative of stronger attractive and the binding interaction (H-bond). The interaction region marked by red color can explain the repulsive and non binding interaction located in the interval [0.02–0.05] a.u. The green color situated between −0.01 and 0.01 a.u in the RDG scatter graphs of C8H12N2O5, which corresponds the weak non covalent interaction (VDW type interaction).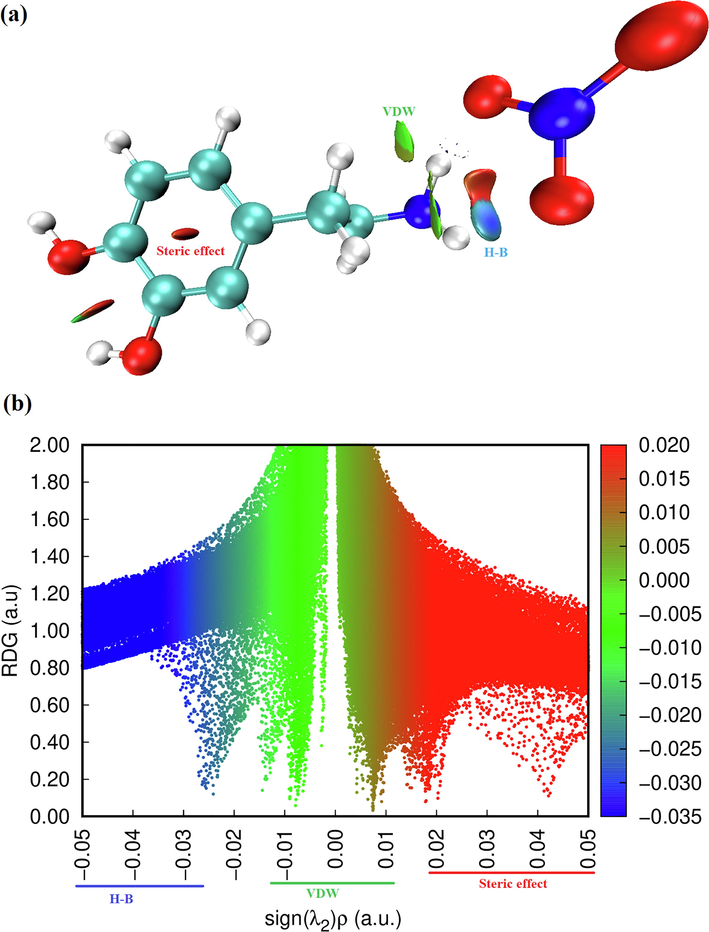
Graphs of RDG versus the sign (λ2) ρ of the molecule title.
5 Mulliken atomic charges
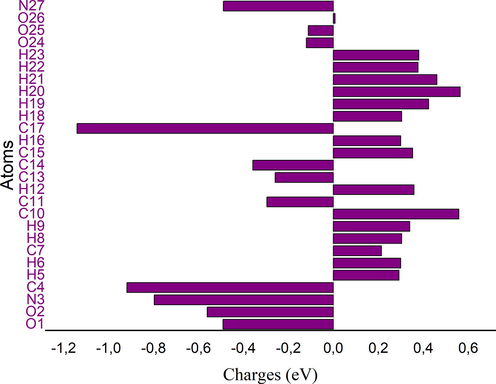
The Mulliken charges is an important tool that analyzes the electronic properties of a molecule, determining atomic charges, dipole moment and more propriety of molecular system (Sagaama et al., 2020). It used to describe the process of electronegativity and charge transfer in reaction (Issa et al., 2020). Mulliken charges describe the load distribution in a molecule and it allows to study the chemical reactivity present in the molecule. The total charge of 2-(3,4-dihydroxyphenyl) ethanaminium nitrate obtained by the Mulliken population analysis with B3LPY / 6–311 ++ G(d, p) is zero. The color range in the scale of positive (green color) and negative (red color) charges and histogram of Mulliken atomic charges in C8H12N2O5 is plotted in Figure S5 and S6. Also, the numerical values of Mulliken charges at various atoms of our molecule are collected in Table S2. Mulliken population analysis shows that the molecule title has many probable sites for electrophilic (presented by red color) and nucleophilic attack (green color). From the listed tabulated values (Table S2) of atomic charge, we can note that most atoms of our compound are positively charged. Moreover, all oxygen and nitrogen atoms (N27, O25, O24, O1 and O2) are negatively charged ones, involved that these centers have the highest electron density, which can interact with the positively charged part of the other atoms easily. The charge distribution of C8H12N2O5 showed that the carbon atom attached with hydrogen atoms (C15, C10 , C17 and C7) is positive, whereas the remaining carbon atoms are negatively charged due to these carbon atoms (C4, C13, and C14) attached with nitrogen and oxygen atom. We noted also that all hydrogen atoms of the molecule title are positively charged and the larger values observed for H20, H19 and H21. These net positive charge values on H atom with the presence of a large negative charge on O and N atoms may suggest the formation of intermolecular interaction (formation of hydrogen bonds) (Kavimani et al., 2017).
6 Natural bond orbital (NBO) analysis
The NBO analysis is a useful method for giving information about intra and intermolecular bonding and interactions among the bonds (Sagaama and Issaoui, 2020). Also, it provides a convenient basis for inspecting charge transfer and conjugative interactions possible in molecular system (Bouacida et al., 2019).This tool helps in examining hydrogen bonding and also hyperconjugative interactions among occupied Lewis type (electron-donors) and unoccupied orbitals (electron-acceptors) (Sethi and Prakash, 2015). In addition, the NBO charge distributions are similar that the Mulliken atomic charges with more or less significant differences in charge values (Romani et al., 2020). In the present compound the natural bond orbital has been calculated at the DFT / B3LYP / 6–311 ++ G(d, p) level and a summary of electron donors (E(i)), electrons acceptors (E(j)) and stabilization energy (E(2)) were presented in Table S3. The energy E (2) characterize the interaction between donor–acceptor, which is determined from the second order perturbation method. Furthermore, larger E(2) values show the intense interactions among electron-donor and acceptor and greater the stability of the molecular system. In our compound C8H12N2O5 hyperconjugative interactions result by the delocalization of electron from σ → σ*, σ → π*, π → π* and LP → σ*. From these results the maximum interactions, which played a vital role in the stabilization of our compound originated from π → π*. The interaction between π (C14-C15) → π *(C11-C13), π (C11-C13) → π *(C10-C17) and π (C14-C15) → π *(C11-C13) of the phenyl ring giving the highest stabilization energy E(2) values around 224.69, 20.95 and 18.97 kcal/mol respectively. Other high energy interactions involving the lone pair (LP) of electron with the anti-bonding σ* electron such as LP2 (O25) → σ*(N3-H20), LP2 (O24) → σ*(N3-H21) and LP1 (O25) → σ*(N3-H20) correspond to stabilizing the molecule with 47.14 kcal/mol, 6.97 kcal/mol and 5.98 kcal/mol, respectively. We remarked also that the NBO calculations illustrate clearly and confirm the existence of hydrogen bonds (LP (O) → σ*(N–H)) as observed in the crystal structure. We can conclude that the H-bond interactions contribute larger to the stabilization of our compound association that Van der Waals interactions.
7 Predicting reactive sites
7.1 Molecular electrostatic potential analysis (MEP)
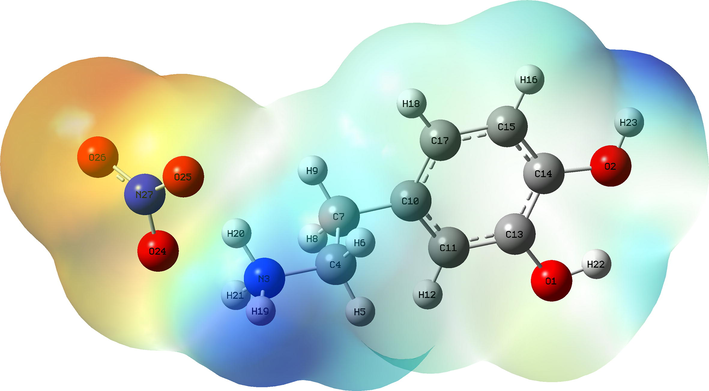
The molecular electrostatic potential (MEP) is a very important propriety for studying and predicting reactive molecular. It is related to the electron density, which is a very useful descriptor for identifying nuleophilic and electrophilic attack sites as well as intermolecular hydrogen bonding (Gatfaoui et al., 2018). We plotted in Figure S7 the three dimensionally map to show the charge distributions of C8H12N2O5. The reactive sites can be located by different color codes situated between −8.480 e-2 a.u (deepest red) to 8.480 e-2 a.u (deepset blue). In general, the red and yellow color is a negative region, which indicates an electron rich site showing nucleophilic reactivity (Noureddine et al., 2021). This region is localized around the oxygen atoms of the nitrate anion. The MEP map shown that the blue color is a positive region promoting the site favorable for electrophilic attacks. This regions is situated around the hydrogen atoms of the organic group (precisely around the NH3+), which indicates an electron deficient sites. According to the figure, we find that the region around the hydrogen atoms of the ammonium group are sites for electrophilic activity (the most positive potential) and oxygen atoms of the NO3− anion group. are sites for nucleophilic attraction (the most negative potential). These sites give details about the region from which the compound can have intermolecular interactions. The MEP analysis affirms the existence of hydrogen bonds between the organic and inorganic groups of our molecule N-H···O type.
7.2 Molecular electrostatic potential (ESP) on the molecular VDW surface
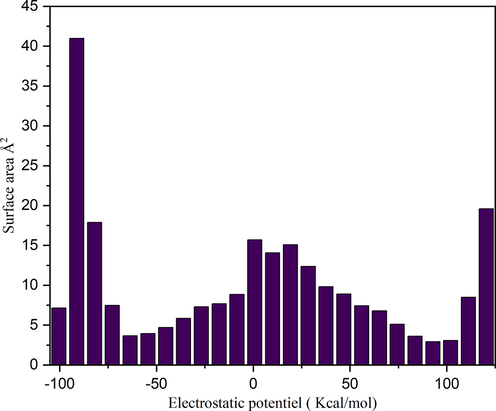
The ESP on the molecular VDW surface helps to investigate the reactive behavior and predict intermolecular interactions. In general, the regions of greatly negative ESP will clearly accept hydrogen bonds, while hydrogen atoms with a positive ESP will be potential donor sites (Noureddine et al., 2021). Furthermore, ESP allows to show that the atom closest to the global ESP minimum on the VDW surface indicating as much as possible to be an active site for electrophilic attack. In this part, we plotted in Figure S8 the molecular structure and surface extrema of our compound and the Summary of surface analysis are given in Table S4. From this figure, the maxima and minima surface are indicated as red and blue spheres respectively. The number of surface maxima 1 has a maximal value equal to 122.86 kcal/mol that correspond to global maximum arising from the positively charged H19. At this point the ESP is much larger than those at other maxima. This is due to the fact of the presence of nitrogen, which attracts the electron from the hydrogen H19. It can be seen that the lone pair of the oxygen O2 of organic group and O25 of the inorganic group leads to a minimum value equal to −89.17 Kcal/mol (minimum 4). Since, it is also the global minimum on the surface, the oxygen's O2 and O25 should be the most favorable site for interacting with positively charged species. The surface area in different ESP ranges is shown in Figure S9. From this graph, it can be seen that there is a large part of the molecular surface having a low ESP value. We also notice the existence of small areas with negative and positive ESP values, corresponding to the regions of maximum and minimum global ESP respectively. In conclusion, we find that the maximum energies as well as the minimum energies located on this surface are derive from the interactions between the two organic and inorganic groups via hydrogen bonds N-H···O, C-H···O and O-H···O.
7.3 Global reactive descriptors: HOMO-LUMO analysis
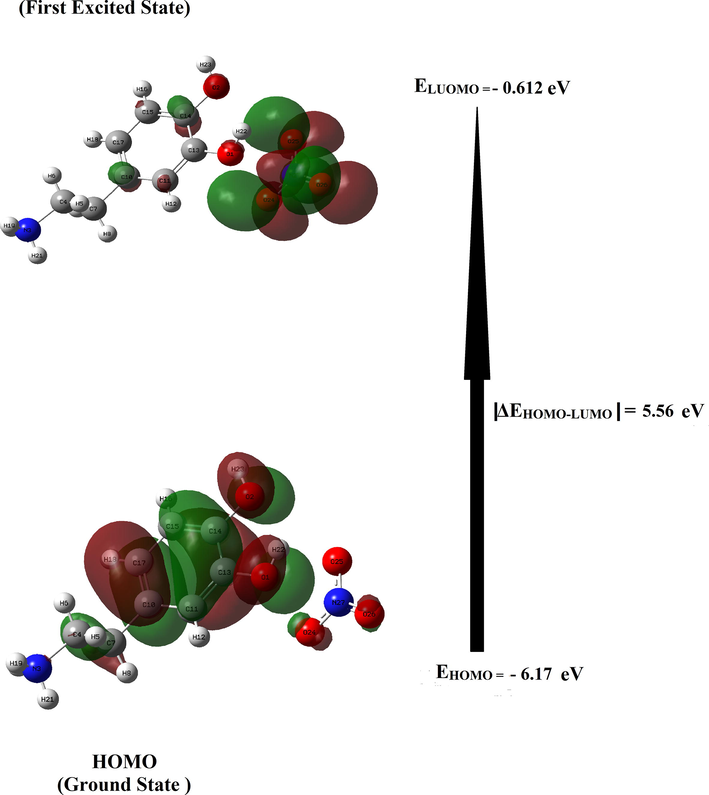
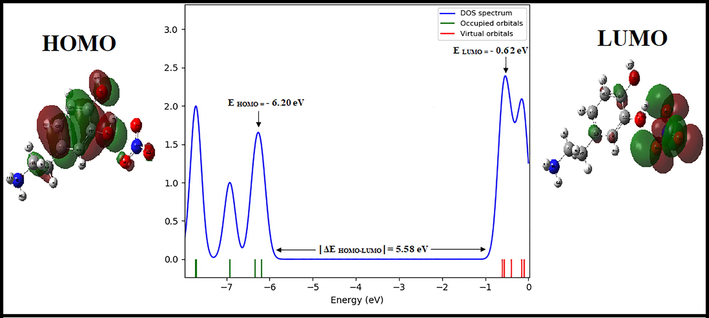
In order to determine the electronic propriety, stability and reactivity of diverse compounds in terms of frontier molecular orbital, a TD-DFT calculation has been realized. HOMO and LUMO are referred to as frontier molecular orbital and acts primarily as an electron donor and acceptor respectively. This energy gap between the highest occupied (HOMO) and the lowest unoccupied molecular orbital (LUMO) plays an important role in the explaining kinetic stability as well as the chemical reactivity of the compound (Noureddine et al., 2021). The literature reveals that molecules having large / small values of energy gap are known as hard / soft molecules (Noureddine et al., 2021). Furthermore, the soft molecules (less reactive) are more polarizable than the hard ones because they need less energy for excitation (Janani et al., 2021). The plot HOMO-LUMO expose the passage of charges within the molecule. Figure S10 displays the HOMO and LUMO for our molecule with the corresponding energies and energy gap ΔEHOMO-LUMO. It can see that the surfaces for FMOs are indicated with two colors; red and green explain positively (electrophilic sites) and negatively (nucleophilic sites) charged surfaces. From this figure, it is clear that the HOMO is entirely localized on the [C8H12NO2]+ cation (precisely in phenyl rings) with a small contribution of two oxygen atoms of the NO3– anion. LUMO is localized on the NO3– anion with a small contribution of a few atoms of the organic group. We are grouped in Table S4 the global reactivity energy descriptor such as EHOMO, ELUMO, electron affinity (A), ionization potential (I), energy gap ΔEHOMO-LUMO, chemical potential (μ), electronegativity (χ), chemical hardness (η), global softness (S) and electrophilicity index (ω) and additional electronic charges (ΔN) (donor and acceptor charge behavior) of our compound. The energy of HOMO and LUMO of the title compound in gas phase ais equal to − 6.17 eV and − 0.61 eV, resp. The absolute value of the energy |ΔE HOMO-LUMO|is calculated using B3LYP / 6–311++G (d, p) method is equal to 5.56 eV. Besides, the large / small value of the chemical potential and electrophilicity index explain the good electrophilic / nucleophilic approach of molecules, respectively (Nageswari et al., 2018). The electrophilicity index is found to be 2.09 eV, which is small and this value confirmed that our molecule has a character nucleophilic. In addition, the value of ω is positive, which indicates that the system can act as an acceptor of electrons and clearly indicating the interactions between filled and unfilled orbital. The chemical hardness is a useful indicator for scaling the stability and reactivity of the molecules. The η is found to be 2.75 eV, therefore the title compound has much stability and rigidity character. Furthermore, if the molecule possessed an elevated electronegativity number, the studied compound will attract electrons to itself. The electronegativity of our molecule is equal to 3.39 eV (this value is>1.7 eV), in this case the character of chemical bond is changed from covalent to ionic. The density of states (DOS) describes the number of available states of molecular orbitals at a particular energy level. The DOS graph was obtained by using TD-DFT Gaussian output file via Gauss-Sum package and it is shown in Figure S11. From the DOS plot, the red and green lines signify the HOMO (occupied orbitals) and the LUMO (virtual orbitals), respectively. The study of the DOS plot and its energy levels (|ΔE HOMO-LUMO|= 5.58 eV) also verify the FMOs analysis.
8 Non linear optical propriety
Nonlinear optics corresponds to optical phenomena involving intense electric fields and playing an important role in many fields of application such as medicine, optoelectronics, and telecommunications (Kirouani et al., 2020). In order to explain the NLO properties, some quantum chemical descriptors which are Polarizability (α) and first order hyperpolarizability (β0) have been used. These parameters have been converted into electrostatic units (esu) and reported in Table S6 and calculated from the following equation such as:
The linear Polarizability describes the capacity of an electric field to distort the electronic distribution of a molecule. The first hyperpolarizability that explain the nonlinear response of atoms and molecules are related to a large range of phenomena variant from nonlinear optics to intermolecular forces, for example the stability of chemical bonds (Akman et al., 2020) as well as the conformation of molecules and molecular aggregates. From the Table S6, the highest value of the dipole moment and hyperpolarizability is observed for direction z (µz = 4.936 Debye and βzzz = 34.9721 esu), while the Polarizability is located along the y-axis (αyy = 2.4740 esu). Urea is one of the reference molecules which is used as a comparison value in the study of the NLO properties of molecular systems (μ of urea is 1.3732 Debye and β is 3.728 × 10-29esu). The μ tot, αtot and βtot values are build to be 14,179 D, 1.9471 × 10-23 esu and 2.3344 × 10-29 esu, respectively. The dipole moment of the title molecule is 10.32 times greater than that of urea and β0 is 0.62 times higher than the reference. Therefore, this result indicates the nonlinearity of the title molecule.
9 Thermal behavior of C8H12NO2 (NO3) compound:
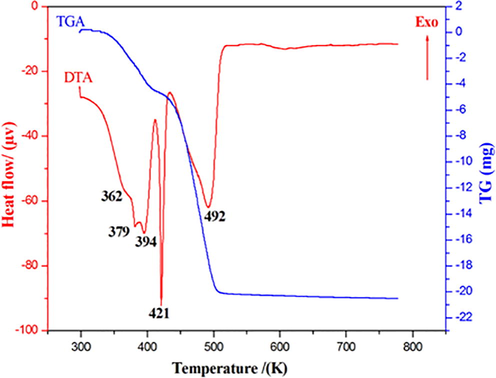
The differential and thermogravimetric thermal analysis curves of dopaminium nitrate on a sample (m = 29.19 mg) carried out under an argon atmosphere in a [298–773 K] temperature range are shown in Figure S12. The TGA curve shows several successive mass losses, the first loss is observed between 350 and 400 K. Differential thermal analysis (DTA) shows that this mass loss corresponds to a series of endothermic peaks, we can think of this stage starting from two water molecules resulting from the condensation of two (OH) hydroxyl groups of the dopaminium entity (experimental% = 16.85 and theoretical% = 17.14). Two endothermic peaks located respectively at 421 K and 492 K accompanied by two successive mass losses, the second of which is the most important, during which the material undergoes in a first stage under the effect of heat a melting and beginning of its decomposition and in a second step it completely decomposes giving gases formed of nitrogen dioxide.
10 Molecular docking
Parkinson, Alzheimer and other disorders are known as neurodegenerative diseases affect the human nervous system. Research from the Institute of Neurodegenerative Diseases revealed that these disorders are commonly seen in the elderly. Dopamine is a key neurotransmitter in Parkinson's disease. In this context, to establish the biological activity of our compound 2-(3, 4-dihydroxyphenyl) ethanaminium nitrate, we performed a docking calculation to discover a new effective treatment for Parkinson disease. Molecular docking is an efficient method to predict the best binding site of protein and ligand.
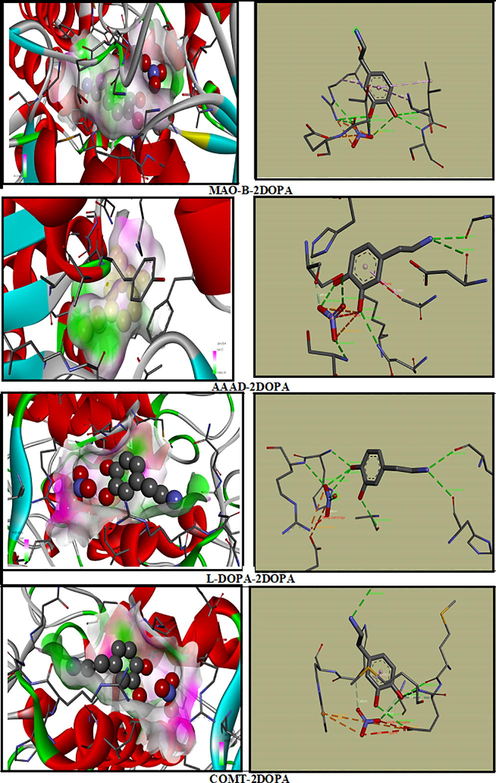
In Parkinson’s disease the neurons that build dopamine are destroyed. To compensate for the dopamine deficit, type B monoamine oxidase inhibitors (MAO-B), C-O-methytransferase (COMT) inhibitors, DOPA decarboxylase (AAAD) and L-DOPA are drugs used to degrade dopamine. In addition, clinical studies prove the effectiveness of MAO-B protein in the development of therapeutics for the treatment of Parkinson's and Alzheimer’s disease (Yamada and Yasuhara, 2004). Our research is to study the inhibition of the different enzymes (COMT, MAO-B, AAAD and L-Dopa) with our molecule 2DOPN (ligand). The structure of the proteins was taken from the Protein Data Bank (PDB). During the calculation, we have determined 10 poses and we presented the best pose which corresponds to lowest binding energies of various proteins with the ligand (Fig. 7). Table 3 summarized molecular docking results of interaction energies (H-bond, VDW and electronic). The complex protein/ ligand with the lowest total energy will exhibit better activity and subsequently better inhibition (Jomaa et al., 2020). From the table, it can be shown that the total energy of the title molecule 2DOPN (ligand) with MAO-B, COMT, L-DOPA and AAAD were established to be −94.31 kcal/mol, −91.99 kcal/mol, −86.32 kcal/mol and −86.08 kcal/mol, respectively. MAO-B has the highest total energy Etot = -94.31 kcal/mol. COMT has the strongest VDW interaction equal to −75.05 kcal/mol. It is also noted that despite the protein L-Dopa has a lower VDW value (-37.99 kcal/mol) but it has the highest value of H-bond (-50.40 kcal/mol). In addition, AAAD has the lowest total energy value, but it has the highest H-Bond value (-41.94 kcal/mol) compared to the others proteins (MAO-B and COMT). As you can see in Figure S13, the ligand 2DOPN is linked to the proteins by several interactions such as; Pi-Alkyl (between the aromatic group and any alkyl group), Pi-Anion, charge-charge. These interactions are important for protein stability (Jomaa et al., 2020). This study indicates that the interactions between ligand / proteins it makes clear that our compound can be an active potential drug and has drug proprieties. Therefore, all this results confirm that 2DOPN may lead to the discovery of new effective treatments against Parkinson's disease.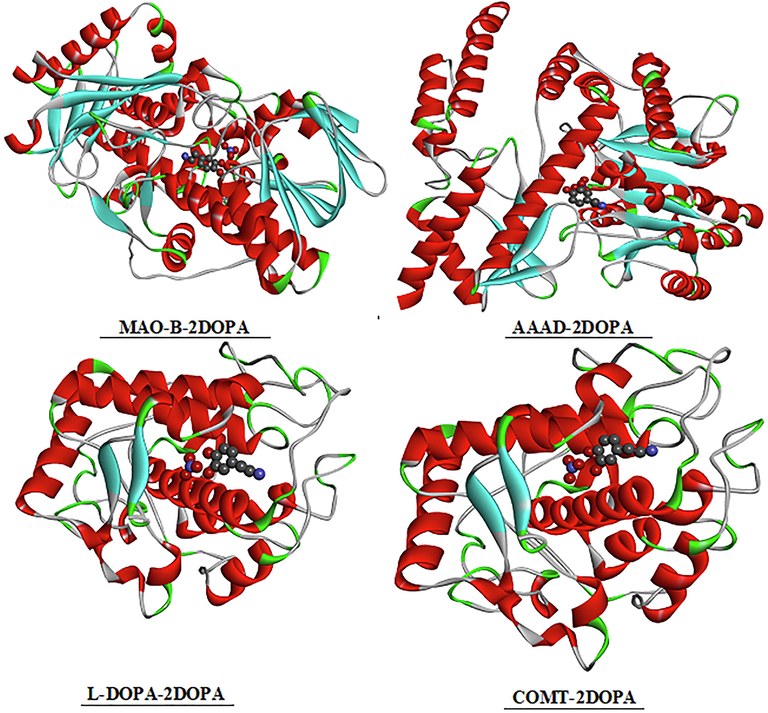
The best docked poses of the ligand 2DOPN with the different proteins.
Ligand
Proteins
code
Total Energy
VDW
H-Bond
Electronic
2DOPN
MAO-B
[2V5Z]
−94.31
−64.11
−31.13
0.92
COMT
[1H1D]
−91.99
−75.05
−16.05
−0.90
L-dopa
[4P6S]
−86.32
−37.99
−50.40
2.07
AAAD
[1JS3]
−86.08
−45.34
−41.94
1.20
11 Conclusion
Computational studies have been carried out with the title compound 2-(3, 4-dihydroxyphenyl) ethanaminium nitrate using DFT methods at B3LYP/6–311++G(d,p) level of theory. A comparison between the experimental and theoretical parameters confirmed that the calculated results can well reproduce the data parameters. The molecular structure of the compound is connected the dopaminium cations by N-H···O, C-H···O and O-H···O hydrogen bonds. In this work, QTAIM, ELF, LOL, RDG as well Hirsfeld surface analyses were performed to confirm the presence of these non covalent interactions. The Mulliken atomic charges and the NBO analysis have been used to study the stability of our molecule. The frontier molecular orbitals were beneficial to evaluate the reactivity of 2DOPN. In addition, the results of Non linear optical propriety conclude that our molecule has a reasonable good NLO behavior. The molecular docking study was performed for four targeted proteins MAO-B, COMT, AAAD and L-DOPA related to Parkinson. These results confirm that our molecule 2DOPN may lead to the discovery of new effective treatments against Parkinson's disease.
Acknowledgements.
Researchers Supporting Project number (RSP-2021/61), King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Dopaminium nitrate. Acta Crystallogr. Sect. E: Struct. Rep. Online. 2014;70(5):o571-o572.
- [Google Scholar]
- Ropinirole for the treatment of early Parkinson's disease. Neurology. 1997;49(2):393-399.
- [Google Scholar]
- New acyclic receptors containing pyridazine units. The influence of π-stacking on the selective transport of lipophilic phenethylamines. Tetrahedron. 2005;61(50):11965-11975.
- [Google Scholar]
- (a) M. Mirzaei H. Eshtiagh-Hosseini Z. Karrabi K. Molčanov E. Eydizadeh J.T. Mague A. Frontera Crystal engineering with coordination compounds of Ni II, Co II, and Cr III bearing dipicolinic acid driven by the nature of the noncovalent interactions CrystEngComm 16 24 2014 5352 5363 (b) H. Eshtiagh-Hosseini M. Mirzaei S. Zarghami A. Bauzá A. Frontera J.T. Mague M. Shamsipur Crystal engineering with coordination compounds of 2, 6-dicarboxy-4-hydroxypyridine and 9-aminoacridine fragments driven by different nature of the face-to-face π⋯ π stacking CrystEngComm 16 7 2014 1359 1377 (c) M. Mirzaei H. Eshtiagh-Hosseini A. Bauzá S. Zarghami P. Ballester J.T. Mague A. Frontera On the importance of non covalent interactions in the structure of coordination Cu (II) and Co (II) complexes of pyrazine-and pyridine-dicarboxylic acid derivatives: experimental and theoretical views CrystEngComm 16 27 2014 6149 6158
- Interaction of hydrology and nutrient limitation in the Ridge and Slough landscape of the southern Everglades. Hydrobiologia. 2006;569(1):37-59.
- [Google Scholar]
- EXPO: a program for full powder pattern decomposition and crystal structure solution. J. Appl. Crystallogr.. 1999;32(2):339-340.
- [Google Scholar]
- Gaussian 09, Revision C.01, Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J.A., Jr.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N.J.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, Ö.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. Gaussian, Inc., Wallingford CT, 2009.
- GaussView, Guassian, Inc. (Carnergie Office Parck-Building6 Pittsburgh PA 151064 USA), Copyright © 2000-2003 Semichem. Inc.
- T. Lu F. Chen 33 5 2012 580 592
- Crystal Explorer (Version 3.1) 2012:UWA.
- J. Comput. Chem.. 2008;29:839.
- Accelrys software inc. Discovery Studio Visualizer. 2005;2
- J.-M. Yang C.-C. Chen GEMDOCK: a generic evolutionary method for molecular docking Proteins Struct. Funct. Bioinforma. 55 2 2004 288 304 http://www.rcsb.org/pdb/
- Vibrational spectroscopic, NBO, AIM, and multiwfn study of tectorigenin: A DFT approach. J. Mol. Struct.. 2020;1217:128443.
- [CrossRef] [Google Scholar]
- Computational study of 3-thiophene acetic acid: Molecular docking, electronic and intermolecular interactions investigations. Comput. Biol. Chem.. 2020;86:107268
- [Google Scholar]
- Electron localisation descriptors in ONETEP: a tool for interpreting localisation and bonding in large-scale DFT calculations. Electronic Structure. 2020;2(2):027001.
- [CrossRef] [Google Scholar]
- Classification of chemical bonds based on topological analysis of electron localization functions. Nature. 1994;371(6499):683-686.
- [Google Scholar]
- Synthesis, experimental, theoretical study and molecular docking of 1-ethylpiperazine-1,4-diium bis(nitrate) Solid State Sci.. 2020;106:106326
- [Google Scholar]
- Molecular docking studies, structural and spectroscopic properties of monomeric and dimeric species of benzofuran-carboxylic acids derivatives: DFT calculations and biological activities. Comput. Biol. Chem.. 2020;87:107311.
- [CrossRef] [Google Scholar]
- A tetrachlorocobaltate(II) salt with 2-amino-5-picolinium: Synthesis, theoretical and experimental characterization. J. Mol. Struct.. 2020;1207:127781
- [Google Scholar]
- Synthesis, experimental and computational study of a non-centrosymmetric material 3-methylbenzylammonium. J. Mol. Struc.. 2020;1225:129132
- [Google Scholar]
- Quantum chemical calculation (RDG) of molecular structural evaluation, Hirshfeld, DSSC and docking studies of 4-nitrophenylacetic acid. J. Mol. Struct.. 2017;1149:69-83.
- [Google Scholar]
- Design, molecular docking analysis of an anti-inflammatory drug, computational analysis and intermolecular interactions energy studies of 1-benzothiophene-2-carboxylic acid. Comput. Biol. Chem.. 2020;88:107348
- [Google Scholar]
- Synthesis, crystal structure, hirshfeld surface analysis, DFT calculations and thermal properties of a new anilinium derivative chlorostannate (IV) hybrid compound. J. Mol. Struct.. 2019;1198:126900.
- [CrossRef] [Google Scholar]
- Novel synthetic ester of Brassicasterol, DFT investigation including NBO, NLO response, reactivity descriptor and its intramolecular interactions analyzed by AIM theory. J. Mol. Struct.. 2015;1083:72-81.
- [Google Scholar]
- Properties and Reactivities of Niclosamide in Different Media, a Potential Antiviral to Treatment of COVID-19 by Using DFT Calculations and Molecular Docking. Biointerface Res. Appl. Chem.. 2020;10:7295-7328.
- [Google Scholar]
- Synthesis and characterization of p-xylylenediaminium bis (nitrate). Effects of the coordination modes of nitrate groups on their structural and vibrational properties. J. Mol. Struct.. 2018;1151:152-168.
- [Google Scholar]
- DFT and molecular docking study of chloroquine derivatives as antiviral to coronavirus COVID-19. J. King Saud University - Science. 2021;33(1):101248.
- [CrossRef] [Google Scholar]
- Quantum chemical studies on molecular structure, AIM, ELF, RDG and antiviral activities of hybrid hydroxychloroquine in the treatment of COVID-19: Molecular docking and DFT calculations. Journal of King Saud University-Science. 2021;33(2):101334.
- [CrossRef] [Google Scholar]
- Quantum chemical calculations, spectroscopic properties and molecular docking studies of a novel piperazine derivative. J. King Saud University - Science. 2021;33(2):101283.
- [CrossRef] [Google Scholar]
- Molecular structure, spectroscopic (FT-IR, FT-Raman, NMR), HOMO-LUMO, chemical reactivity, AIM, ELF, LOL and Molecular docking studies on 1-Benzyl-4-(N-Boc-amino) piperidine. J. Mol. Struct.. 2021;1230:129657.
- [CrossRef] [Google Scholar]
- Electronic and vibrational spectroscopic (FT-IR and FT-Raman) investigation using ab initio (HF) and DFT (B3LYP and B3PW91) and HOMO/LUMO/MEP analysis on the structure of l-serine methyl ester hydrogen chloride. J. Mol. Struct.. 2018;1166:422-441.
- [Google Scholar]
- Effect of the phosphonomethylene moiety on the structural, vibrational, energetic, thermodynamic and optical proprieties of ((Phenylcarbamoylmethyl-phosphonomethyl-amino)-methyl)-phosphonic acid: DFT investigation. J. Mol. Struct.. 2020;1215:128193.
- [CrossRef] [Google Scholar]
- F. Akman, N. Issaoui, A. S. Kazachenko, Intermolecular hydrogen bond interactions in the thiourea/water complexes (Thio-(H2O)n) (n = 1, …, 5): X-ray, DFT, NBO, AIM, and RDG analyses, J. Mol. Modeling, 26(61),(2020) 161.
- Clinical pharmacology of MAO inhibitors: safety and future. Neurotoxicology. 2004;25:215-221.
- [Google Scholar]
- Experimental computational and in silico analysis of (C8H14N2) 2 [CdCl6] compound. J. Mol. Struct.. 2020;1213:128186.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2021.101616.
Appendix A
Supplementary data
The following are the Supplementary data to this article: