

Molecular docking studies on the interaction of NCI anticancer analogues with human Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit
⁎Corresponding author. eadavid@abu.edu.ng (David Ebuka Arthur)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The Research entails the screening of 119 NCI anticancer compounds on lipid kinase P13K-gamma (4FA6) in order to identify chemical agent that best inhibits P13K-gamma a class IB phosphoinositide which is a pro-survival signaling pathway critical in the development of cancer in white blood cells (leukocyte). The Result reported included binding energy (Kcal/mol), inhibition constant and pictorial representation of the docked poses for the most active compounds. The significance of the interaction types involved were highlighted as well as the influence of their frequency on the value of their binding energy calculated using the monte Carlo algorithm from ICM-Pro molsoft program.
Keywords
NCI
P13kgamma
Leukemia
SAR
Molecular docking

1 Introduction
Cancer encompasses a group of diseases categorized by uncontrolled growth and spread of abnormal cells (Lopez et al., 2014; Wu et al., 2010). Presently, there has been an increase in cancer treatment through a targeted method of inhibiting protein kinases, since inappropriate activation protein kinase activity causes cancer (Downward, 2003; O'Reilly et al., 2006). One group of kinases that has become a recent focus of drug discovery is the phosphoinositide 3-kinase (P13K) family (Guo et al., 2009; Le et al., 2012). Members of the P13K family catalyze the transfer of the gamma-phosphate from ATP to the 3′-hydroxyl group of phosphatidylinositol and its derivatives, collectively they are termed phosphoinositides (Toker and Cantley, 1997) (Janku, 2017).
The phosphoinositide 3kinase (PI3K) pathway is a key signal transduction system that links oncogenes and multiple receptor classes to many essential cellular functions, including those essential to cell proliferation, cell growth, apoptosis, cell differentiation, chemotaxis, cytoskeletal rearrangement, phagocytosis vesicle trafficking, secretion, protein synthesis and metabolic pathways (Guo et al., 2009; Massacesi et al., 2016; Simoncini et al., 2000).
Phosphatidylinositol 3-kinases (PI3Ks) phosphorylate lipid molecules, rather than proteins, and are consequently known as lipid kinases. Specifically, PI3Ks phosphorylate the 3′-OH position of the inositol ring of phosphatidyl inositides (Simoncini et al., 2000). According to their structure and substrate specificity, PI3Ks belong to three distinct categories called Class I, II and III. Class I is further divided into Class IA and IB. Among the different classes of PI3K, Class IA PI3K seems to play the predominant role in cancer (Martini et al., 2014; Stark et al., 2015). Growth factor receptor tyrosine kinases activate Class IA PI3Ks, and G-protein coupled receptors activate Class IB, which contains only one catalytic subunit named p110gamma or P13K gamma. P13K gamma in addition to its lipid kinase activity exhibits also a serine/threonine protein kinase activity as demonstrated by autophosphorylation. P13K gamma was reported to be involved in a number of aspects concerning leukocyte activation (Rommel et al., 2007; Rückle et al., 2006). other members (isoforms) of the P13K family that have been isolated from mammalian cells and grouped according to their primary structure and substrate specificity includes (class II: PI3KC2 alpha, beta and gamma; class III: Vps34 yeast homologue) (Cantley, 2002; Fruman et al., 1998)
Targeting signaling pathways that are deregulated in human cancer has been a promising approach in cancer therapy (Wong et al., 2010). The initial success of imatinib in chronic leukemia has validated this approach and has encouraged the development of additional therapies (Michor et al., 2005; Talpaz et al., 2002), hence the need for a new drug that target mutated proteins such as PI3K-gamma that plays a crucial role in the control of diverse immune modulatory and vascular functions like respiratory burst, mast cell reactivity, platelet aggregation, endothelial activation as well as smooth muscle contractility, have therefore presented both an opportunity and a challenge for understanding the molecular behavior of chemical active agents at the binding site of the receptor. We hope to achieve this aim by docking some potent anticancer compounds to screen cytostatic inhibitors that target PI3K isoforms and give a more elaborate reaction mechanism for their activity.
2 Experimental section
The hardware and software used in this work for molecular optimization, molecular dynamics and molecular docking study includes: Computer (HP pavilion Intel(R) core i5-4200U with 1.63 Hz and 2.3 Hz processor and windows 8.1 operating system), Spartan 14 (Hehre and Huang, 1995), ChemBio Ultra 12.0 (Evans, 2014; Li et al., 2004), Molsoft.icm-pro.v3.8.3 software (Abagyan and Totrov, 1994; An et al., 2005).
3 Ligand preparation
In this study, a dataset of 119 compounds were used to study their inhibitory properties on P13K through molecular docking simulations. The chemical structures of the data set, NSC and CAS number were taken from the drug discovery and development arm of the National Cancer Institute (NCI). Eligible compounds were determined by reviewing and curating the raw data collected from the literature (NCI database), which is openly available to the general public on the DTP web site (https://wiki.nci.nih.gov/display/NCIDTPdata/Molecular + Target + Data), while the anti-cancer screening method and assay used to measure the biological activities are reported in the DTP-NCI web site (https://dtp.cancer.gov/default.htm). The data contains aminopterin and camptothecin derivatives, colchicine analogues and so on.
The structures were imported into Spartan 14 graphic user interphase (GUI) and was subsequently converted into 3D structure by using the view option on Spartan 14. From the build option on Spartan 14 the structures were clean by checking minimize using molecular mechanic force field (MM+) option in order to remove all strain from the molecular structure. In addition this will ensure a well define conformer relationship among compounds of the study (Viswanadhan et al., 1989). From the set-up calculation option on Spartan 14, the calculation was set to equilibrium geometry at the ground state using density functional theory at B3LYP (Becke88 three-parameter hybrid exchange potentials with Lee-Yang-Parr correlation potential) level of theory and 6-311G (d) basis set for the geometrical optimization of the cleansed structures i.e. B3LYP/6-311G (d) level of theory.
4 Receptor preparation
The receptor employed in this study is a human Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform. The PDB file of the receptor was download from RCSB PDB (www.rcsb.org), the PI3K unit complexed with 2-amino-8-cyclopentyl-4-methyl-6-(1H-pyrazol- 4-yl)pyrido[2,3-d]pyrimidin-7(8H)-one (PDB ID: 4FA6) (Le et al., 2012), was an x-ray diffraction crystal structure of a human Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K with a resolution of 2.7 Å.
The receptor file was imported into the Molsoft.icm-pro.v3.8.3 GUI (Abagyan and Totrov, 1994; An et al., 2005) and the complexed ligand 2-amino-8-cyclopentyl-4-methyl-6-(1H-pyrazol- 4-yl)pyrido[2,3-d]pyrimidin-7(8H)-one was deleted from the object structure before converting the PDB file into an ICM-object by deleting the excess water molecules contained in the x-ray structures, optimizing the hydrogens and other amino acids such as Histidine, proline, glycine, cysteine etc., missing side chains were also treated before the receptor was used for the docking process.
5 Docking process
The displayed receptor after treatment was docked with the ligands, the search algorithm Monte-Carlo was used for to locate the global minimum on the receptor extremely rugged energy landscape, the ligand position and internal torsions optimized by stochastic monte-Carlo search in the frame work of the Internal Coordinates Mechanics (ICM) registers the best fit for the ligands. The ICM scoring function is weighted according to the following parameters (i) internal force-field energy of the ligand, (ii) entropy loss of the ligand between bound and unbound states, (iii) ligand-receptor hydrogen bond interactions, (iv) polar and non-polar solvation energy differences between bound and unbound states, (v) electrostatic energy, (vi) hydrophobic energy, and (vii) hydrogen bond donor or acceptor desolvation. The lower the ICM score, the higher the chance the ligand is a binder.
The 2D and 3D interactions of the docking result were gotten by importing our result into the Discovery studio visualizer, hence enabling us in identifying significant interaction between the ligands and the receptor binding site.
Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Thioguanine is shown in Fig. 1. Thioguanine binds firmly at the target site of 4FA6 with three conventional hydrogen bonds (Val882, Glu880) and one C-H interaction with Ile881. The ICM score for the best interaction pose is reported in Table 1, as −22.026 Kcal/mol and Ki = 0.963.
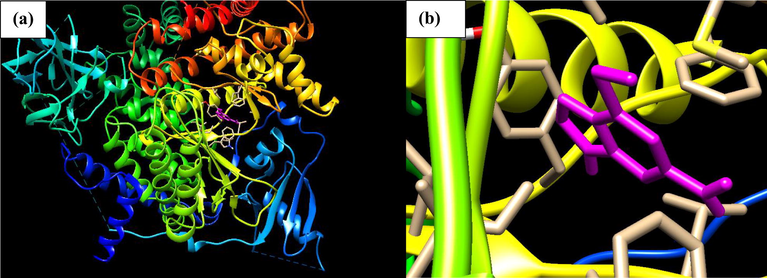
- (a) Docked poses of Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Thioguanine ligand (stick figure in Purple colour); (b) enlarged view of Thioguanine with 4FA6 Pose.
| Index | Name | Binding Energy (ΔG = Kcal/mol) | KI | Nflex | Hbond | Hphob | VwInt |
|---|---|---|---|---|---|---|---|
| 1 | AMINOPTERIN DERIVATIVE3 | –33.089 | 0.946 | 8 | −10.269 | −5.721 | −36.111 |
| 2 | METHOTREXATE DERIVATIVE | −31.447 | 0.948 | 10 | −8.545 | −5.681 | −40.271 |
| 3 | AN ANTIFOL | −29.475 | 0.951 | 10 | −10.982 | −7.129 | −37.09 |
| 4 | MITOXANTRONE | −27.852 | 0.954 | 12 | −9.727 | −7.868 | −30.45 |
| 5 | 11-HYDROXMETHYL-20(RS)-CAMPTOTHECIN | −24.829 | 0.959 | 4 | −9.123 | −6.816 | −20.147 |
| 6 | BREQUINAR | −24.209 | 0.96 | 1 | −4.912 | −7.594 | –23.276 |
| 7 | ANTHRAPYRAZOLE DERIVATIVE | –23.229 | 0.962 | 9 | −8.689 | −7.744 | −27.939 |
| 8 | 11-FORMYL-20-RS-CAMPTOTHECIN2 | –22.077 | 0.963 | 2 | −7.719 | −6.23 | −21.812 |
| 9 | THIOGUANINE | –22.026 | 0.963 | 1 | −5.857 | −2.194 | −18.947 |
| 10 | 3-HP | −21.587 | 0.964 | 1 | −6.854 | −2.778 | −16.258 |
| 11 | ALPHA-TGDR | −21.38 | 0.965 | 4 | −8.217 | −3.157 | −24.535 |
| 12 | CAMPTOTHECIN Na SALT | −21.141 | 0.965 | 4 | −2.588 | −6.334 | –22.831 |
| 13 | THIOPURINE | −21.055 | 0.965 | 0 | −4.412 | −1.911 | −17.771 |
| 14 | PYRAZOLOACRIDINE | −21.05 | 0.965 | 5 | −2.076 | −7.854 | –32.928 |
| 15 | METHOTREXATE | −20.988 | 0.965 | 9 | −9.084 | −5.957 | –33.422 |
| 16 | PYRAZOFURIN | −20.575 | 0.966 | 5 | −8.751 | −2.679 | −19.819 |
| 17 | OXANTHRAZOLE | −20.511 | 0.966 | 10 | −11.028 | −7.249 | −25.222 |
| 18 | MELPHALAN | −20.002 | 0.967 | 9 | −5.555 | −6.422 | −24.405 |
| 19 | 5HP | −18.933 | 0.969 | 1 | −3.853 | −3.228 | −18.207 |
| 20 | BISANTRENE HCl | −18.803 | 0.969 | 2 | −6.342 | −7.057 | −27.121 |
| 21 | COLCHICINE | −18.646 | 0.969 | 1 | −3.98 | −6.643 | −25.752 |
| 22 | GLYCINATE | −18.184 | 0.97 | 6 | −3.784 | −7.89 | −25.611 |
| 23 | MITOZOLAMIDE | −18.143 | 0.97 | 2 | −4.942 | −3.968 | −20.115 |
| 24 | FTORAFUR | −17.664 | 0.971 | 4 | −5.465 | −3.806 | −19.954 |
| 25 | BAKERS ANTIFOL | −17.297 | 0.971 | 3 | −2.585 | −8.681 | −30.976 |
| 26 | DICHLORALLYL LAWSONE | −17.235 | 0.971 | 2 | −1.979 | −5.354 | −21.806 |
| 27 | 9-AMINO-20-RS-CAMPTOTHECIN | −17.203 | 0.971 | 2 | −4.382 | −6.574 | −25.77 |
| 28 | INOSINE GLYCODIALDEHDE | −16.446 | 0.973 | 7 | −8.261 | −3.477 | −21.042 |
| 29 | GUANAZOLE | −16.181 | 0.973 | 0 | −5.87 | −1.179 | −13.23 |
| 30 | MACBECIN | −15.751 | 0.974 | 4 | −4.26 | −7.533 | −26.23 |
| 31 | CAMPTOTHECIN HYDROX | −15.431 | 0.974 | 2 | −4.659 | −6.672 | −24.326 |
| 32 | 7-CHLOROCAMPTOTHECIN | −15.283 | 0.975 | 2 | −4.871 | −6.682 | −19.515 |
| 33 | VINBLASTINE SULFATE | −14.859 | 0.975 | 8 | −1.819 | −11.104 | −29.373 |
| 34 | DOXORUBICIN | −14.774 | 0.975 | 8 | −4.203 | −7.954 | −35.804 |
| 35 | AMINOPTERIN DERIVATIVE | −14.704 | 0.975 | 9 | −5.15 | −6.875 | −34.253 |
| 36 | m-AMSA | −14.7 | 0.975 | 2 | −0.46 | −8.18 | −27.799 |
| 37 | 5-FLUOROURACIL | −14.501 | 0.976 | 0 | −2.235 | −2.211 | −15.564 |
| 38 | 14-CHLORO-20(S)-CAMPTHOTHECIN HYDRATE | −14.39 | 0.976 | 2 | −4.94 | −6.946 | −17.762 |
| 39 | HYCANTHONE | −14.363 | 0.976 | 5 | −2.048 | −8.145 | −28.87 |
| 40 | COLCHICHINE DERIVATIVE | −14.36 | 0.976 | 2 | −1.267 | −9.044 | −35.139 |
| 41 | AMONAFIDE | −14.171 | 0.976 | 3 | −2.678 | −6.237 | –22.285 |
| 42 | CYCLOCYTIDINE | −14.05 | 0.977 | 3 | −6.286 | −3.433 | −14.897 |
| 43 | TRIMETREXATE | −13.984 | 0.977 | 2 | −1.478 | −7.771 | −26.584 |
| 44 | BETA-TGDR | −13.984 | 0.977 | 4 | −5.993 | −3.19 | –23.74 |
| 45 | FLURODOPAN | −13.457 | 0.978 | 4 | −3.219 | −4.975 | −21.405 |
| 46 | URACIL NITROGEN MUSTARD | −13.362 | 0.978 | 4 | −3.094 | −5.085 | −21.295 |
| 47 | RUBIDAZONE | −13.329 | 0.978 | 7 | −3.862 | −9.62 | −39.789 |
| 48 | ARA-C | −13.21 | 0.978 | 5 | −5.157 | −3.422 | −21.5 |
| 49 | ACIVICIN | −13.198 | 0.978 | 4 | −5.451 | −2.784 | −14.249 |
| 50 | 5-AZACYTIDINE | −12.996 | 0.978 | 5 | −6.848 | −2.539 | −16.645 |
| 51 | HYDROXYUREA | −12.812 | 0.979 | 2 | −5.615 | −0.43 | −10.028 |
| 52 | CAMPTOTHECIN ANALOG | −12.445 | 0.979 | 3 | −4.862 | −7.173 | −20.725 |
| 53 | AMINOPTERIN DERIVATIVE | −12.29 | 0.979 | 8 | −5.063 | −6.682 | –33.846 |
| 54 | THIOCOLCHICINE | −12.277 | 0.979 | 1 | −2.647 | −7.658 | −28.759 |
| 55 | PYRAZOLOIMIDAZOLE | −12.22 | 0.98 | 0 | −2.126 | −3.307 | −11.13 |
| 56 | TETRAPLATIN | −12.17 | 0.98 | 2 | −5.949 | −3.108 | −8.602 |
| 57 | DAUNORUBICIN | −12.169 | 0.98 | 6 | −2.956 | −7.985 | –32.129 |
| 58 | ALLOCOLCHICINE | −11.968 | 0.98 | 1 | −3.149 | −8.413 | –23.915 |
| 59 | CAMPTOTHECIN | −11.912 | 0.98 | 2 | −4.927 | −6.459 | −16.949 |
| 60 | CAMPTOTHECIN 9-METHOX | −11.73 | 0.98 | 2 | −1.661 | −7.204 | −21.386 |
| 61 | CAMPTOTHECIN GLUTAMATE1 | −11.691 | 0.98 | 8 | −6.512 | −6.743 | –23.633 |
| 62 | CAMPTOTHECIN ANALOG3 | −11.67 | 0.98 | 2 | −4.904 | −6.75 | −17.174 |
| 63 | METHYL CCNU | −11.603 | 0.981 | 5 | −1.489 | −5.614 | −19.757 |
| 64 | TRITYL CYSTEINE | −11.366 | 0.981 | 9 | −7.091 | −6.724 | −19.347 |
| 65 | TRIETHYLENEMELAMINE | −11.205 | 0.981 | 0 | 0 | −5.951 | −17.337 |
| 66 | MENOGARIL | −11.123 | 0.981 | 5 | −3.213 | −7.239 | −24.427 |
| 67 | HEPSULFAM | −11.038 | 0.982 | 12 | −2.463 | −4.564 | −18.664 |
| 68 | CAMPTOTHECIN 20-O-(N,N-DIMETHYL)GLYCINATE | −10.879 | 0.982 | 4 | −2.229 | −7.33 | −26.22 |
| 69 | CAMPTOTHECIN,20O | −10.821 | 0.982 | 7 | −3.882 | −8.166 | –32.044 |
| 70 | CAMPTOTHECIN ANALOG2 | −10.682 | 0.982 | 4 | −2.764 | −7.561 | –23.92 |
| 71 | CARBOPLATIN | −10.425 | 0.983 | 0 | −4.996 | −2.614 | −12.68 |
| 72 | BCNU | −10.321 | 0.983 | 6 | 0 | −4.669 | −18.198 |
| 73 | APHIDICOLIN GLYCINATE | −10.194 | 0.983 | 8 | −5.056 | −7.069 | –22.803 |
| 74 | TEROXIRONE | −10.045 | 0.983 | 6 | −3.131 | −5.741 | −18.813 |
| 75 | PIPOBROMAN | −9.763 | 0.984 | 6 | −1.357 | −6.527 | −18.947 |
| 76 | CAMPTOTHECIN PHOSPHAT | −9.602 | 0.984 | 5 | −3.228 | −7.216 | −20.546 |
| 77 | 5-AZA-2-DEOXYCYTIDINE | −9.535 | 0.984 | 4 | −5.09 | −2.796 | −15.777 |
| 78 | CAMPTOTHECIN ACETATE | −9.453 | 0.984 | 2 | −1.468 | −7.505 | −24.132 |
| 79 | 5,6-DIHYDO-5-AZACYTIDINE | −9.17 | 0.985 | 5 | −5.395 | −3.117 | −19.315 |
| 80 | DIANHYDROGALACTITOL | −9.112 | 0.985 | 5 | −4.079 | −3.192 | −7.471 |
| 81 | MORPHOLINODOXORUBICIN | −8.947 | 0.985 | 8 | −2.778 | −9.342 | −37.806 |
| 82 | VM-262 | −8.771 | 0.985 | 6 | −5.66 | −8.88 | –22.74 |
| 83 | PCNU | −8.755 | 0.985 | 5 | −2.444 | −4.211 | −19.583 |
| 84 | PORFIROMYCIN | −8.746 | 0.985 | 3 | −3.149 | −5.9 | −20.082 |
| 85 | SIPIROHYDANTOIN | −8.571 | 0.986 | 7 | −1.834 | −7.913 | −20.819 |
| 86 | MITOMYCIN1 | −8.125 | 0.986 | 3 | −2.094 | −5.194 | −26.068 |
| 87 | CAMPTOTHECIN ETHYLGLYCINATE ESTER | −8.047 | 0.987 | 5 | −2.119 | −7.187 | −27.815 |
| 88 | CCNU | −7.972 | 0.987 | 5 | 0 | −5.695 | −16.801 |
| 89 | CHLORAMBUCIL | −7.614 | 0.987 | 9 | −3.014 | −6.928 | −15.12 |
| 90 | CIS-PLATINUM | −7.529 | 0.987 | 0 | −4.179 | −0.87 | −7.386 |
| 91 | CAMPTOTHECIN HEMISUCCINATE SODIUM SALT | −7.102 | 0.988 | 5 | −1.32 | −7.774 | −29.282 |
| 92 | CAMPTOTHECIN LYSINATE HCl | −6.57 | 0.989 | 9 | −3.998 | −6.894 | −31.317 |
| 93 | BUSULFEX (busulfan) | −6.328 | 0.989 | 7 | 0 | −5.052 | −18 |
| 94 | CYANOMORPHOLINODOXORUBICIN | −6.289 | 0.989 | 8 | −5.814 | −9.022 | −34.354 |
| 95 | NITROGEN MUSTARD | −6.162 | 0.99 | 4 | 0 | −5.16 | −11.191 |
| 96 | PIPERAZINE DRUGSMAINATOR | −5.892 | 0.99 | 5 | 0 | −6.516 | −15.032 |
| 97 | CAMPTOTHECIN BUTYLGLYCINATE ESTER | −5.872 | 0.99 | 7 | −2.368 | −7.45 | −26.85 |
| 98 | N,N-DIMENZYL DAUNOMYCIN | −5.733 | 0.99 | 10 | −1.209 | −11.9 | −45.659 |
| 99 | DEOXYDOXORUBICIN | −5.186 | 0.991 | 7 | −2.207 | −8.28 | –32.606 |
| 100 | RHIZOXIN | −4.887 | 0.992 | 5 | −2.168 | −11.306 | –33.339 |
| 101 | CHLOROZOTOCIN | −4.825 | 0.992 | 10 | −4.637 | −4.219 | −14.688 |
| 102 | 2-DEOXY-5-FLUOROURIDINE | −4.822 | 0.992 | 13 | −3.211 | −8.909 | −28.752 |
| 103 | YOSHI | −4.177 | 0.993 | 10 | 0 | −5.692 | −16.948 |
| 104 | PIPERAZINEDIONE | −3.901 | 0.993 | 2 | −1.817 | −6.647 | −15.247 |
| 105 | AZQ | −3.635 | 0.994 | 4 | −0.842 | −7.851 | –23.672 |
| 106 | CHIP | −3.573 | 0.994 | 2 | −5.33 | −1.091 | −3.754 |
| 107 | CYCLODISONE | −3.572 | 0.994 | 0 | 0 | −2.734 | −9.45 |
| 108 | CLOMESONE | −3.564 | 0.994 | 5 | 0 | −4.248 | −12.296 |
| 109 | L-ALANOSINE | −3.368 | 0.994 | 6 | −4.09 | −0.683 | −7.597 |
| 110 | MAYTANSINE | −2.715 | 0.995 | 6 | −6.412 | −10.729 | −24.95 |
| 111 | THIO-TEPA | −2.262 | 0.996 | 3 | −0.327 | −4.515 | −10.408 |
| 112 | ASALEY | −1.731 | 0.997 | 13 | −1.95 | −9.634 | −27.415 |
| 113 | VINCRISTINE SULFATE | −1.678 | 0.997 | 9 | −3.073 | −10.024 | –22.579 |
| 114 | HALICHONDRI | 0.9 | 1.002 | 8 | −7.333 | −11.178 | –23.944 |
| 115 | TAXOL | 0.99 | 1.002 | 13 | −4.527 | −10.472 | −35.197 |
| 116 | PALA | 1.023 | 1.002 | 10 | −4.629 | −1.338 | −11.965 |
| 117 | VP-162 | 1.46 | 1.002 | 5 | −1.11 | −9.404 | –33.24 |
| 118 | CARBOXYPHTHALATOPLATINUM | 4.229 | 1.007 | 1 | −2.275 | −5.371 | −15.395 |
| 119 | DOLASTINE | 8.036 | 1.014 | 23 | −2.857 | −14.258 | −34.604 |
Ki = inhibition Constant, Nflex = Number of Flexible Bonds, Hbond = Hydrogen bond interaction, Hphob = hydrophobic interaction.
There are eight amino acids involved in a hydrophobic interaction with Thioguanine (Trp812, Ile881, Ile831, Met953, Ile879, Tyr867, Ile963, Glu880) which is reported in Table 2 and can be seen in Fig. 2, thioguanine though a small molecule, was found to have a high binding energy and hence inhibiting P13K (4FA6) better than 95% of all the other drugs in this study. The unique stability of thioguanine in the binding site is attributed to the large number of pi-interactions such as Pi-Pi interaction with Phe961 and Tyr867, Pi-alkyl interaction with Ile963, Ile879, Ile881 and finally Pi-Sulphur interaction with Met953 resulting from the number of Nitrogen atom in the hetero-aromatic ring. The large number of Pi-sigma interactions (Pi-alkyl and Pi-Sulphur) which largely involves charge transfer helps in intercalating the drug in the binding site of the receptor (4FA6).
| Name | Hydrogen Bond (HB) interaction | Bond Length (Å) for HB interaction | Hydrophobic interaction | Pi-sigma and amide interaction | Alkyl Interaction | Pi-sulphur | Carbon-Hydrogen Interaction |
|---|---|---|---|---|---|---|---|
| Thioguanine | Val882, Glu880 | (2.87, 2.88), 3.21 | Trp812, Ile881, Ile831, Met953, Ile879, Tyr867, Ile963, Glu880 | – | Phe961, Ile963, Ile879, Tyr867 | Met953 | Ile881 |
| Mitoxantrone | Val882, Lys890, Ser806, Lys807, Glu880 | 3.14, 3.06, (3.21, 2.78), 2.67, 3.13 | Met953, Ile831, Glu880, Ile879, Ile963, Pro810, Trp812, Met804, Lys833, Asp964 | – | Pro810, Ile831, Ile963 | Met804 | Asp964 |
| Methotrexate Derivative | Glu880, Val882, Gln893, Lys807, Ala805 | 2.89, 3.14, 2.85, 2.74, 2.31 | Ile879, Tyr867, Trp812, Met953, Ile881, Ile963, Ile831, Lys890, Asp950, Ala889, Tyr757 | – | Ile831, Ile963 | Met804, Met953 | Asp950, Tyr867 |
| Brequinar | Ala805, Ser806 | 3.07, 2.49 | Asp950, Thr887, Met953, Glu880, Ile879, Val882, Tyr867, Ile963, Met804, Ile831 | – | Pro810, Ile831, Ile963, Ile879, Tyr867, Met804, Met953 | – | Thr887 |
| Anthrapyrazole Derivative | Glu880, Lys890, Asp950, Ser806, Lys807 | 2.76, 2.65, 3.31, 3.20, 2.54 | Phe961, Val882, Met953, Tyr867, Ile831, Ile963, Trp812, Met804, Lys883, Asp964 | – | Pro810, Ile963, Ile831 | Met804 | Asp964 |
| An Antifol | Ser806, Asn951, Thr887, Lys890, Val882, Glu880 | 2.39, 2.59, 3.03, 2.62, (2.70, 2.89), 2.81 | Met804, Asp950, Lys808, Asp964, Ala805, Trp812, Lys833, Ile963, Ile879, Ile831, Met953, Ala885, Glu880, Ile881 | – | Phe961, Ile881, Ile963, Ile831, Met804, Ala805 | Met953 | – |
| Aminopterin Derivative 3 | Asp950, lys807, Ser806, Glu880, Val882 | 2.76, 3.04, (2.92, 2.55), 2.73, (3.03, 2.89) | Asn951, Ile963, Lys808, Ile831, Lys833, Pro810, Met953, Ile881, Tyr867, Ile879 | – | Met953, Phe961, Ile963, Ile831, Pro810 | – | – |
| 11-Hydroxmethyl-20(Rs)-Camptothecin | Lys807, Asp950, Glu880, Val882, Ser806 | (2.71, 3.49), 3.37, 3.77, (3.01, 2.80), 2.32 | Ser806, Lys833, Ile831, Pro810, Tyr867, Ile963, Met953, Ile879, Ile881 | – | Lys833, Pro810, Ile963, Ile831, Ile881, Phe961 | Met963 | – |
| 11-Formyl-20-Rs-Camptothecin | Tyr867, Ser806, Ala805, Lys890 | 3.11, (3.29, 2.82), 2.30, 2.59 | Ile879, Ile963, Ile831, Asp964, pro810, Trp812, Asp950, Met804 | Ile831 | Ile831, Ile963 | Met804 | Asp964 |
| 3HP | Asp884, Val882, Glu880 | 3.00, (2.70, 2.75), 3.04 | Lys883, Ala885, Ile881, Trp821, Met953, Ile831 | – | Ile831 | Met953, Trp812 | – |
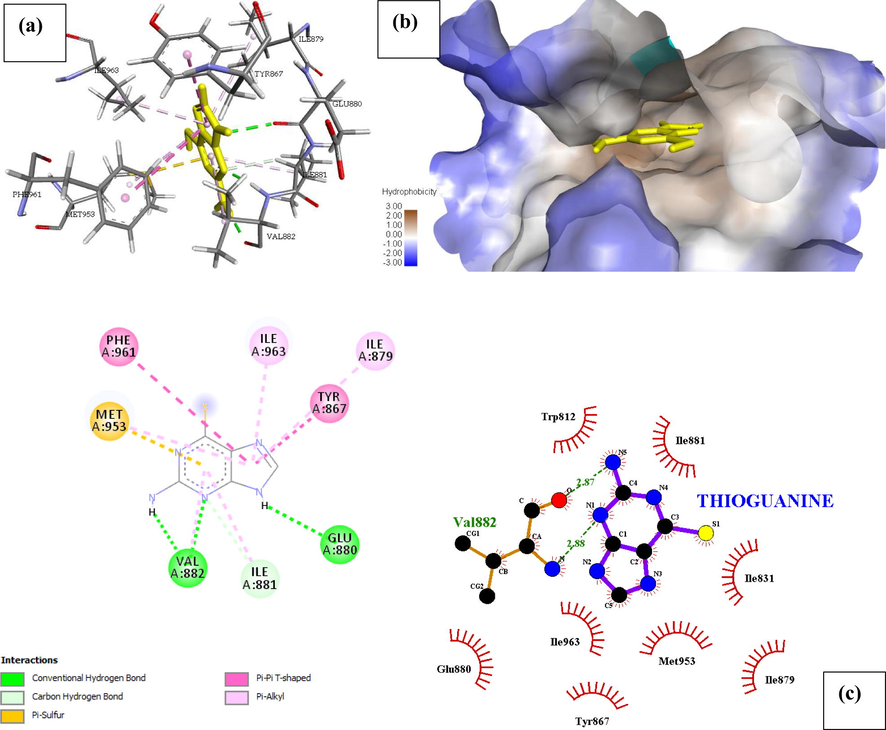
- Docked poses of Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Thioguanine ligand (stick figure) (a) 3D Thioguanine with surrounding amino acids of 4FA6; (b) Thioguanine with 4FA6 (hydrophobicity surface) at the active binding site; (c) 2D view of interaction type of Thioguanine with surrounding amino acids of 4FA6.
Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform (PDB ID: 4FA6) with mitoxantrone is shown in Fig. 3, the binding energy and inhibition constant of this interaction is reported in Table 1, to be −27.852 Kcal/mol and Ki = 0.954 respectively making it the 4th most active drugs with the ability to inhibit the receptor P13K (4FA6). The docked result of the displayed in Fig. 3, indicates the drugs has six hydrogen bond interaction with five amino acids reported with their interatomic distance from Mitoxantrone on Table 2. Other important interactions such as Hydrophobic interaction, Pi-alkyl, Pi-Sulphur interactions were also reported in Table 2. Mitoxantrone high affinity was also associated with the presence of Van der waal forces created on the backbone of the amide substituents with the respective amino acids Ile879, Lys808, Lys833, Asp836, Ser806, Asn951, Phe961, Met953, Trp812 and Ile 881, which undoubtedly created a strong cohesive environment, thereby stabilizing the complex formed.
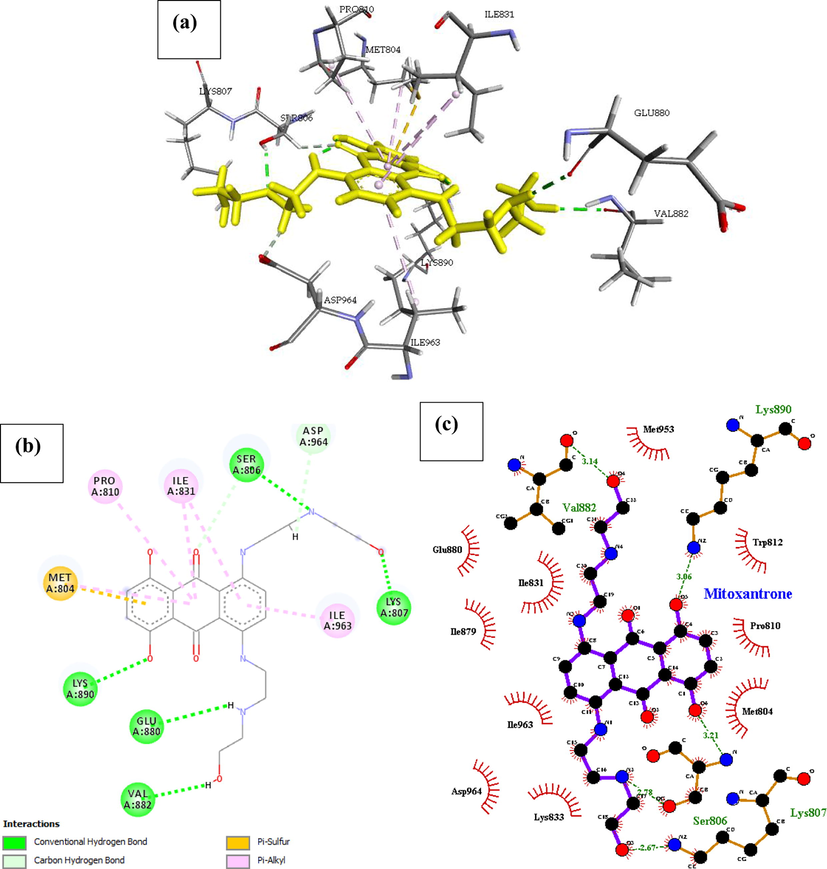
- Docked poses of Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Mitoxanthrone ligand (stick figure) (a) 3D Mitoxanthrone with surrounding amino acids of 4FA6; (b) 2D view of interaction type of Mitoxanthrone with surrounding amino acids of 4FA6; (c) 2D view of internuclear distance for hydrogen bond interaction of Mitoxanthrone with surrounding amino acids of 4FA6.
Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Methotrexate derivative. The drug binds to the protein molecule satisfactorily with an estimated free binding energy of −31.447 kcal/mol and Ki of 0.948 making it the second most active drug. It shows binding with five H-bonds between five different amino acids as captured in Fig. 4, further enquiry indicates the presence of a pi-Sulfur interaction with Met804, and pi-alkyl interaction with a pair of isoleucine amino acid (Ile 963 and Ile831). Conformational stability of the drug associated with other Pi-stacked interaction includes Pi-cation interaction with Lys890. The compound also displayed many hydrophobic contacts mediated by the aliphatic amino acids Ile879, Tyr867, Trp812, Met953, Ile881, Ile963, Ile831, Lys890, Asp950, Ala889, Tyr757.
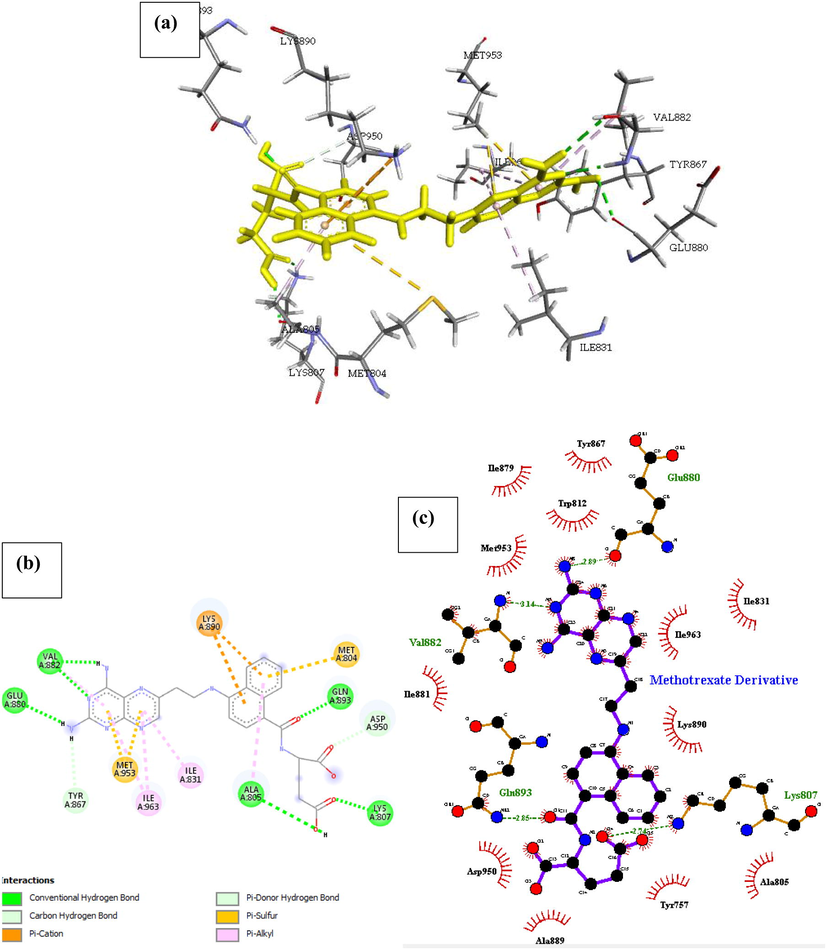
- Docked poses of Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Methotrexate Derivative ligand (stick figure) (a) 3D Methotrexate Derivative with surrounding amino acids of 4FA6; (b) 2D view of interaction type of Methotrexate Derivative with surrounding amino acids of 4FA6; (c) 2D view of internuclear distance for hydrogen bond interaction of Methotrexate Derivative with surrounding amino acids of 4FA6.
Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Brequinar. Binding ability of Brequinar is good, there are two hydrogen bonds formed with the amino acids Ala805 and Ser806 respectively, the binding energy of the complex is reported as −24.209 kcal/mol in Table 1. The conformational energy of the ligand is minimized by the presence of Pi-cation interaction with Lys890 balanced by a 187° angle of Pi-anion interaction with Asp950 on the next adjacent ring of the compound. Other noticeable pi interaction is with Ile879, Ile831, Ile963, Tyr867 and Met953 forming a Pi-alkyl interaction with a substituent of the compound. There are about nine hydrophobic interactions with the drug at the binding site, shown in Fig. 5 and reported in Table 2.
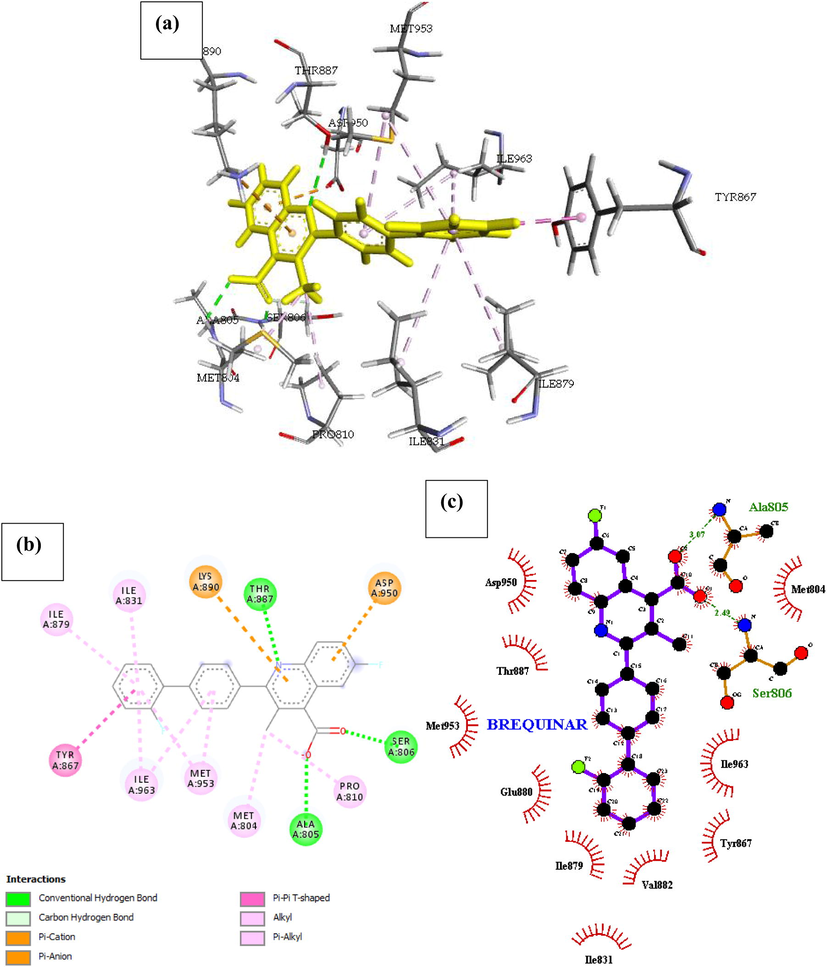
- Docked poses of Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Brequinar ligand (stick figure) (a) 3D Brequinar with surrounding amino acids of 4FA6; (b) 2D view of interaction type of Brequinar with surrounding amino acids of 4FA6; (c) 2D view of internuclear distance for hydrogen bond interaction of Brequinar with surrounding amino acids of 4FA6.
Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Anthrapyrazole derivative expressed in Fig. 6. Anthrapyrazole derivative being primarily a kinase inhibitor docks well with the P13K (4FA6) domain with a free binding energy of –23.229 kcal/mol and inhibition constant of 0.962. Seven H-bonds were identified between the protein and ligand molecule, five of which shows conventional H-bonding with Glu880, Lys890, Asp950, Ser806, Lys807 of the receptor with an interatomic distance of 2.76, 2.65, 3.31, 3.20 and 2.54 Å respectively from the drug. In addition, Pro810, Ile963 and Ile831 formed pi-alkyl bonds with the drug, while Met804 pi-sulfur bond with the cyclohexane backbone of the ligand. Hydrophobic interactions are mediated by Phe961, Val882, Met953, Tyr867, Ile831, Ile963, Trp812, Met804, Lys883, Asp964.

- Docked poses of Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Anthrapyrazole Derivative ligand (stick figure) (a) 3D Anthrapyrazole Derivative with surrounding amino acids of 4FA6; (b) 2D view of interaction type of Anthrapyrazole Derivative with surrounding amino acids of 4FA6; (c) 2D view of internuclear distance for hydrogen bond interaction of Anthrapyrazole Derivative with surrounding amino acids of 4FA6.
Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with An Antifol. The docked structure presented in Fig. 7 shows a negative free energy of binding (-29.475 Kcal/mol) suggesting that binding is practicable as most of the interaction energies are of H-bond type with amino acids (Ser806, Asn951, Thr887, Lys890, Val882, Glu880) and more than twenty hydrophobic interactions with fourteen amino acids reported in Table 2, therefore ensuing in an overall negative value. The result of the complex stability can be linked to the with extra Pi-sigma interaction associated with Pi-cation (Lys833), Pi-Sulfur (Met953) and Pi-alkyl interactions (Ile881, Ile963, Ile831, Ala805 and Met804).
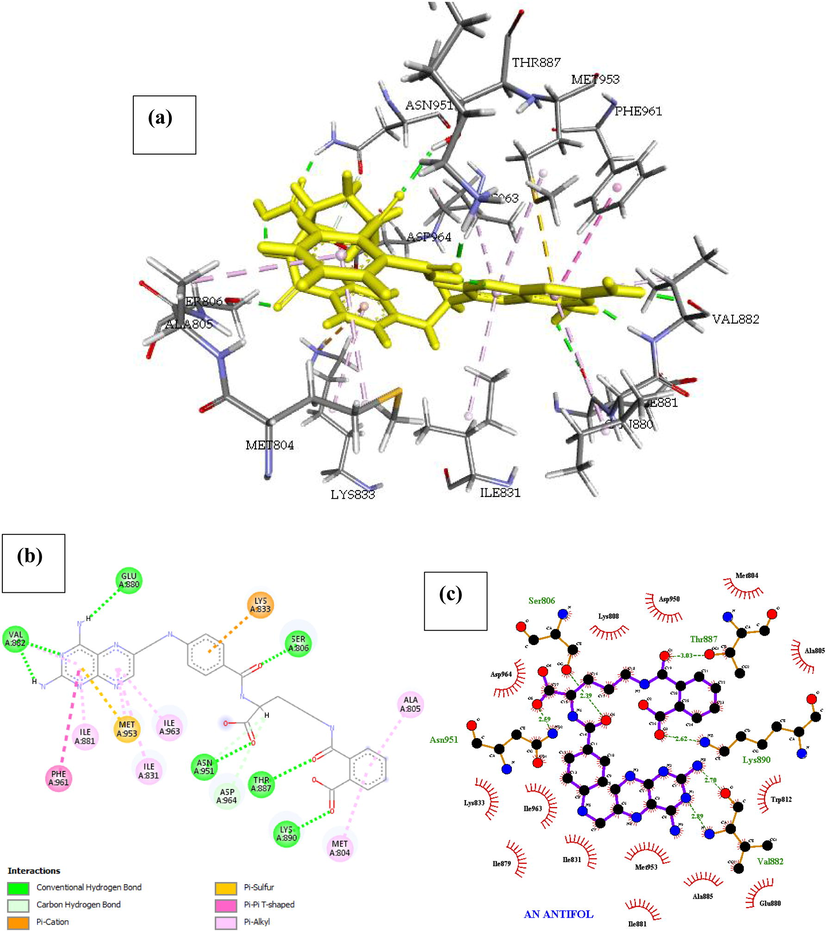
- Docked poses of Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with An Antifol ligand (stick figure) (a) 3D An Antifol with surrounding amino acids of 4FA6; (b) 2D view of interaction type of An Antifol with surrounding amino acids of 4FA6; (c) 2D view of internuclear distance for hydrogen bond interaction of An Antifol with surrounding amino acids of 4FA6.
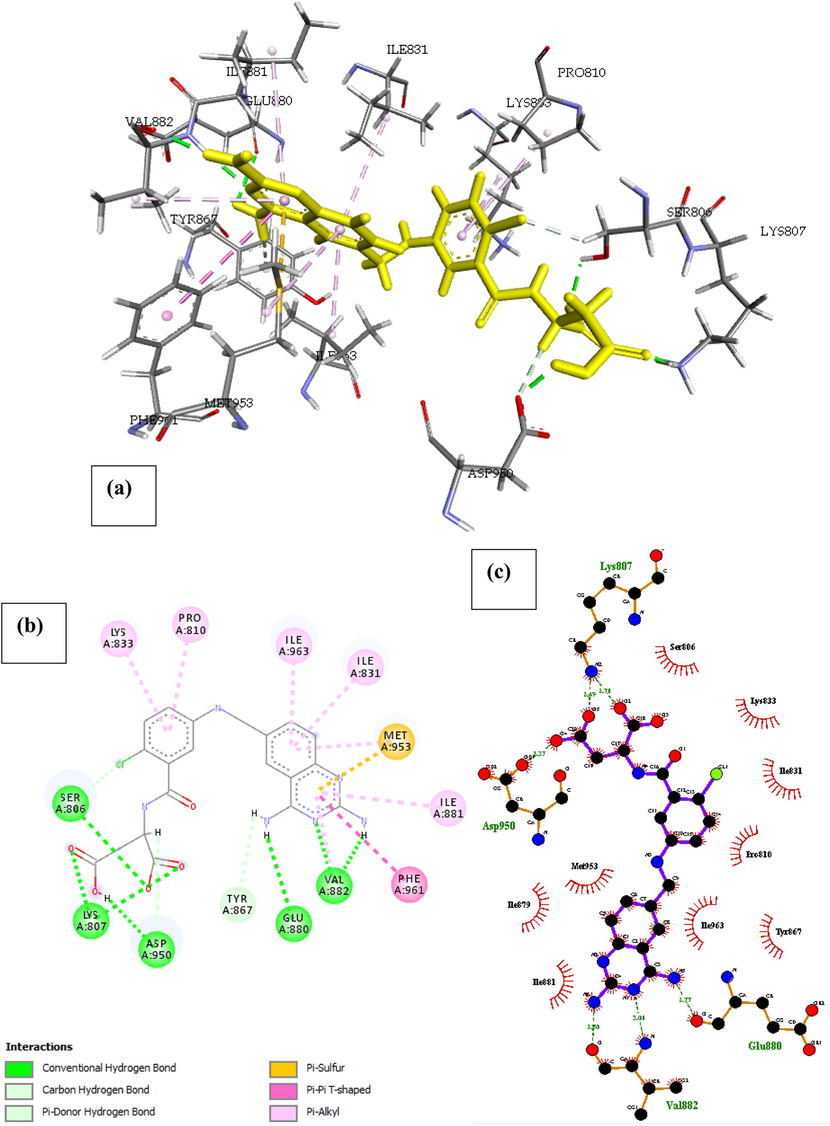
Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Aminopterin Derivative 3. The interaction within the complex can be seen in Fig. 8., Aminopterin Derivative 3 inhibited P13k more than any drug we studied. Our computational studies indicated that the Binding energy of Aminopterin Derivative 3 with the receptor is –33.089 Kcal/mol, this interaction was achieved by 8 flexible bonds on the drug and Pi-sigma interaction introducing stabilizing charges responsible for intercalating the drug within the P13k protein (4FA6). There were seven (7) H-bonds identified in the complex and more than twelve hydrophobic interaction with ten (10) amino acids (Table 2). The Pi-Cation interaction with Lys833 was balanced the chlorine atom in the Drug, which formed a Pi-donor hydrogen bond with the OH of Ser806 in the binding site of the receptor. other noticeable interactions are pi-alkyl interaction with Pro810, Ile831, Ile963, Met953 and Pi-Pi interaction with Phe961 which is responsible for the shape of the complex.
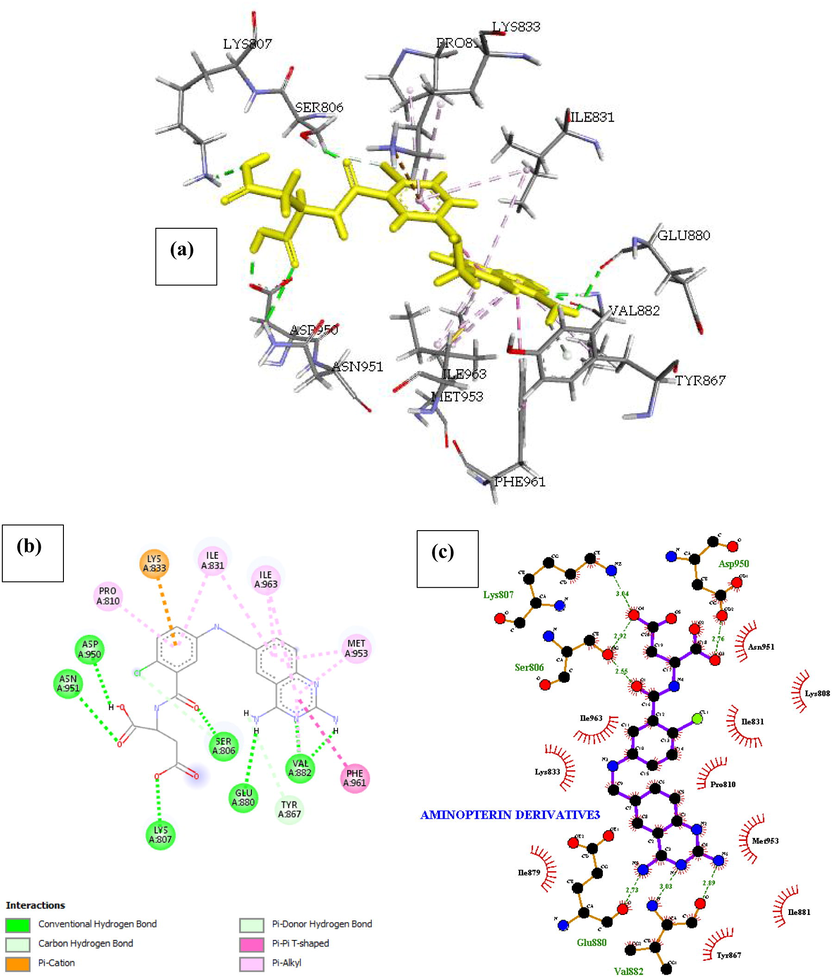
- Docked poses of Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with Aminopterin Derivative 3 ligand (stick figure) (a) 3D Aminopterin Derivative 3 with surrounding amino acids of 4FA6; (b) 2D view of interaction type of Aminopterin Derivative 3 with surrounding amino acids of 4FA6; (c) 2D view of internuclear distance for hydrogen bond interaction of Aminopterin Derivative 3 with surrounding amino acids of 4FA6.
P13K (PDB ID: 4FA6) with 11-Hydroxmethyl-20(RS)-Camptothecin is presented in Fig. 9, the binding energy and inhibition constant of this interaction is reported in Table 1, to be −24.829 Kcal/mol and Ki = 0.959 respectively making it the 5th most active NCI molecule with the ability to inhibit P13K (4FA6). This high binding affinity is as linked with the presence of seven H-bond with the amino acids Lys807, Asp950, Glu880, Val882 and Ser806 (interatomic distance of (2.71, 3.49), 3.37, 3.77, (3.01, 2.80) and 2.32 Å respectively). Other interactions such as hydrophobic-type with Ser806, Lys833, Ile831, Pro810, Tyr867, Ile963, Met953, Ile879 and Ile881, the Pi-alkyl interaction with Lys833, Pro810, Ile963, Ile831, Ile881, Pi-sulfur bond with Met953 and Pi-Pi T-shaped interaction with Phe961 results from the high number of heteroaromatic atom primarily Nitrogen which is responsible for the conformation of the drug within the receptor.
- Docked poses of Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with 11-Hydroxmethyl-20(Rs)-Camptothecin ligand (stick figure) (a) 3D 11-Hydroxmethyl-20(Rs)-Camptothecin with surrounding amino acids of 4FA6; (b) 2D view of interaction type of 11-Hydroxmethyl-20(Rs)-Camptothecin with surrounding amino acids of 4FA6; (c) 2D view of internuclear distance for hydrogen bond interaction of 11-Hydroxmethyl-20(Rs)-Camptothecin with surrounding amino acids of 4FA6.
P13K (PDB ID: 4FA6) with 11-Formyl-20-(RS)-Camptothecin. The result of the docked complex was illustrated in Fig. 10. The free energy of the interaction was reported in Table 1, as –22.077 Kcal/mol and the inhibition constant as Ki = 0.963 making it the 8th most active compound in our list. The decrease in the binding energy of 11-Formyl-20-(RS)-Camptothecin when compared with 11-Hydroxmethyl-20(RS)-Camptothecin was linked to the rigid back bone of the structure and less number of hetero atoms on the benzene rings. Four amino acids namely Tyr867, Ser806, Ala805, Lys890 formed five conventional hydrogen bond interaction with 11-Formyl-20-(RS)-Camptothecin and one carbon-hydrogen bond with Asp964. About 15 hydrophobic interactions with Ile879, Ile963, Ile831, Asp964, pro810, Trp812, Asp950 and Met804 were identified within the complex. A Pi-sigma (Ile879), Pi-Sulfur (Met804) and Pi-Alkyl (Ile831, Ile963) interactions were also seen in the complex, we identified their presence as a direction strain in the backbone of the drug responsible for normalizing the dipole moment of the drug through charge transfer with its neighboring amino acids.
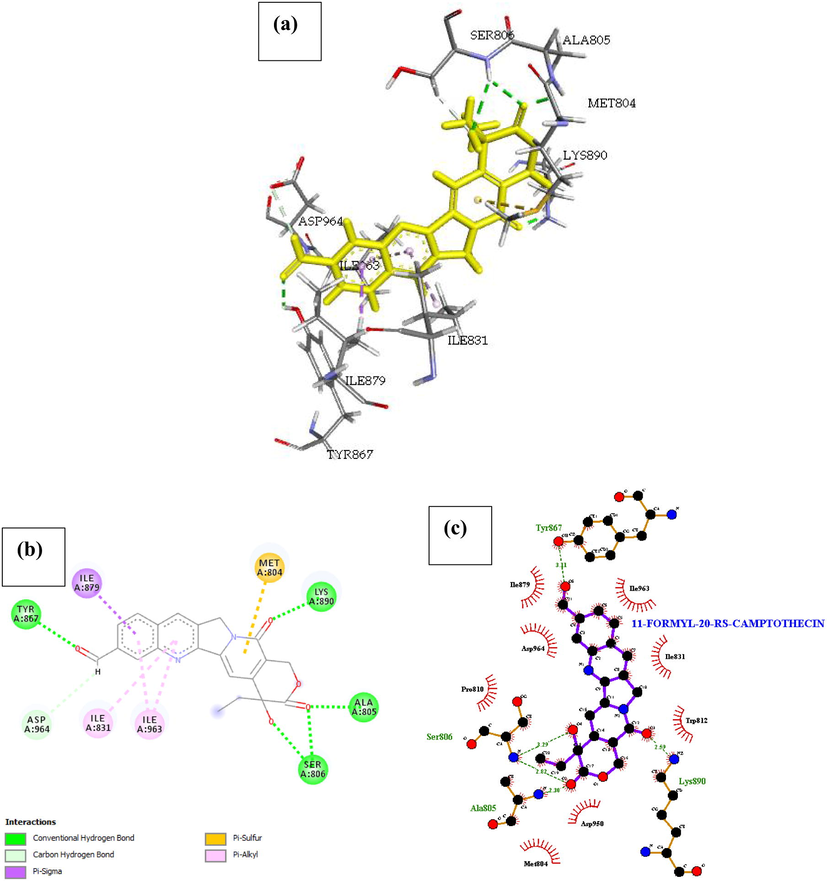
- Docked poses of Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with11-Formyl-20-Rs-Camptothecin ligand (stick figure) (a) 3D 11-Formyl-20-Rs-Camptothecin with surrounding amino acids of 4FA6; (b) 2D view of interaction type of 11-Formyl-20-Rs-Camptothecin with surrounding amino acids of 4FA6; (c) 2D view of internuclear distance for hydrogen bond interaction of 11-Formyl-20-Rs-Camptothecin with surrounding amino acids of 4FA6.
Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with 3HP. The docked structure presented in Fig. 11 shows a negative free energy of binding (−21.585 Kcal/mol) suggesting the possibility a stable interaction between the drug and the receptor. There are three conventional hydrogen bonds identified between the ligand and the binding site of the receptor. The extra stabilizing energy associated with the binding affinity of the drug can be linked to the Pi-Sulfur interaction of the pyridine component of the drug with Met953 competing with an opposing interaction with a Pi-alkyl bond (Ile831) at an angle of 344°. Other interaction includes the Pi-Sulfur (Met953) interaction with the amide substituent of 3HP.
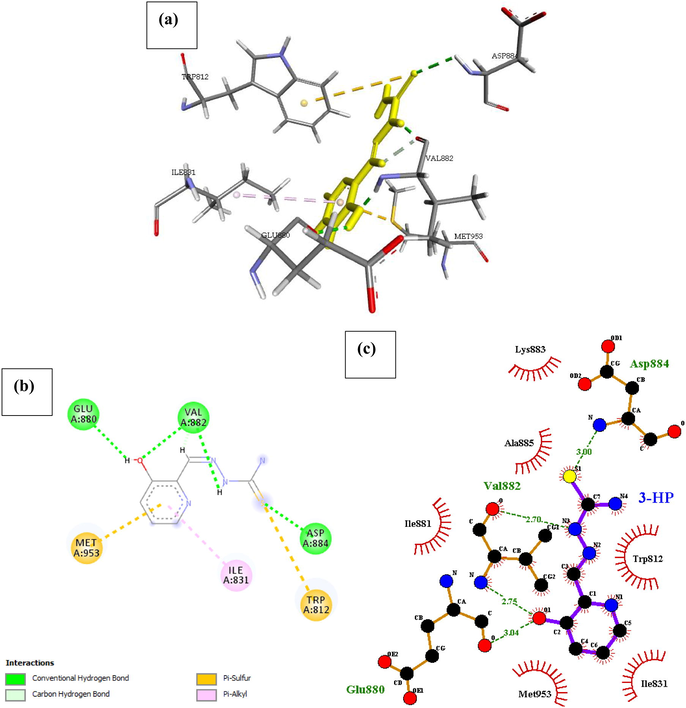
- Docked poses of Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform P13K (PDB ID: 4FA6) with3HP ligand (stick figure) (a) 3D 3HP with surrounding amino acids of 4FA6; (b) 2D view of interaction type of 3HP with surrounding amino acids of 4FA6; (c) 2D view of internuclear distance for hydrogen bond interaction of 3HP with surrounding amino acids of 4FA6.
The extra stabilizing energy associated with the binding affinity of the drug can be linked to the Pi-Sulfur interaction of the pyridine component of the drug with Met953 competing with an opposing interaction with a Pi-alkyl bond (Ile831) at an angle of 344°. Other interaction includes the Pi-Sulfur (Met953) interaction with the amide substituent of 3HP.
Fig. 12 shows that a strong hydrogen bond was formed between the ligand and Val882 and Asp964, this strong interaction was because of the Nitrogen atom on the compound. The existence of a P1-Sulfur bond helps in the intercalation of the ligand in the binding site. Other bond types such as alkyl and pi-alkyl bond helps to improve the hydrophobic interaction of the ligand in the binding pocket of the receptor.
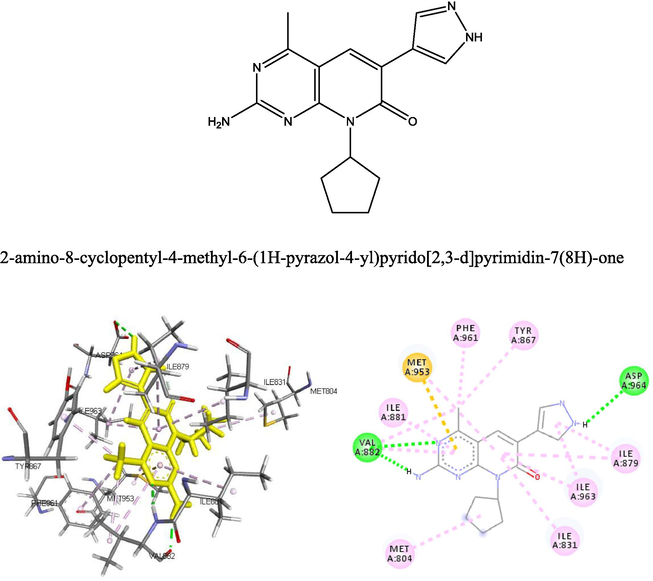
- The 2D and 3D structure of the natural ligand docked with the receptor p13k.
IPI-549 with a CAS number (1693758–51-8) and IUPAC/Chemical Name: (S)-2-amino-N-(1-(8-((1-methyl-1H-pyrazol-4-yl)ethynyl)-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide, is a potent and selective phosphoinositide-3-kinase (PI3Kγ) Inhibitor as an Immuno-Oncology Clinical Candidate (Kd = 0.29 nM). Bioactivity data of IPI-549 has been reported, biochemcial IC50 (nM) for PI3K isoform: 3200 (α); 3500 (β); 16 (γ); and >8400 (δ) respectively. Cellar IC50 (nM) of IPI549 for PI3K isoform: 250 (α); 240 (β); 1.6 (γ); and 180 (δ) respectively. The binding energy of IPI-549 (32.554 KCal) in the binding site after it was docked and evaluated by ICM pro was reported to be higher than all the 119 compounds in the NCI data base, with an exception to aminopterin derivative 3, which has a binding affinity of 33.088 Kcal. The docking result in Fig. 13 shows the presence of hydrogen bond between the IPI-554N-atom with Val 882, this interaction was found to be consistent with the interaction of the natural ligand at that binding site, as well as other ligands found to have high binding affinity with the receptor. This suggest that the presence of this bond type coupled with other molecular descriptor type are responsible for inhibition of P13k.
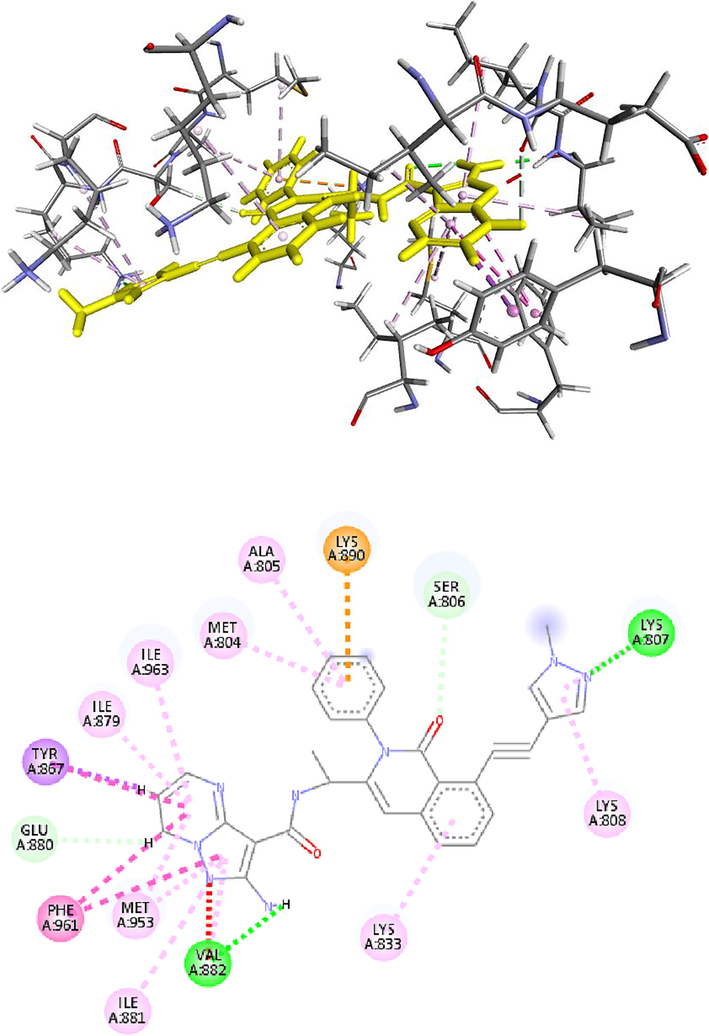
- The 2D and 3D structure of IPI59 docked with the receptor p13k.
The compound were subjected to a QSAR study, by calculating their molecular descriptors, performing a GA-MLR genetic algorithm multiple linear regression study. The result presented in Table3, was used to evaluate the molecular descriptors responsible for the behavior of these compounds at the binding site.
| Friedman LOF | 82.895 |
| R-squared | 0.618 |
| Adjusted R-squared | 0.601 |
| Cross validated R-squared | 0.543 |
| Significant Regression | Yes |
| Significance-of-regression F-value | 36.279 |
| Critical SOR F-value (95%) | 2.325 |
| Replicate points | 0 |
| Computed experimental error | 0 |
| Lack-of-fit points | 112 |
| Min expt. error for non-significant LOF (95%) | 4.076 |
6 Conclusion
The growing interest in developing PI3K inhibitors clearly indicates the central role of PI3Ks in cancer treatment, all the compounds were found to inhibit the receptors by completely occupying the active sites in the target receptors. In the figures, most of the inhibitors were found to involve in both the hydrophobic interactions and hydrogen bonding with the receptors, as strong inhibitor binding is also reflected by the frequency of hydrogen bonds as shown in Table 2. In our docking study, the best overall binding was exhibited in terms of estimated free energy of binding and Ki value by Aminopterin Derivative 3 followed by Methotrexate Derivative and at least by Dolastine. The molecular docking results shown in figures confirmed that the hydrophobic interactions and the hydrogen bonding with these targets have pivotal contributions to the binding structures and binding free energies, although the van der Waals and Pi-interactions contributed to the stabilization of the binding structures. We hope the comprehensive structural understanding, binding modes and the vital factors affecting the binding free energies gotten from the present computational studies will provide valuable insights for future rational structure-based design of novel and potent inhibitors.
Acknowledgement
The authors would like to thank Andrew Orry, for granting us the license for ICM-Pro molsoft program we adopted for this docking study.
References
- Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol.. 1994;235(3):983-1002.
- [Google Scholar]
- Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol. Cell. Proteomics. 2005;4(6):752-761.
- [Google Scholar]
- Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer. 2003;3(1):11-22.
- [Google Scholar]
- History of the Harvard ChemDraw project. Angewandte Chemie International Edition. 2014;53(42):11140-11145.
- [Google Scholar]
- A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion–resistant growth. Cancer Res.. 2009;69(6):2305-2313.
- [Google Scholar]
- Chemistry with Computation: An introduction to SPARTAN. Wavefunction Inc.; 1995.
- Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: from laboratory to patients. Cancer Treat. Rev.. 2017;59:93-101.
- [Google Scholar]
- Design and synthesis of a novel pyrrolidinyl pyrido pyrimidinone derivative as a potent inhibitor of PI3Kα and mTOR. Bioorg. Med. Chem. Lett.. 2012;22(15):5098-5103.
- [Google Scholar]
- Personal experience with four kinds of chemical structure drawing software: review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. J. Chem. Inf. Comput. Sci.. 2004;44(5):1886-1890.
- [Google Scholar]
- Borderline resectable pancreatic cancer: definitions and management. World J. Gastroenterol. WJG. 2014;20(31):10740.
- [Google Scholar]
- PI3K/AKT signaling pathway and cancer: an updated review. Ann. Med.. 2014;46(6):372-383.
- [Google Scholar]
- PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. OncoTargets Therapy. 2016;9:203.
- [Google Scholar]
- mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res.. 2006;66(3):1500-1508.
- [Google Scholar]
- PI3Kδ and PI3Kγ: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat. Rev. Immunol.. 2007;7(3):191-201.
- [Google Scholar]
- PI3Kγ inhibition: towards an'aspirin of the 21st century'? Nat. Rev. Drug Discovery. 2006;5(11):903-918.
- [Google Scholar]
- Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407(6803):538-541.
- [Google Scholar]
- PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol.. 2015;23:82-91.
- [Google Scholar]
- Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood. 2002;99(6):1928-1937.
- [Google Scholar]
- Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387(6634):673-676.
- [Google Scholar]
- Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Comput. Sci.. 1989;29(3):163-172.
- [Google Scholar]
- Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev.. 2010;20(1):87-90.
- [Google Scholar]
- Curcumin induces apoptosis in human non-small cell lung cancer NCI-H460 cells through ER stress and caspase cascade-and mitochondria-dependent pathways. Anticancer Res.. 2010;30(6):2125-2133.
- [Google Scholar]




