

Translate this page into:
Molecular level interaction of solvents (water, benzene and DMSO) analysis of the 2-Bromo-6-nitrotoluene's reactive charge transfer, docking, and spectroscopic properties
⁎Corresponding author. issaoui_noureddine@yahoo.fr (Noureddine ISSAOUI)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University. Production and hosting by Elsevier.
Abstract
The chemical structure of 2-Bromo-6-nitrotoluene was optimized using the B3LYP/6–311++G (d, p) basis set and density functional theory. Calculations were made on the molecules of the substance 2-Bromo-6-nitrotoluene's geometrical-parameters. A variety of DFT techniques, including border atomic orbitals, essential binding orbitals, local reaction descriptors, and molecular electrostatic potentials, are worn to test the reactivity of molecules. Several solvents (water, benzene and DMSO) were examined by UV–vis spectroscopy and vapor phase electronic transition. Estimated efficiency for the most heavily inhabited molecular orbital and even the least vacant molecular orbital describes electron excitation properties. Environmental toxicity, pharmacological similarity, localized orbital location, and electron localized function were assessed. To evaluate the material's metabolic processes, chemical interaction modelling was used. In conclusion, this paper offers a thorough investigation combining spectroscopic and quantum computational methods to evaluate the metabolic function and solvation impact of selected therapeutic substances. The information gathered is therefore very useful in planning for future study.
Keywords
DFT
MEP
ELF
Molecular docking

1 Introduction
Several nitro derivatives of toluene or methylated derivatives of nitrobenzene have a number of industrial and pharmaceutical uses (Booth, 2015; Allangawi et al., 2023a, 2023b, 2023c, 2023d, 2023d, 2023e). Nitrotoluene is commonly employed in the production of pigments, anti - oxidants, agro - reagents, and pictorial chemical products. Drug intermediate for substances including thioacetazone, para-aminobenzoic acid, benzocaine, procaine hydrochloride, and folic acid, among others. N-acetyl methyl ester of (±)-clavicipitic acids (Harrington et al, 1987; Allangawi et al., 2023a, 2023b, 2023c, 2023d, 2023d, 2023e) and carbazomadurin, a highly oxygenated carbazole alkaloid that protects neuronal cells (Knölker and Knöll, 2003; Allangawi et al., 2023a, 2023b, 2023c, 2023d, 2023d, 2023e; Kosar et al., 2023), have both been completely synthesized using 2-bromo-6-nitrotoluene as a starting reagent. It serves as a common structural system for a variety of naturally existing chemicals and physiologically active substances. (Segneanu et al., 2017; Atanasov et al.,2021). Heterocyclic aromatic and pyrroles indicator containing bridgehead nitrogen have recently piqued the interest of drug research activities. (Constantinos et al., 2019; Markus et al., 2016; Sun et al., 2020). The preparation of dyes is its main use. Variants derived from 2-Bromo-6-nitrotoluenes have been shown to be useful in a variety of cellular mechanisms, comprising cytotoxic, antimicrobial activity, and anti-inflammatory actions. (Asif, 2016; Ata et al., 2004).
The chemical formula (C7H6BrNO2) and molecular weight (216.03 g/mol) of 2-Bromo-6-nitrotoluene salt are given. The results of a thorough investigation show that substantial research has been done on the synthesis of 2-Bromo-6-nitrotoluene and substituted derivatives as well as their biological features, never received attention on precise pharmacological predictions. It stimulates as to conduct a detailed exploration of 2B6NT. The current study investigates the optimized molecule's chemical structures, solvation effects, and biological activities (Frisch and Fox, 2009). One can transform a collection of digital time samples using the DFT into its frequency domain representation also wide applications in drug development. By accounting for all bonds, the Potential Energy Distribution is generated. Pre-Adme (Lee, 2004) and Gusar (Zakharov, 2010) are online software programs that were used to assess ADMI, drug similarity, and toxicity characteristics also evaluations of UV (ultraviolet) for various solvents have been performed. The interaction of the molecule with various proteins is revealed by molecular docking studies, which explains why it has effective anti-inflammatory capabilities (Sagaama et al., 2020). In the final analysis, the present dissertation gives a thorough evaluation of the solvation effects and pharmacological functionality of selected pharmaceutical substances utilizing quantum computational and spectroscopic techniques. As an outcome, the data gathered are very useful for planning upcoming study.
2 Evaluation of computation
DFT assessment and assimilation for 2B6NT have been performed in this work using the Gaussian 09 W (Trucks et al., 2009) software platforms. The recipient associations in the NBO paradigm were investigated using the new package F O C K matrices. (Reed et al.,1988) Computed parameters included global hardness (g), softness (S), energy gap (DE), chemical potential (m) energies, RDG and molecular orbitals with the most and least occupancy (Arjun et al., 2020). Using Multiwfn (Tian and Chen, 2012; Malar Wezhli et al., 2022) ELF and LOL assessments are conducted. Likewise, the chemical's streamlined shape is studied and therapeutic similarity and ADMET properties have been characterized. The structural modelling tool Auto-Dock (Sangeetha et al., 2021) has been deployed to determine the ideal contractual configuration and affinities.
3 Findings & discussion
3.1 Geometry - optimization
Title compound's optimized structural parameters were determined employing 6–311++G(d,p) basis set with atom numbering system at B3LYP levels. Fig. 1 displays the outcomes. Bond-length and bond-angles of the compound with the title are provided in Table 1. The number of bonds in this molecule is 8 CC, 6 CH, 2 ON and 1 OH. It is discovered that is (O9-H15) is 2.364 Å, which is an extremely large band length. The average length of CC bonds is 1.419 Å, and the O—N (B-L) is 1.224 Å. O—H (B-A) is 2.364 Å while C—H (B-L) is 1.091 Å on average. Compared to other bond angles, N8-O9-H15has the lowest bond angle (88.10 0 *) and O9-N8-O10 has the highest bond angle (124.9 0) were determined employing D.F.T technique. While the investigative XRD (Piers et al., 1962; Foster et al., 1950) was performed on the compound in its solid state and the theorized examination was carried out on the compound in its gaseous form, there are only tiny variations among theorized and experiential results.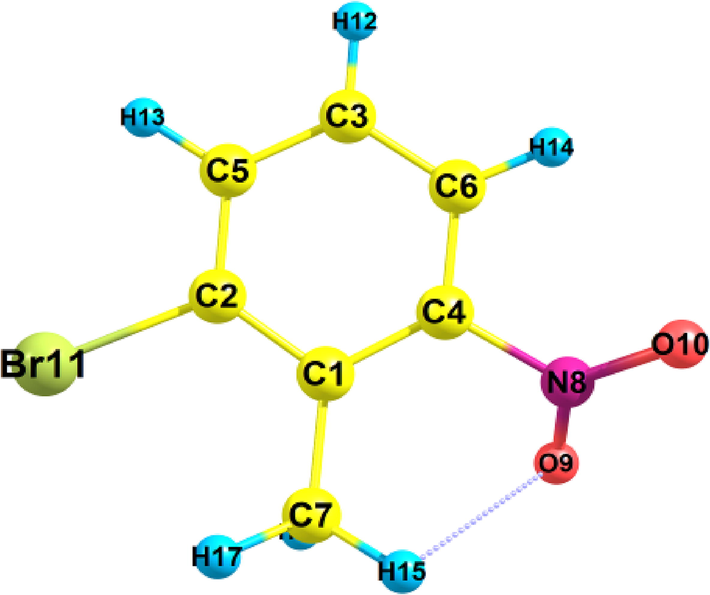
Optimized geometric structure with atom numbering of 2-Bromo-6-nitrotoluene.
Parameter
B3LYP/6–311 + G(d,p)
Experimental*
Parameter
B3LYP/6–311 + G(d,p)
Parameter
B3LYP/6–311 + G(d,p)
Experimental*
Bond length(Å)
C1-C2
1.405
1.410
C3-C6
1.387
C7-H15
1.087
1.080
C1-C4
1.405
1.410
C3-H12
1.083
C7-H16
1.094
1.090
C1-C7
1.506
1.505
C4-C6
1.391
C7-H17
1.09
C1-C5
1.392
C4-C8
1.482
N8-O9
1.224
C2-Br11
1.921
1.925
C5-H13
1.082
N8-O10
1.224
1.225
C3-C5
1.391
1.390
C6-H14
1.081
O9-H15
2.364
2.360
Bond angle(°)
C2-C1-C4
114.5
114.5
C5-C2-Br11
116.7
C4-C6-H14
119.3
119.5
C2-C1-C7
121.8
12.1.7
C2-C5-C3
119.8
C4-N8-O9
117.9
117.5
C1-C2-C5
123
123.1
C5-C3-H13
119.7
C4-N8-O10
117.2
C1-C2-Br11
120.2
C5-C3-H12
119.6
H15-C7-H16
107.2
107.5
C4-C1-C7
123.7
123.8
C4-C5-C6
120.1
H15-C7-H17
109.1
109.1
C1-C4-C6
124
124
C3-C5-H15
120.5
C7-H15-O9
105.2
105.5
C1-C4-N8
120.5
120.1
C6-C3-H12
120.3
H16-C7-H17
107.7
107.5
C1-C7-H15
112.1
111.1
C5-C6-C4
119
O9-N8-O10
124.9
C1-C7-H16
110.6
C3-C6-H14
121.7
N8-O9-H15
88.10
88.1
C1-C7-H17
109.9
109.5
C6-C4-N8
115.5
4 Acceptor-Donar interaction
A complete comprehension of the interactions between molecules on an interorganizational & intra - molecular interconnections is provided by N.B.O investigation. (Weinhold and Landis, 2005; Sidir et al., 2010). Employing the second order Fock matrix donor–acceptor correlations will be calculated. Because of the links, the concentrated Lewis structuring contributes to the utilization of the unoccupied non– Lewis orbitals. (Jayabharathi et al., 2012; Sangeetha et al., 2022a ). In order to elucidate the inter - molecular electron transport, rehybridization as well as depolarization of charge distribution NBO experiments were performed on the title molecule (Issaoui et al., 2016). Table S1 lists the stabilization energy of the donor& acceptor. For non-Lewis’s orbital, use an asterisk. E(2) denotes an intensity of maximal integrins interactions. Once the amount of E(2) increases, the coupling across electron donors and acceptors intensifies (Muthu et al., 2015). Conjugative interactions cause e-n delocalization of (C 1 – C 2) spread to antibonding (C4-N8), π (C 1 – C 2) with antibonding π*(C3-C5), π*(C4-C6), resulting in stabilization energies of 3.24,20.78,15.98 correlatively. A significant e-n shift were seen as antibonding orbital (N8-O10) to the oxygen atoms lone pair LP(2), which has a large stabilization energy of 29.99 kcal/mol. Low stabilization Energy 3.84 and 1.78 kcal/mol for the Br(11) lone pair (LP1) on the anti-bonding σ * (C1-C2) observed correlatively.
Table S2 displays the interactions of electrons for various solvents. At 20.67 and 18.54 kcal/mol, LP (2) O9 with (C4-N8) and π (C1-C2) with π *(C3-C5), in water have substantially greater stabilization energies. Minimal energy was discovered in LP (1) -O9 to σ * (C4-N8) & (C2-C5) with σ *(C1-C2) at 4.78 and 3.08 k cal/mol correlatively. The stabilizing energies of benzene LP (3) Br(11) with π * (C 1 – C 2) and π (C 4 – C 6) with π * (C 1 – C 2), however, are significantly higher, coming in 24.74 and 11.13 kcal/mol, respectively. Enough less energy, measured as 4.23 and 3.84 -k- c a l / m o l, is related towards σ (C3–C6) belongs σ * (C4–N8) and LP(2) of Br(11) to σ * (C 1–C 2) correlatively. Additionally, it was discovered that the stabilizing energy of 30.56,23.74 kcal/mol at LP (2) O9 with σ *(N8-O10) and π(C3-C5) with π*(C4-C6) corresponds to DMSO is significantly higher. A very little amount of stabilizing energy 4.33 and 4.53 kcal/mol was discovered from the reactions (C 7 – H16) corresponds to σ* (C1 - C2) as well as (C7 – H17) to σ*(C 1 – C 4) correlatively.
5 MEP analysis
With expertise of the molecule's changeable charging regions, this was attainable to figure out whether atoms associate with each other and the sort of chemical bond. It also employs color grading to demonstrate the chemical structure, shape, negativity, positively, & unbiased electrically charged possibilities. Consequently, the physiochemical characteristics were examined (Scrocco and Tomasi, 1978). Figure S1 depicts different possible electrochemical ranges solely over the inter-face. Ordered from red to orange to yellow to green to blue, the electrostatic potential rises. The maps' colour code was discovered to be between −4.937 eV (deepest red) and 4.937 eV (blue), whereas red indicates repulsion (electrophilic assault) as well as blue indicates attack (nucleophilic attack). It is used to predict specific activation centers of the molecule indicated in the label in Figure S1, in addition to the hydrogen - bonded relation (Murry et al., 1996; Sebastin and Sundarganesan, 2010; Okulik and Jubert, 2005).
5.1 Investigation of FMO
Each exciton absorbed energies when it is propelled to the ground level to the excited level. These are the identical orbitals since they primarily play the roles of electron acceptor and donor, respectively, in a molecule. Table 2 lists multiple properties of the investigation compound in numerous solvents. Figure S2 depicts the simulated FMOs of various solvents. Head compound energy gap is determined as 4.564 e V (gas), 4.191(water), 4.351(benzene) and 4.194(DMSO) of various solvents (Joseph et al., 2013). It is possible to identify the electron density index, chemical prospective & softening, and electron configuration (Parr et al., 1999). The biological activity of the molecule is indicated by the electrophilicity index calculation, which has different values of 5.834(gas), 6.187(water), 5.977(benzene) 6.181(DMSO) respectively. EA's lower value (2.878 eV) indicates that the compound rapidly forms a title takes in electrons to create connections.
Basis set
Gas
Water
Benzene
DMSO
HOMO(eV)
−7.442
−7.187
−7.274
−7.189
LUMO(eV)
−2.878
−2.997
−2.924
−2.995
Ionization potential
7.442
7.187
7.274
7.189
Electron affinity
2.878
2.997
2.924
2.995
Energy gap(eV)
4.564
4.191
4.351
4.194
Electronegativity
5.160
5.092
5.099
5.092
Chemical potential
−5.160
−5.092
−5.099
−5.092
Chemical hardness
2.282
2.095
2.175
2.097
Chemical softness
0.219
0.239
0.230
0.238
Electrophilicity index
5.834
6.187
5.977
6.181
Table 3 lists numerous properties of the research compound in various solvents. Table 4a and Table 4b shows that water, benzene, and DMSO have the theoretically predicted wavelengths of 296.58 nm, 297.91 nm, and 296.71 nm, respectively. The concordance of theoretically reported peaks for different solvents is seen in Figure S3. Based on examination, there were no discernible differences in band-gap or wavelength for the different solvents used. Three different types of solvents—water, benzene, and DMSO—are used to examine fluid impact, molecules bio-chemical. Hence, research is done on the brand molecule's electrical shift. For the purpose of studying compounds, scientists get a thorough understanding of electron transitions. Water, Benzene, DMSO, and Gas Phase are some examples of liquids where the (Selvakumari et al., 2022) M062X method has been used, will earn & inquire about electron shift on 2B6NT. Investigating inter-action processes was done using the IEFPCM (Sangeetha et al., 2022b) technique. The pharmacological aspect of 2B6NT was investigated using electro-philicity indices findings (Parthasarathi, et al., 2004; Sangeetha et al., 2021; Selvakumari et al., 2021).
Solvents
Wavelength (nm)
Band gap(eV)
Energy(cm-1)
Osc. Strength(f)
Symmetry
Major Contribution
Water
332.50
3.73
30075.61
0.0102
Singlet-A
H-5->LUMO (16%), H-1->LUMO (11%)
282.03
4.40
35457.75
0.0507
Singlet-A
HOMO->LUMO (82%)
275.20
4.51
36337.70
0.0049
Singlet-A
H-6->LUMO (56%), H-5->LUMO (12%)
Benzene
339.33
3.65
29469.89
0.0084
Singlet-A
H-3->LUMO (54%), H-1->LUMO (12%)
278.53
4.45
35902.16
0.0012
Singlet-A
H-5->LUMO (68%)
275.87
4.49
36248.98
0.0598
Singlet-A
HOMO->LUMO (79%)
DMSO
332.74
3.73
30053.83
0.0106
Singlet-A
H-5->LUMO (16%), H-1->LUMO (11%)
282.05
4.40
35455.33
0.0537
Singlet-A
HOMO->LUMO (82%)
275.33
4.50
36319.95
0.0051
Singlet-A
H-6->LUMO (56%), H-5->LUMO (13%)
Atom
Mulliken
atomic
charges
fr+
Fukui
functions
Δf(r)
local
Softness
sr0 fr0
0,1(N)
N + 1(-1,2)
N-1(1,2)
fr-
fr0
sr + fr+
sr-fr-
1C
0.8698
0.8662
0.0326
−0.0035
0.8371
0.4168
−0.8406
−0.0008
0.1833
0.0913
2C
0.3438
0.3489
−0.1362
0.0051
0.4800
0.2426
−0.4749
0.0011
0.1051
0.0531
3C
−0.0465
−0.0482
−0.2012
−0.0017
0.1547
0.0765
−0.1564
−0.0004
0.0339
0.0167
4C
−1.0912
−1.0860
0.0811
0.0052
−1.1723
−0.5835
1.1775
0.0011
−0.2567
−0.1278
5C
−0.1892
−0.1912
−0.1813
−0.0020
−0.0079
−0.0050
0.0060
−0.0004
−0.0017
−0.0011
6C
−0.2794
−0.2812
−0.1712
−0.0018
−0.1082
−0.0550
0.1065
−0.0004
−0.0237
−0.0120
7C
−0.4645
−0.4657
−0.6227
−0.0011
0.1582
0.0785
−0.1594
−0.0003
0.0346
0.0172
8 N
−0.1672
−0.1695
0.4896
−0.0023
−0.6568
−0.3295
0.6545
−0.0005
−0.1438
−0.0722
9O
0.0813
0.0804
−0.2840
−0.0009
0.3653
0.1822
−0.3662
−0.0002
0.0800
0.0399
10O
−0.0693
−0.0763
−0.4481
−0.0069
0.3787
0.1859
−0.3856
−0.0015
0.0829
0.0407
11 Br
−0.1565
−0.1438
0.1183
0.0126
−0.2747
−0.1310
0.2874
0.0028
−0.0602
−0.0287
12H
0.1919
0.1915
0.2239
−0.0005
−0.0320
−0.0162
0.0315
−0.0001
−0.0070
−0.0036
13H
0.2143
0.2185
0.2275
0.0041
−0.0131
−0.0045
0.0172
0.0009
−0.0029
−0.0010
14H
0.2315
0.2268
0.2395
−0.0047
−0.0080
−0.0064
0.0033
−0.0010
−0.0018
−0.0014
15H
0.2038
0.2022
0.2262
−0.0016
−0.0224
−0.0120
0.0208
−0.0004
−0.0049
−0.0026
16H
0.2077
0.2061
0.2451
−0.0016
−0.0373
−0.0195
0.0358
−0.0003
−0.0082
−0.0043
17H
0.1197
0.1212
0.2261
0.0015
−0.1064
−0.0524
0.1079
0.0003
−0.0233
−0.0115
Atom
Mulliken
atomic
charges
Fukui
functions
fr 0
local
softness
sr0 fr0
0, 1 (N)
N + 1(-1,2)
N-1(1,2)
fr+
fr-
sr + fr+
sr-fr-
Water
1C
0.9509
0.9768
0.9005
0.0259
0.0504
0.0381
−0.0244
0.0062
0.0120
2C
0.3152
0.5403
0.2240
0.2251
0.0912
0.1582
0.1339
0.0538
0.0218
3C
−0.0944
−0.0211
−0.1034
0.0733
0.0089
0.0411
0.0644
0.0175
0.0021
4C
−1.0997
−1.1789
−1.0770
−0.0791
−0.0228
−0.0510
−0.0564
−0.0189
−0.0054
5C
−0.1994
−0.1949
−0.2641
0.0045
0.0647
0.0346
−0.0602
0.0011
0.0155
6C
−0.2931
−0.1794
−0.3323
0.1137
0.0392
0.0765
0.0745
0.0272
0.0094
8 N
−0.0848
−0.0516
−0.1440
0.0332
0.0591
0.0462
−0.0259
0.0079
0.0141
9O
0.0526
0.0732
−0.1997
0.0207
0.2523
0.1365
−0.2316
0.0049
0.0603
11 Br
−0.1530
0.1228
−0.1957
0.2758
0.0426
0.1592
0.2332
0.0659
0.0102
12H
0.2258
0.2805
0.1946
0.0547
0.0312
0.0430
0.0235
0.0131
0.0075
Benzene
1C
0.899
0.854
0.949
−0.045
−0.050
−0.048
0.004
−0.010
−0.011
2C
0.330
0.523
0.213
0.193
0.117
0.155
0.075
0.044
0.027
3C
−0.068
−0.025
−0.051
0.043
−0.017
0.013
0.060
0.010
−0.004
4C
−1.087
−1.143
−1.141
−0.056
0.054
−0.001
−0.110
−0.013
0.012
5C
−0.193
−0.189
−0.264
0.004
0.071
0.038
−0.066
0.001
0.016
6C
−0.287
−0.172
−0.318
0.115
0.031
0.073
0.083
0.026
0.007
7C
−0.479
−0.454
−0.474
0.025
−0.005
0.010
0.031
0.006
−0.001
8 N
−0.135
−0.107
−0.180
0.028
0.045
0.036
−0.017
0.006
0.010
9O
0.069
0.105
−0.136
0.036
0.206
0.121
−0.170
0.008
0.047
10O
−0.109
−0.038
−0.334
0.070
0.225
0.148
−0.154
0.016
0.052
DMSO
1C
0.949
0.973
0.902
0.024
0.048
0.036
−0.024
0.006
0.011
2C
0.316
0.540
0.224
0.224
0.092
0.158
0.132
0.053
0.022
3C
−0.094
−0.021
−0.102
0.073
0.008
0.040
0.064
0.017
0.002
4C
−1.099
−1.178
−1.079
−0.079
−0.021
−0.050
−0.058
−0.019
−0.005
5C
−0.199
−0.195
−0.264
0.004
0.065
0.035
−0.060
0.001
0.015
6C
−0.293
−0.179
−0.332
0.114
0.039
0.076
0.075
0.027
0.009
7C
−0.500
−0.492
−0.468
0.008
−0.032
−0.012
0.040
0.002
−0.008
8 N
−0.086
−0.053
−0.145
0.033
0.059
0.046
−0.026
0.008
0.014
9O
0.053
0.074
−0.198
0.021
0.251
0.136
−0.230
0.005
0.060
10O
−0.158
−0.115
−0.424
0.043
0.266
0.155
−0.223
0.010
0.063
5.2 Population Analyses and reactivity of molecules:
Molecular values are calculated using the NBO methods (Mulliken, 1995) as well as the findings presented in Table 4a. This is depicted visually in Figure.S4 for different solvents. Mulliken atomic charge of 2B6NT is estimated utilizing unique B3LYP systemization (Parr and Yang, 1980). This demonstrates that Br11 and C3 carbon atoms have stronger negatives charges than H17, which exhibits a greater, positively charge. Oxygen-atoms C6, C7, and N8 have negative charges, hydrogen atoms H12 have positive charges. It can be used to ascertain the electron's structure, polarizability, and molecular electrostatic potential (Ayers and Parr, 2000).
Table 4b shows Population Analyses and reactivity of molecules for different solvents. In comparison to other carbon atoms, the carbon atom C1 in water has a stronger + ve charge, while the carbon atoms C4 and N8 have a bigger -ve charge. In comparison to all other atoms, C 2 and O9 have a greater + ve charge, but C4 and C6 have a major -ve charge in benzene. The carbon atom C1 and C2 in DMSO has bigger (+) charges, while Carbon C4 and C5 had greater (-) charges compared with remaining all.
5.3 Studies of E.L.F and L.O.L
They display the areas of chemical region where gaining a significant electrons combination was more prone to happen via ELF and LOL maps. A surface simulation was also created using predictions focused on covalently bonding (Sangeetha et al., 2021). Since localized orbitals overlap, LOL believes that localized orbital gradients should be strengthened, in contrast to ELF, which sees electron pair density (Silvi and Savin, 1994). In Fig. 2(a), (b) ELF and LOL maps are displayed & which runs from 0.0 to 1.0 and includes stronger ordering around 0.5 and 1.0, indicates areas including both non-bonded and bonding regionalized atoms; delocalization of particles was proved utilizing slight size of 0.5, correspondingly (Savin et al., 1997). When electron localization has an impact on electron density, LOL reaches huge - values (>0.5) (Sangeetha et al., 2022c).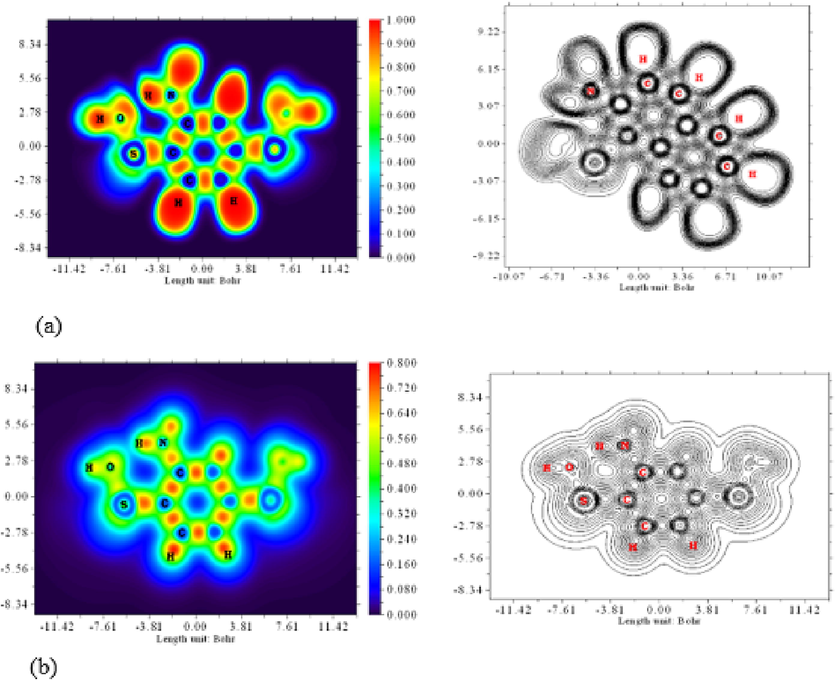
(a) Electron Localization Function, (b) Localized Orbital Locator colour filled and contour map of 2-Bromo-6-nitrotoluene.
6 Biological and pharmacological research
The drug likeness of the reference chemical is investigated to primarily evaluate its ability to be employed as a main ingredient in a novel medication dosage form. Numerous drug similarity parameters that were explored in this work. It must adhere to Lipinski's rule in order to be taken into consideration as a prospective drug (Lipinski et al., 1997; 2004). The anticipated medication similarity for 2A4BSA is shown in Table 5. It demonstrates that these indicators have values that are appropriate for the drug that is being evaluated. The Lipinski's rule of five suggests a cut - off point of 5, while the AlogP, among the most key metrics which reveals how hydrophobic or lipophilic a molecule is, is equivalent to 3.06, which is considerably lesser.
Descriptor
Value
Hydrogen Bond Donor(HBD)1
3
Hydrogen Bond Acceptor (HBA)1
1
AlogP1
3.06
Topological polar surface area(TPSA)1 (A2)
45.82
Number of atoms1
11
Number of rotatable bonds1
1
6.1 Toxicity
The title molecule's anticipated values in several toxicity assays were, respectively, 0.01926, 0.04578, 0.02146, and 0.1239 demonstrates enhanced bioactivity of head molecule. Carcinoo-test findings are negative, demonstrating that 2B6NT is not a variant that can't pass Ame's analysis. While 100 T A R L I 1 0, N A 1 5 3 5 T A, and R L I − 1 5 3 5 1 0 T A I all derive positive data, TA100 NA derives negative values. The impacts of ecological toxicity have enhanced bioactivity (Zakharov et al., 2010). 1.21, 0.27, 0.38, 0.113, 0.08, and 0.16 (enzyme kinase, nuclear receptor, ligand. Ion channel in proteas. The proper amounts of the inhibitor—G P C R ligand have been established.
6.2 Reduced density gradient (RDG):
Focusing on electron abundance, evaluation on RDG is utilised to assess authentic area weak correlations. RDG analysis was carried out using a color-filled map and colour scale bar utilizing Multiwfn 3.8 (Noureddine et al., 2020). The results are displayed in Figure.S5, where ring dia-zepinyl denotes greater adhesion, and the blue spot encircled on neighbouring element of carbon are displayed. Header composite, produces the steric effect, is shown by the isosurface region in red. The colour green in a map of an isosurface filled with colours denotes weak interactions like vander- waals adhesion (Nkungli and Ghogomu, 2017). Inferences are made regarding the correlations of concentration on the surface of the protein, a crucial parameter in biological research.
6.3 Analysis of rama-chandran plot
Using Ramachandran graphing, researchers are examining a unique characteristic of biomolecules. Also, it is employed to identify empirical transmission couple for amino acids. The research outcome (Thomas et al., 2020) and the online PASS predictor (Reddy et al., 2016) were used to collect this data. The Ramachandran plot (Saji et al., 2020), which is one of the molecular docking evaluations, is used to conceal protein molecules because of their homogeneity and durability. The majority of the vestiges that are thus on the green zone are shown in Fig. 3 (a, b and c) to be within the permissible red zone, with only a few minor remnants found close to its banned region. The results show that some proteins (4ZQY, 5 V47 and 6YPE) are structurally stable.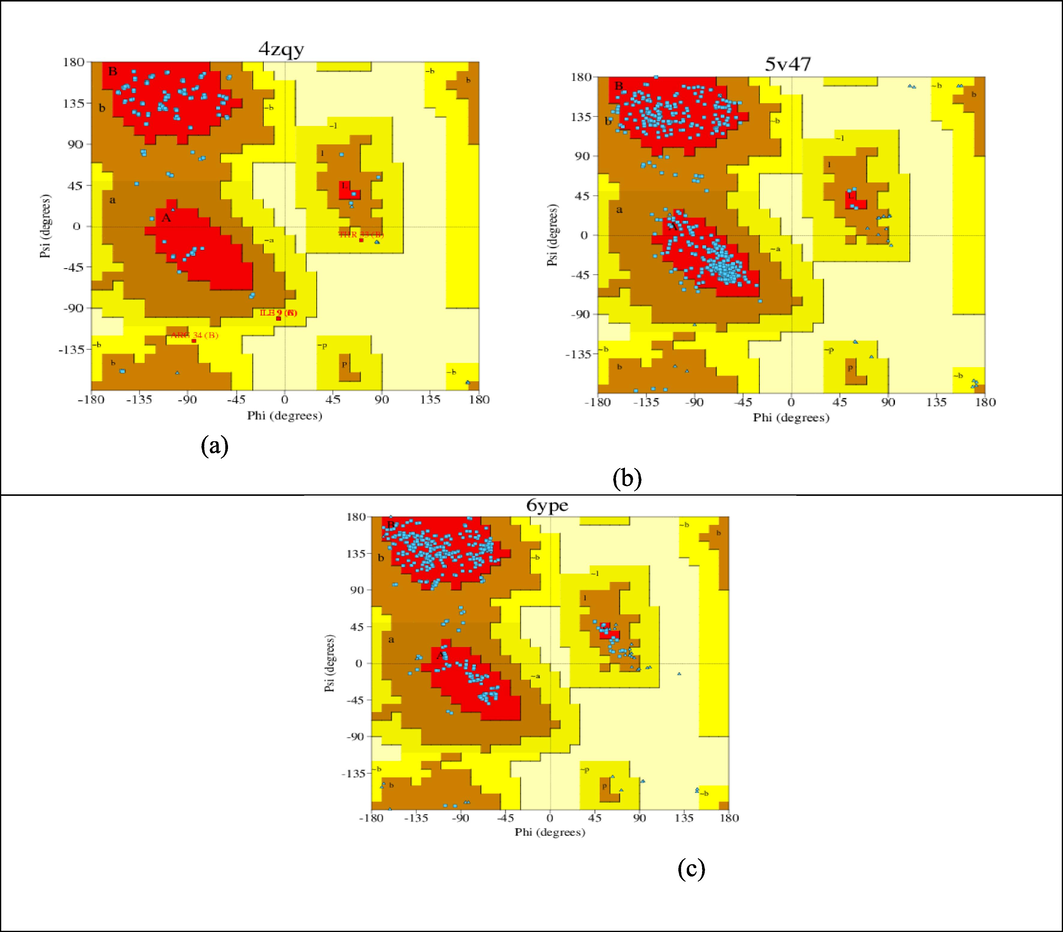
Ramachandran plot for receptor protein (a) 4ZQY, (b) 5V47 and (c) 6YPE.
6.4 Exploration of docking aspects
Major innovations in clinical applications involve molecular docking, a formidable computer technique for calculating the enforceable propensity of a receptor with enzymes. Precise binding position of the ligand and protein may be studied using this new approach (Kragh-Hansen, 1985). Through this work, we can comprehend how ligands, which are tiny molecules, are transported across biological systems. Auto docking experiments are a great way to get understanding of the underlying chemical processes that enable ligands to bind to receptors with well-known three-dimensional structures. Given that the substance in question is an anti-inflammatory drug, its capacity to lessen inflammation is further investigated through docked its receptor with the high affinity of distinct biological objectives. The protein 4ZQY, which is important for anti-inflammatory disorders, has the title chemical docked into its active site. This protein data bank was retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) website. The receptor was lodged in the distinct categories of the selected molecule to ascertain the least interaction potential value. For the reason of analyzing the strategy of authoritative, the docked compliance with the least official vitality was chosen. The obtained docking characteristics for the chemical with regard to the focused-on proteins are appeared in Table 6. Fig. 4(a, b and c), it is depicted how the title chemical interacts with the proteins 4ZQY, 5 V47 and 6YPE when used as a ligand. With regard to these, 4ZQY exhibits minimal binding-energy at −5.32 kcal/mol, overall large percentage of docked antagonists reacted with ligand. The LYS'26/HN hydrogen bonds in this protein's four residues have a bond distance of 2.5A0. Anti-intemperate protein 4ZQY is successfully bound to 2B6NT, as evidenced by the decreased binding energy.
Protein
(PDB ID)Bonded residues
Bond distance
Estimated inhibation constant(μm)
Binding Energy(kcal/mol)
Intermolecular energy(kcal/mol)
Reference RMSD (AO)
4ZQY
ARG’65/NH2
ARG’65/NB2
LYS’26/CE
LYS’26/HD22.2
2.1
1.8
2.5207.17
−5.32
−5.19
27.54
5 V47
ARG’149/1HH2
2.5
118.0.66
−6.16
−6.45
30.70
ARG’149/1HH2
2.3
6YPE
ARG’152/1HH2
2.12.2
110.07
−6.06
−6.12
29.55
LYS’29/HD2
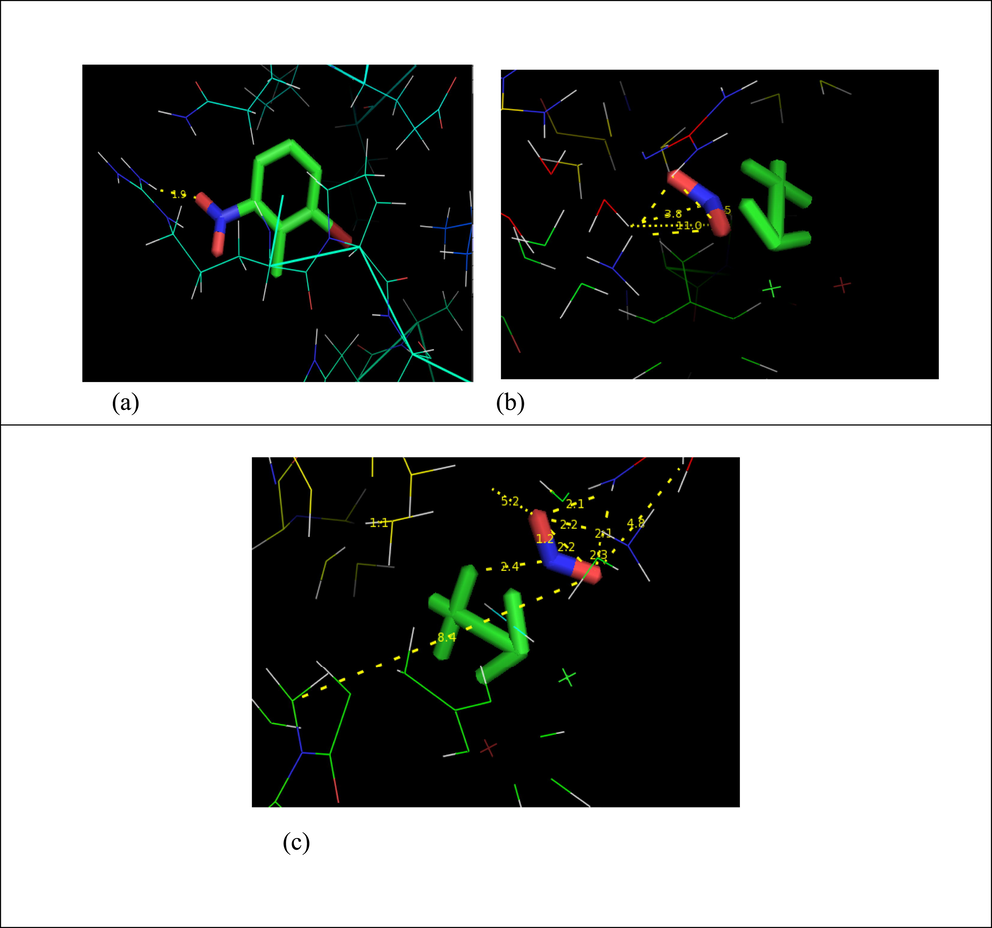
Docking of 2-Bromo-6-nitrotoluene with (a)4ZQY, (b)5V47 and (c)6YPE protein.
7 Conclusion
D F T and the 6 – 311 G + + (d, p) premise set were used to carefully examine the vibrations of 2B6NT. The calculated tool geometry provided a reliable approximation and a basis for determining the other variables in the proposed exploration. In addition to assessing architectural characteristics like bond-angle and bond -length, other metrics for the current investigation are determined. Conjugative interactions cause delocalization for σ (C1-C2) spread to antibonding σ*(C4-N8), π (C 1-C2) with antibonding π*(C3-C5), π*(C4-C6), resulting in stabilization energies of 3.24, 20.78, 15.98 correlatively. A significant interaction were, electron density shift from the antibonding orbital π * (N8-O10) to the oxygen atoms lone pair LP(2), which has a large stabilization energy of 29.99 kcal/mol. Low stabilization Energy 3.84 and 1.78 kcal/mol for the Br(11) lone pair (LP1) on the anti-bonding σ * (C1-C2) observed correlatively. Additionally, the FMO study gave the requisite theoretical justification for believing that the molecule is both biologically active and non-toxic. A significant affinity efficiency of −5.32 k cal / mol was discovered during molecular modelling experiments involving its 4ZQYY protein coupled and the protein receptor antagonist, revealing the usefulness to the therapeutic sector. UV–vis spectra were used to reveal the electrical characteristics of various solvents. Also reported for 2B6NT are L.O.L, E.L.F, R.D.G, pharmacological similarity, A D M E correlatively. The pharmaceutical precautionary properties of the research molecule, which reveal anti-viral, anti-fungal, and anti-oxidant properties, were studied and substantiated by structural modelling computations. In conclusion, the present dissertation offers a thorough investigation employing quantum computational and spectroscopic methodologies to evaluate the solvation effects and physiological functionality of selected pharmaceutical substances. In order to prepare for future study, the data gathered are highly valuable.
Acknowledgements
Researchers Supporting Project number (RSP2023R61), King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Investigation of the cyclo carbon nano ring and respective analogues (Al6n6 And B6n6) as support for the single atom catalysis of the hydrogen evolution reaction, materials science in semiconductor processing. J. Mater. Sci. Semiconductor Process.. 2023;162(107544)
- [CrossRef] [Google Scholar]
- Removal of methylene blue by using sodium alginate-based hydrogel; validation of experimental findings via DFT calculations. J. Mol. Graph. Model.. 2023;122(108468)
- [CrossRef] [Google Scholar]
- Anchoring the late first row transition metals with B12P12 nanocage to act as single atom catalysts toward oxygen evolution reaction (OER) J. Mater. Sci. Semicond. Process.. 2023;153(107164)
- [CrossRef] [Google Scholar]
- High drug carrying efficiency of boron-doped Triazine based covalent organic framework toward anti-cancer tegafur; a theoretical perspective. J. Comput. Theor. Chem.. 2023;1220(113990)
- [CrossRef] [Google Scholar]
- Chemical sensing ability of aminated graphdiyne (GDY-NH2) toward highly toxic organic volatile pollutants. J. Comput. Theor. Chem.. 2023;1222(114079)
- [CrossRef] [Google Scholar]
- Crystal structure, DFT and Hirshfeld Surface Analysis of (E)-N′-[(1-chloro-3,4-dihydronaphthalen-2-yl) methylidene] benzohydrazide monohydrate. J. Acta Crystallographica. 2020;76:132-136.
- [CrossRef] [Google Scholar]
- Biologically active compounds from natural and marine natural organisms with antituberculosis, antimalarial, leishmaniasis, trypanosomiasis, anthelmintic, antibacterial, antifungal, antiprotozoal, and antiviral activities. Cellmed Orthocellular Med. Pharm. Assoc.. 2016;622(4):22.1-22.19.
- [CrossRef] [Google Scholar]
- Biologically active compounds from natural and marine natural organisms with antituberculosis, antimalarial, leishmaniasis, trypanosomiasis, anthelmintic, antibacterial, antifungal, antiprotozoal, and antiviral activities. Helv Chim Acta.. 2004;87:1090-1098.
- [CrossRef] [Google Scholar]
- Nat. Rev. Drug Discov.. 2021;20:200-216.
- [CrossRef]
- Quantitative Structure-Activity Relationship Study of a Benzimidazole -Derived Series Inhibiting Mycobacterium tuberculosis H37Rv. J. Am. Chem. Soc.. 2000;122:2010-2201.
- [CrossRef] [Google Scholar]
- The Monoacetylation of 2,4-Diamino-6-nitrotoluene. J. Am. Chem. Soc.. 1950;72(9):3959.
- [CrossRef] [Google Scholar]
- Frisch, M.J., Fox, D.J., 2009,Gaussian 09.Revision E.01, Gaussian, Inc, Wallingford CT. https://gaussian.com/glossary/g09/.
- J. Am. Chem. Soc.. 1987;109(14):4335-4338.
- [CrossRef]
- Molecular docking studies, structural and spectroscopic properties of monomeric and dimeric species of benzofuran-carboxylic acids derivatives: DFT calculations and biological activities. Acta Mol. Biomol. Spectosc.. 2016;136:1227-1242.
- [CrossRef] [Google Scholar]
- Vibrational spectroscopy, quantum computational and molecular docking studies on 2-chloroquinoline-3-carboxaldehyde. Acta Mol. Bromol. Spectrosc.. 2012;97:131-136.
- [CrossRef] [Google Scholar]
- Spectroscopic (FT-IR, FT-Raman), first order hyperpolarizability, NBO analysis, HOMO and LUMO analysis of N-[(4-(trifluoromethyl)phenyl]pyrazine-2-carboxamide by density functional methods. J. Chem. 2013;10:S2281-S2294.
- [Google Scholar]
- Photochemical approaches to complex chemotypes: applications in natural product synthesis. J. Chem. Rev. 2016;116(17):9683-9747.
- [CrossRef] [Google Scholar]
- Knölker, H.J., Knöll, J., 2003, Chemical communications (Cambridge, England), 10 (10), 1170-1171 (2003-06-05). https://doi.org/10.1039/B301979A.
- J. Inorg. Organomet. Polym Mater.. 2023;33(515–528)
- [CrossRef]
- Relations between high-affinity binding sites of markers for binding regions on human serum albumin. Pharmacol. Rev.. 1985;225(3):629-638.
- [CrossRef] [Google Scholar]
- Lee, S.K., Chang, G.S., Lee, I.H., Chung, J.E., Sung, K.Y., No, K.T., 2004., The PreADME: PC-based program for batch prediction of adme properties, EuroQSAR 2004, 9.5-10, Istanbul, Turkey. https://preadmet.bmdrc.kr/.
- Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol.. 2004;1(4):337-341.
- [CrossRef] [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 1997;23(1–3):3-25.
- [CrossRef] [Google Scholar]
- Multi wave function analysis. J. Mater. Today: Proc.. 2022;50:2826-2835.
- [CrossRef] [Google Scholar]
- Theoretical Study of the Structureand Vibrational Spectrum of [Zn(2-Aminothiazole)2Cl2] J. Chem Phys. Met.. 1995;23:1833-1840.
- [CrossRef] [Google Scholar]
- Murry, J.S, Sen, K., 1996, Molecular Electrostatic Potentials: Concepts and Applications, Theoretical and Computational Chemistry Book Series, vol. 3. Elisevier, Amesterdam . https://www.elsevier.com/books/molecular-electrostatic-potentials/murray/978-0-444-82353-3.
- Synthesis, structure, spectroscopic studies (FT-IR, FT-Raman and UV), normal coordinate, NBO and NLO analysis of salicylaldehyde p. J. Mol. Sruct.. 2015;1081:400-412.
- [CrossRef] [Google Scholar]
- Tetrazoles via multicomponent reactions. J. Chem. Rev. 2019;119(3):2042.
- [CrossRef] [Google Scholar]
- Theoretical analysis of the binding of iron(III) protoporphyrin IX to 4-methoxyacetophenone thiosemicarbazone via DFT-D3, MEP, QTAIM, NCI, ELF, and LOL studies. J. Mol. Model.. 2017;23(7):1-20.
- [CrossRef] [Google Scholar]
- Experimental and DFT studies on the molecular structure, spectroscopic properties, and molecular docking of 4-phenylpiperazine-1-ium dihydrogen phosphate. J. Mol. Struct.. 2020;1207:17762.
- [Google Scholar]
- Theoretical study on the structure and reactive sites of non-steroidal anti-inflammatory drugs. J. Mol. Des.. 2005;4:17-30.
- [Google Scholar]
- Parr, R.G., Yang, W., 1980, Theoretical Study of the Reaction of (2, 2)-Dichloro (Ethyl) Arylphosphine with Bis (2, 2)-Dichloro (Ethyl) Arylphosphine by Hydrophosphination Regioselective by the DFT Method, Oxford University press, New York. https://doi.org/10.4236/cc.2017.53010.
- Computational analysis of theacrine, a purported nootropic and energy-enhancing nutritional supplement. J. Arun Phys Soc.. 1999;121:1922-1994.
- [CrossRef] [Google Scholar]
- Intermolecular reactivity through the generalized philicity concept. Chem. Phys. Lett.. 2004;394:225-230.
- [Google Scholar]
- The Monoacetylation of 2,4-Diamino-6-nitrotoluene. Can. J. Chem.. 1962;40:3.
- [CrossRef] [Google Scholar]
- Synthesis, biological evaluation and QSAR studies of a novel series of annelated triazolo [4, 3-c] quinazolines. Indian J. Chem. B Org.. 2016;55B:898-911.
- [CrossRef] [Google Scholar]
- Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev.. 1988;88:899-926.
- [CrossRef] [Google Scholar]
- Molecular docking studies, structural and spectroscopic properties of monomeric and dimeric species of benzofuran-carboxylic acids derivatives: DFT calculations and biological activities. J. Comput. Biol. Chem.. 2020;87:107311
- [CrossRef] [Google Scholar]
- Spectroscopic and quantum computational study on naproxen sodium. Spectochem. Acta -part A Mol. Biomol. Spectrosc.. 2020;226:117614
- [CrossRef] [Google Scholar]
- Electronic properties of solvents (Water, Benzene, Ethanol) using IEFPCM model, spectroscopic exploration with drug likeness and assessment of molecular docking on 1-Octanesulfonic Acid Sodium Salt. J. Mol. Liquids. 2021;344:117719
- [CrossRef] [Google Scholar]
- Electronic properties (in different solvents), spectroscopic progression and evaluation on 4-morpholinepropane sulfonic acid along with molecular docking analysis. J. Mol. Liq.. 2022;349:118107
- [CrossRef] [Google Scholar]
- Electronic properties (in different solvents), spectroscopic progression and evaluation on 4-morpholinepropane sulfonic acid along with molecular docking analysis. J. Mol. Liq.. 2022;349:118107
- [CrossRef] [Google Scholar]
- Investigation of Spectroscopic (FT-IR, FT-Raman), Reactive Charge Transfer and Docking Properties of (1S)-(+)-10- Camphorsulfonic acid by density functional method. J. Mater. today Proc.. 2022;50:2768-2776.
- [CrossRef] [Google Scholar]
- ELF: the electron localization function. Angew. Chem. Int. Ed. Emgl.. 1997;36:3-25.
- [CrossRef] [Google Scholar]
- Electronic molecular structure, reactivity and intermolecular forces: an euristic interpretation by means of electrostatic molecular potentials. Adv. Quant. Chem. 1978;11:115-193.
- [CrossRef] [Google Scholar]
- Molecular structure, vibrational analysis (FT-IR, FT-Raman), NMR, UV, NBO and HOMO–LUMO analysis of N, N-Diphenyl Formamide based on DFT calculations. Spectrochim Acta A. 2010;75:941-952.
- [Google Scholar]
- Bioactive Molecules Profile from Natural Compounds. 2017;10 P-200-208
- [CrossRef]
- Evaluation of electronic properties in different solvents, spectroscopic exposition (FT-IR, FT-Raman), and molecular docking studies of 5-Chloro-2-hydroxypyridine - insulysin inhibitor. J. Mol. Liquids. 2021;341:117304
- [CrossRef] [Google Scholar]
- Donor acceptor groups effect, polar protic solvents influence on electronic properties and reactivity of 2-Chloropyridine-4-carboxylic acid. J. Indian Chem. Soc.. 2022;99(6):100478
- [CrossRef] [Google Scholar]
- academia.edu alanından [PDF] Ab initio Hartree-Fock and density functional theory investigations on the conformational stability, molecular structure and vibrational spectra of 7-acetoxy-6-(2, 3. J. Mol. Struct.. 2010;964:134-151.
- [CrossRef] [Google Scholar]
- Silvi, B., Savin, A., 1994, A topological classification, Nature 371 ,683–686. https://www.nature.com/articles/371683a0.
- J. Bioorganic Medicinal Chem.. 2020;28(23):115811.
- [CrossRef]
- Detailed quantum mechanical, molecular docking, QSAR prediction, photovoltaic light harvesting efficiency analysis of benzil and its halogenated analogues. J. Mol. Struct.. 2020;1181:455-466.
- [CrossRef] [Google Scholar]
- Multiwfn: Aamultifunctional wavefunction analyzer. J. Comput. Chem.. 2012;33(5):580-592.
- [CrossRef] [Google Scholar]
- Gaussian 09, Revision E.01. Wallingford, CT: Gaussian Inc; 2009.
- Valency and Bonding: a Natural Bond Orbital Donor-acceptor Perspective. Cambridge University Press; 2005.
- Zakharov, A., Poroikov, V., Associates, GUSAR - Prediction of values for substances copyright (C) 2010, http://pharmaexpert.ru/GUSAR/environmental.html.
- Zakharov, A., Poroikov, V., Associates. GUSAR- Prediction of Values of Substances Copyright© 2010 http://www.way2drug.com/gusar/references.html.
Further reading
- Insight into non-covalent interactions in a tetrachlorocadmate salt with promising NLO properties: Experimental and computational analysis. J. Mol. Struc. 2021;124215:130730.
- [Google Scholar]
- Non-covalent interactions and molecular docking studies on morphine compound. J. King Saud University - Science. 2021;33(8):101606.
- [CrossRef] [Google Scholar]
- Study of a new piperidone as an anti-Alzheimer agent: Molecular docking, electronic and intermolecular interaction investigations by DFT method. J. King Saud University - Science. 2021;33(8):101632.
- [CrossRef] [Google Scholar]
- Theoretical and experimental study of guar gum sulfation. Journal of Molecular Modeling 2021
- [CrossRef] [Google Scholar]
- Kazachenko, A.S., Issaoui, N., Sagaama, A., Malyar, Y.N., Al-Dossary, O., Bousiakou, L.G., Kazachenko, A.S., Miroshnokova, A.V., Xiang, Z., Hydrogen bonds interactions in biuret-water clusters: FTIR, X-ray diffraction, AIM, DFT, RDG, ELF, NLO analysis, (2022) J. King Saud. Univ. Sci., 34 (8), art. no. 102350. DOI:https://doi.org/10.1016/j.jksus.2022.102350
- Khodiev, M., Holikulov, U., Jumabaev, A., ISSAOUI, N., Nikolay Lvovich, L., Al-Dossary, O.M., Bousiakoug, L.G.; Solvent effect on the self-association of the 1,2,4-triazole: A DFT study,(2023) J. Mol. Liq. , 382, art. no. 121960. DOI: https://doi.org/10.1016/j.molliq.2023.121960
- Nitro Compounds, Aromatic. Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH; 2000.
- Rajagopalan, N.R.,, Krishnamoorthy, P., Jayamoorthy, K., Austeria M, Bis(chiourea), Investigation of spectroscopic (FT-IR, FT-Raman), reactive charge transfer and docking properties of (1S)-(+)-10-Camphorsulfonic acid by density functional method, J. Mod. Sci 2(4) 219–225. https://doi.org/10.1016/j.matpr.2020.08.674.
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2023.102789.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




