

Molecular characterization and evolutionary history analysis of a novel Kudzu mosaic virus (KMV) infecting the Kudzu (Pueraria montana)
⁎Corresponding authors. muhammadarif@subu.edu.tr (Muhammad Arif), aczhou@zafu.edu.cn (Aicun Zhou)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Objectives
Plant viruses are a significant contributor to the economic losses experienced in numerous plant species. Begomoviruses exhibit a wide range of host species, encompassing both cultivated and non-cultivated plants. The kudzu plant, scientifically known as Pueraria montana, is indigenous to the Southeast Asian region. Moreover, it exhibits varied attributes with regards to its utilization in the fields of medicine, food, cosmetics, and the herbal industry.
Methods
During a routine survey, some P. montana plants having typical symptoms of yellowing, mosaic, and mottle were observed in areas including Fort-Minro (FM) and Anari of district Dera Ghazi Khan and Maari (MA) of district Rajanpur. The genomic DNA was extracted from the collected samples. The polymerase chain reaction (PCR) and rolling circular amplification (RCA) was carried out to isolate the DNA and DNA- β from the collected plant specimens. An evolutionary research was conducted to assess the lineage of this virus inside a novel geographical region.
Results
A 500-bp amplicon size was obtained from the samples of the FM and MA, while the approximately 2.7-kb amplicon size of its full genome was obtained from the samples of both locations. The results of the sequence analysis demonstrated conclusively that the plants in question are infected with Kudzu mosaic virus (KMV). The evolutionary history analysis has confirmed the virus's lineage in a new region, and the nucleotide frequencies of obtained sequences were variable.
Conclusion
The presence of a novel bipartite begomovirus in the tropical region has been substantiated using a phylogenetic and evolutionary investigation. The observation of a white fly population on infected plants has also suggested that the transmission of this virus through the migration of this insect vector can be a viable mechanism.
Keywords
Kudzu
Begomoviruses
Phylogenetic analysis
Nucleotide comparison
MEGA11

1 Introduction
Kudzu (Pueraria montana, family; Fabaceae.) is a common weed and is usually found all over the world, especially in east Asian countries including China, Japan, and Korea (Harrington et al., 2003, Jewett et al., 2003). This plant is vulnerable to many pathogens, including fungi, bacteria, nematodes, and viruses. Many abiotic factors are also responsible for reducing the crop quality of this plant. There are 3 types of viruses, which are continuously affecting the quality and quantity of this plant (Zhang and Wu 2013, Igori et al., 2022).
Viruses are directly or indirectly responsible for reducing crop yield and quality all over the world and leading to economic crises. This type of damage leads to extensive molecular and cellular-level research on these plant pathogens and also the interactions of viruses with their plant hosts and vectors. Beyond the damage caused by these viruses, some plant viruses, for example, Tobacco mosaic virus (TMV), is used as a model organism to recognize the viruses’ expression and replication of genes (Caranta 2011, Arif et al., 2022).
Viruses have been identified as a significant factor, both directly and indirectly, in the decline in agricultural yield and quality on a global scale, hence contributing to economic crises. This form of harm necessitates in-depth investigation at the molecular and cellular levels concerning these plant infections, as well as the examination of the relationships between viruses and their plant hosts and vectors. In addition to the detrimental effects induced by these viral agents, certain plant viruses, such as the Tobacco mosaic virus (TMV), serve as model organisms for studying the expression and replication of viral genes (Arif et al., 2020).
There are two ways that mixed infection modifies the onset of symptoms, leading to either positive or negative interactions. Different reactions in plants, the blocking of a particular virus by another, or, whereas, the escalation of symptoms are all impacted by certain circumstances. The host plant may influence how a viral infection manifests in a mixed infection (Liang et al., 2022).
In South Korea, the Kudzu Virus D (KuVD) was discovered infecting Pueraria montana. This virus has a complete genome sequence of almost 7922 nucleotides without poly tail A and also five ORFs, which are mostly encoded from 5 to 3. The viral genome consists of several key components, including a replicase known as ORF1, three triple block gene proteins referred to as TGB1-3, and ORF2-ORF4. Additionally, there is a single CP-like ORF5 present in the virus. This organization of the genome is unique in the species of subfamilies including Quinivirinae and Betaflexiviridae (Rubino et al., 2012, Akram et al., 2022, Igori et al., 2022).
The member of the Betaflexiviridae family belongs to the + ve ssRNA viruses, which are highly responsible for damaging many groups of plants. These viruses have been identified in many herbaceous and woody species of plants. Viruses in this family have a typical RNA genome size of 5.9–9.0 kb. It has non-enveloped, filamentous, and flexuous virions with a size of 600–1000 nm in length and 12–13 nm in diameter and mostly depends on species and genus. This family consist of two sub-families including Quinivirinae (genera: Foveavirus, Robigovirus, Banmivirus, Carlavirus and Sustrivirus) and Triniviridae (genera Capulavirus, Divavirus, Ravavirus, Wamavirus, Citrivirus, Trichovirus, Chordovirus, Prunevirus, Tepovirus, Vitavirus) (Lefkowitz et al., 2018, Siddell et al., 2019). In the groups of Betaflexiviridae family, the CP gene is trailed by single encoding putative nucleic acid binding protein (Adams et al., 2016). The objective of this study was to investigate the exact viral pathogen and to confirm the presence of new begomoviruses and their satellites infecting kudzu plants diversity of Dera Ghazi Khan Division.
2 Material and methods
2.1 Sample collection
In 2020, during a survey in Dera Ghazi Khan to find out the new hosts of begomoviruses and their associated betasatellites, some Kudzu plants showing disease like symptoms were observed in a hilly area of the Koh-Suliman range: Fort Minro (FM), which is approximately 6470 feet above from the sea level. Yellow veins and mosaic symptoms were also observed on infected plants from the location of Anari, having coordinates of 29°55′48″N 69°59′05″En/29.9300°N 69.9848°En/29.9300; 69.9848. Meanwhile, in Maari hill (MA) station of Rajanpur, same type of symptoms was also noticed in the same plant. Infected plants had also shown whitefly infestation. Based on visible observations, it was indicated that the plants infected with Kudzu may be afflicted with begomovirus or another related helper virus. A total of 20 samples exhibiting symptoms and 10 samples from without symptoms were taken from each location.
2.2 DNA extraction from the samples
The genomic plasmid was obtained both from collected symptomatic and healthy leaves by using Cetyl trimethyl ammonium bromide method (CTAB) as described by Biswas et al. (2013). The symptomatic leaves from each sample were pulverized into a fine powder by crushing them in liquid nitrogen with a pre-chilled pestle and mortar. The tissue powder obtained was promptly transferred into a 50-ml autoclaved polypropylene centrifuge tube containing 20 ml of CTAB extraction buffer that had been pre-warmed to 65 °C. The experimental protocol involved the utilization of an extraction buffer comprising the following components: cetyltrimethylammonium bromide (CTAB) at a concentration of 1.5 %, Tris hydrochloride (HCl) at a pH of 8.0 and a concentration of 100 millimolar (mM), sodium chloride (NaCl) at a concentration of 1.4 M (M), ethylenediaminetetraacetic acid (EDTA) at a pH of 8.0 and a concentration of 20 mM, β-mercaptoethanol at a concentration of 2 %, and polyvinylpyrrolidone (PVP) at a concentration of 2 %.
The entire tissue powder was thoroughly suspended in the extraction buffer through a process of rotation and inversion of the mixing tubes. The homogenate mixture was incubated at a temperature of 65 °C for a duration of one hour in the water bath. The aforementioned blend was consistently arranged during the duration of incubation. After the incubation period, a solution consisting of chloroform and isoamyl alcohol at a ratio of 24:1 was added to each tube in a volume of 15 ml. The combination undergoes centrifugation until it undergoes a transformation resulting in the formation of a dark green emulsion. Subsequently, the mixture tubes underwent agitation for a duration of 30 min utilizing a rotary shaker. Following the first shaking, all tubes were subjected to centrifugation at a speed of 4000 revolutions per minute (RPM) for a duration of 20 min using a centrifugable shaking machine. The supernatant mixture was transferred to a new, sterile 50-ml tube. A volume of 2 µl of RNase solution, with a concentration of 10 mg/ml, was introduced into each tube. Subsequently, the tubes were subjected to incubation in a water bath at a temperature of 37 °C for a duration of roughly 1 h. This particular phase was implemented on an optional basis and can alternatively be carried out immediately following the purification procedures. The repetition of chloroform, isoamyl alcohol, and centrifugation protocols was conducted after RNAseq treatment.
2.3 PCR amplification, cloning, and sequencing
Mainly, plant viruses are amplified by applying begomovirus-specific primers (Shahid et al., 2007). which are proficient at amplifying all begomoviruses and betasatellites. A PCR was carried out for amplifying the conserved region of begomoviral DNA-A with the help of universal primers (Table 1) having the sequence AV494 /Dep3-(5ʹ-GANGSATGHGTRCADGCCATATA-3ʹ/5ʹ-GCCYATRTAYAGRAAGCCMAG-3ʹ) (Zi-Fu et al., 2007). The reaction for amplification was added in a volume of 25 µl having template 1 µl, enzyme 0.5 µl, dNTPs 0.5 µl, 2X buffer 12.5 µl, reverse primer 1 µl, forward primer 1 µl, and double distilled water 8.2 µl to amplify the begomoviruses and betasatellites with the help of primer sets. DNA fragment were obtained from infected plant samples but not from healthy samples after the PCR reaction. The PCR products underwent electrophoresis at a voltage gradient of 5 V cm-1, passing through agarose gels with a concentration of 1 % w/v. This process was conducted in electrophoresis buffer consisting of 1x Tris Acetic acid ethylenediaminetetraacetic acid (EDTA). An agarose gel was used to elute the amplified DNA, and a purification kit (QuikGene, Qiagen Inc., USA) was utilized. Before performing the ligation reaction, T4 competent cells (Escherichia coli) cells were prepared. The purified PCR product was ligated into the Pgem-t Esay vector (Promega, Madison, WI, USA). The 200 µl from each ligated product was transferred into each Petri plate. All the petri plates were incubated at 37 °C for a period of 16 h (Birnbaum et al., 2005). Selected colonies were double-checked by PCR with the help of M13F and M13R primers, and positive colonies were recovered with the help of a purification kit (QuikGene, Qiagen Inc., USA).
| Host | Location | Virus genome | Sequence of used primers 5ʹ–3ʹ | Total no. of sampleΩ |
|---|---|---|---|---|
| Kudzu (Pueraria montana) |
Fort-Minro, Mari | KuMV DNA A | AV494 /Dep3α | 9 |
| Begomo-F-(5′-GGATCCTTTGTTGAACGCCTTTCC-3′)* | ||||
| Begomo-R-(5′ GGATCCCACATGTTTAAAGTAAAGC-3′)* | ||||
| Kudzu (Pueraria montana) |
Fort-Minro | KuMV DNA- β | β01/β02∞ | 5 |
The PCR products were allowed to be digested with the help of Begomovirus-beta-BamHI-F GGATCCATGACGAGGAGCAAAACAA and Begomovirus-beta-PstI-R CTGCAGAACGGTGAACTTCTTATTGA. The confirmation of all cloned genes was achieved through the restriction release of fragments, and subsequent sequencing of selected clones carried out by the commercial company Beijing Genomics Institute (BGI) located in Shenzhen, China. For double-checking the obtained sequences, sequence-specific primers were designed with the help of Primer 6 software. The sequences that were acquired were combined and subsequently subjected to analysis using Lasergene 10, a software for sequence analysis developed by DNASTAR Inc. located in Madison, WI, USA. All of the acquired sequences were compared to previously discovered begomoviruses and betasatellites sequences that were submitted to GenBank using the Blastn search of the NCBI non-redundant nucleotide database (https://www.ncbi.nlm.nih.gov/BLAST).
2.4 PCR/RCA amplification of DNA β
The RCA is widely used for detecting the betasatellites of begomoviruses (Briddon et al., 2002). A PCR/RCA was conducted to amplify the DNA β of each infected plant sample with the help of universal abutting primer pairs β01/β02. For amplifying the full-length DNA β, primer pairs were designed with the help of Primer 6 software from the obtained partial DNA β sequences of each plant sample. PCR/RCA was performed in a volume of 25 µl solution containing template 1 µl, enzyme 0.5 µl, dNTPs 0.5 µl, 2X buffer 12.5 µl, reverse primer 1 µl, forward primer 1 µl, and double distilled water 8.2 µl. After PCR reaction, all the products were digested with sequence-specific endonucleases using RCA. These restriction enzymes were selected from NEBcutter V2.0 (New England BioLabs, Inc.). The digestion reaction was held in a volume 20 µl comprising of 10X green enhancer 2 µl, selected enzyme one 1 µl, selected enzyme two 1 µl, and 16 µl plasmid with the optional addition of ø29 polymerase DNA and random hexamers in 50 % glycerol by using the Templiphi DNA amplification Kit (Dean et al., 2001). The RCA products of putative begomoviral DNA β were eluted from an agarose gel with the help of a purification kit (QuikGene, Qiagen Inc., USA). All obtained products were cloned and sent to the commercial company Beijing Genomics Institute (BGI, Shenzhen, China) for Sanger sequencing. The sequences that were acquired were compiled and examined using Lasergene 10 sequence analysis software, developed by DNASTAR Inc. in Madison, WI, USA. A Blastn search was conducted on the NCBI non-redundant nucleotide database to compare the acquired sequences with previously discovered betasatellite sequences in GenBank (https://www.ncbi.nlm.nih.gov/BLAST).
2.5 Phylogenetic analysis
In order to get insight into the lineage of begomoviral species and their satellites, the relevant sequences were retrieved from the non-redundant database of the National Center for Biotechnology Information (NCBI). A phylogenetic tree was constructed for the virus by utilizing aligned viral genomes. The neighbor joining method was utilized in order to conclude this phylogenetic tree. From the 1,000 replicates obtained, the bootstrap consensus tree was inferred to represent the transformational history of the taxa under analysis (Felsenstein 1985, El-Adawy et al., 2023). The utilization of this transformational analysis proved to be beneficial in the application of MEGA7 software (Kumar et al., 2016). The nucleotide comparison was conducted using the methodology outlined in the study by (Kumar et al., 2016) and implemented in MEGA11 software. The frequencies of all the acquired sequences were compared based on their percentage values (Tables 2 and 3).
| Virus/Isolate name | T(U) | C | A | G | Total(bp*) |
|---|---|---|---|---|---|
| Kudzu mosaic virus Yg3 isolate | 33.5 | 18.6 | 25.1 | 22.8 | 2677 bp* |
| Kudzu mosaic virus China isolate | 29.7 | 21.6 | 25.4 | 23.3 | 2729 bp* |
| Kudzu mosaic virus Vietnam isolate | 29.9 | 21.7 | 25.7 | 23.1 | 2731 bp* |
| Kudzu mosaic virus isolate CQ isolate | 33.2 | 18.8 | 25.1 | 23.0 | 2666 bp* |
| Kudzu mosaic virus isolate CQ01‘ | 29.4 | 21.9 | 25.6 | 23.1 | 2731 bp* |
| Kudzu mosaic virus isolate Fujian | 32.9 | 19.4 | 25.3 | 22.3 | 2669 bp* |
| Kudzu mosaic virus isolate YG | 29.6 | 21.9 | 25.4 | 23.2 | 2730 bp* |
| Kudzu mosaic virus FM isolate | 33.1 | 19.5 | 25.7 | 21.7 | 2672 bp* |
| Kudzu mosaic virus MA isolate | 29.6 | 21.9 | 25.2 | 23.3 | 2731 bp* |
| Average | 31.3 | 20.5 | 25.4 | 22.8 | 2701 bp* |
Nucleotide comparison of our sequences which are in bold form, with other sequences retrieved from the NCBI database. All this nucleotide comparison was obtained from MEGA11. All frequencies are given in a percent. These are the nucleotide frequencies of the Kudzu mosaic virus.
| Virus/Isolate name | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Kudzu mosaic virus Yg3 isolate | 0.0504 | 0.0086 | 0.0514 | 0.0128 | 0.0514 | 0.0101 | 0.0537 | 0.0083 | 0.0498 | |
| Kudzu mosaic virus China isolate | 1.2581 | ID | 0.0556 | 0.0054 | 0.0490 | 0.0048 | 0.0552 | 0.0055 | 0.0527 | 0.0053 |
| Kudzu mosaic virus Vietnam isolate | 0.1966 | 1.2476 | ID | 0.0532 | 0.0125 | 0.0534 | 0.0106 | 0.0565 | 0.0046 | 0.0519 |
| Kudzu mosaic virus isolate CQ isolate | 1.2559 | 0.0791 | 1.2085 | ID | 0.0488 | 0.0040 | 0.0556 | 0.0054 | 0.0504 | 0.0028 |
| Kudzu mosaic virus isolate CQ01‘ | 0.1908 | 1.0885 | 0.1885 | 1.0449 | ID | 0.0467 | 0.0126 | 0.0481 | 0.0127 | 0.0470 |
| Kudzu mosaic virus isolate Fujian | 1.2671 | 0.0753 | 1.224 | 0.0453 | 1.0680 | ID | 0.0559 | 0.0046 | 0.0512 | 0.0036 |
| Kudzu mosaic virus isolate YG | 0.1920 | 1.2712 | 0.2140 | 1.2246 | 0.2012 | 1.2341 | ID | 0.0579 | 0.0102 | 0.0535 |
| Kudzu mosaic virus FM isolate* | 1.2855 | 0.0946 | 1.2517 | 0.0724 | 1.0753 | 0.0695 | 1.2694 | ID | 0.0538 | 0.0048 |
| Kudzu mosaic virus MA isolate* | 0.2089 | 1.2327 | 0.0464 | 1.1972 | 0.1936 | 1.2137 | 0.2250 | 1.2441 | ID | 0.00494 |
| Kudzu mosaic virus Hanoi isolate | 1.2543 | 0.0754 | 1.2071 | 0.0189 | 1.0415 | 0.0395 | 1.2260 | 0.0712 | 1.1958 | ID |
3 Results
This study was carried out to determine the existence of KDV infecting kudzu plants, which were collected from different areas of Dera Ghazi Khan division. This area has versatile vegetation in terms of flora and fauna. It was observed during a routine survey that some plants have mosaic and mottle-like symptoms, which clearly indicate the infection of any plant virus, like begomovirus, as shown in Fig. 1. The presence of a white fly population also confirmed the existence of a new plant virus. There was a huge difference in the phenology of healthy and symptomatic plants. Healthy plant leaves were free from the typical symptoms of plant virus and no sign of a white fly population was available.

- Symptoms exhibited by Kudzu (Pueraria montana) from which KuMV was detected and PCR detection of KuMV with M, DL 2000 Maker (Takara). The 1-kb ladder was used during gel electrophoresis to check the actual band size of amplicons.
A total of twenty samples were collected from the spot for further analysis. These twenty samples include both symptomatic and asymptomatic samples. These twenty samples were stored at −20 °C. DNA was obtained both from healthy and infected samples by the mean of CTAB method as described in the methodology section. A PCR reaction was carried out to check the presence of begomovirus infection in symptomatic plants. The samples containing a higher concentration of DNA were selected for further PCR reactions. The samples collected from the Fort Minro area have a higher concentration of DNA. The samples from the Fort Minro and Mari areas were selected for further analysis because they had higher concentrations of DNA.
A PCR reaction was carried out to check the presence of begomovirus in the samples from Fort Minro and Mari. The pairs of degenerate primers AV494/Dep3 were selected for the amplication of the conserved region of begomoviral DNA-A. After PCR, all the samples were loaded on 1 % gel-electrophoresis to obtain the size of amplicons. A 500-bp amplicon size was obtained from the symptomatic samples of the Fort Minro and Mari areas as shown in Fig. 1. All these samples were further tested by PCR reaction to re-check the authenticity of the amplicon size. The amplified products were eluted with the help of a purification kit (QuikGene, Qiagen Inc., USA). All the eluted products were sent to commercial company for proper sequencing Beijing Genomics Institute (BGI), Shenzhen, China.
The obtained sequencing results revealed that the amplicon clone possessed a size of 500 base pairs, which closely resembled the previously submitted sequences of KuMV in the NCBI database.
The obtained sequences have confirmed the presence of a 500-bp amplicon. A Blastn search of the NCBI non-redundant nucleotide database was applied for comparing all the obtained sequences with already identified begomoviruses and betasatellites sequences submitted to GenBank (http://www.ncbi.nlm.nih.gov/BLAST).
To obtain the full-length genome sequence of DNA β, abutting primer pairs were selected. The RCA reaction was carried out for this purpose as described in the material and methods. RCA yielded the expected size of products. RCA products from both locations were allowed for digestion with SphI endonuclease and yielded almost 2.7-kb with more or less than 2.8 kb fragments from both locations. The Fort-Minro products were selected for sequencing. The Fort-Minro sample was comprised of 2672 nucleotides with a 1–2 nucleotide difference, whereas the sequence obtained from the Maari sample consisted of 2731 nucleotides. The obtained sequence was blasted at NCBI to check the nucleotide sequence identity. It shared 78 % nucleotide sequence identity with the KuMV DNA B of Vietnam isolate (GenBank accession number DQ641691) and shared 76 % with the Mungbean yellow mosaic virus (MYMV) with GenBank accession number AJ132574. It was revealed from sequence analysis that the putative stem sequences of both KuMV DNA A and KuMV DNA B were different.
The overall mean distance of this virus with other related databases was also obtained from MEGA11. During analysis, it was confirmed that the mean distance of this virus is 0.72, while the standard error is 0.03.
To understand the origin of this virus and its associated satellites, related sequences were assembled from NCBI data base. A phylogenetic tree was constructed both for this virus. The tree was inferred with the help of the neighbor-joining method. The evolutionary analysis was carried out in MEGA11 software. It was observed that our sequence of this virus has followed the criteria of ICTV for a new virus. By following the ICTV standards for the new begomovirus nucleotide sequence identity limit, which is <89 %, the Fort-Minro isolate was distinguished as a new isolate of bipartite KuMV infecting Pueraria montana in Fort-Minro, Dera Ghazi Khan. There is no other report of this virus in Pakistan, but this virus has already been identified in Vietnam, China, and Thailand. It is suggested that this virus might transfer with long-distance migration of whitefly from one place to another.
The maximum-likelihood composite was carried out in MEGA11. We have retrieved the suggested sequences of KDV from NCBI and compared all the obtained nucleotide sequences with our sequences. Kudzu mosaic FM isolate has the highest similarity index with other submitted databases in NCBI, while Kudzu mosaic virus MA isolate has almost less similarity with other submitted databases (Fig. 2).
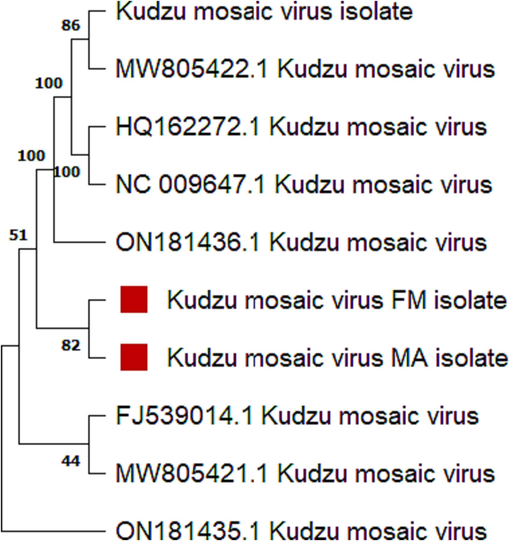
- Phylogenetic tree representing the evolutionary information of the pedigree of this virus. MA isolate and FM isolate represent to our sequences. Kudzu mosaic virus Fort-Minro isolate and Kudzu mosaic Maari isolate were the obtained sequences, which were compared with all other related sequences obtained from the database of NCBI.
4 Discussion
A routine survey to check the diversity of begomoviruses and their betasatellites was held in the Fort-Minro and Mari areas of Dera Ghazi Khan. These areas have versatile natural flora and fauna in terms of cultivated and non-cultivated plants. A virus-like symptom, including vein clearing, mosaic, mottle yellowing, and growth stunting, was observed in some kudzu plants. The population of white fly was clearly visible on these plants, which boosted us to collect and characterize these symptoms. Crops and weeds are a major source of begomoviruses, providing virus inoculum from which these viruses infect other major crops. Frequent incidences of mixed infection in weeds and other plants proceed to the epidemic of a new type of virus. It is now understood fact that recombination imparts genetic modification to begomoviruses in the universe (Lefeuvre et al., 2007, Moriones et al., 2007, Moriones and Navas-Castillo 2008) and recombination has been described to take place in many begomovirus-infecting plants (Kirthi et al., 2002, Rojas et al., 2005, Davino et al., 2012). Classification of begomoviruses depends on genome organization; monopartite have only one ssDNA, while bipartite have two ssDNA consisting of DNA A and DNA-β (Fauquet and Stanley 2005). The classification of begomoviruses is dependent on certain standards sanctioned by ICTV, which comprise: a distinctive begomovirus full-length nucleotide sequence identity that should be more or equal to 89 % and ORFs arrangement (Fauquet et al., 2008).
Begomoviruses originating from various geographical regions have the capacity to interact with non-cognate betasatellites and alphasatellites, resulting in varying degrees of deficiencies. The occurrence of satellite exchange among begomoviruses is quite plausible. Similar to the cases of CoYV and CoGMV, which were initially detected in Vietnam and China respectively, these viral strains have now spread globally, leading to the emergence of serious diseases worldwide. The cotton leaf curl virus (CLCV) has been identified as a significant impediment in the agricultural sectors of India and Pakistan. Furthermore, its prevalence has extended beyond these regions, affecting a multitude of malvaceous plant species in various countries worldwide (Barreto et al., 2013).
The introduction of new crops is recognized as a contributing factor to the emergence of novel epidemics of geminiviruses in a given region (Nawaz-ul-Rehman and Fauquet 2009). Wild plants serve as reservoirs for the generation of novel viruses and their variants through their function as “melting pots” for the exchange and recombination of genetic material with begomoviruses (Azhar et al., 2010, Mubin et al., 2010). The outcome of the interaction between begomoviruses and host plants is contingent upon the virus's capacity to effectively counteract the plant's defense mechanisms (Moissiard and Voinnet 2004, Vanitharani et al., 2004).
The International Committee on Taxonomy of Viruses (ICTV) has established a complete nomenclature for organizing and mentioning the newly originated plant viruses (Zerbini et al., 2017). According to this criteria, if the complete genome sequence distinctiveness of a new plant virus like begomovirus is less than 89 % via the Clutrral V algorithm, then this plant virus can be added as a new species but if the sequence similarity is more than 89 %, then this can lead to a member of the same species. It was confirmed from the sequence similarity that our species is a new plant virus in this area because its similarity index was less than 89 %. This kudzu mosaic virus has confirmed the presence of a new strain of begomovirus in this tropical area.
5 Conclusion
In this study, we have characterized the KuMV-infecting plants from the areas of Fort-Minro and Mari in Dera Ghazi Khan division. This plant is considered invasive, and this virus can be transferred to other natural flora and fauna in this natural area. This virus is native to Asia and has been detected in many other tropical and sub-tropical countries. There is no other report yet available for the presence of this virus in this area and also in surrounding areas. It is assumed that the migration of white fly can be the successful cause of virus in this area.
Studies in humans and animals
This work does not involve the study of humans and animals.
Funding
The authors extend their appreciation to the Researchers supporting project number (RSP2023R173), King Saud University, Riyadh, Saudi Arabia.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016) Arch. Virol. 2016;161:2921-2949.
- [Google Scholar]
- Species diversity, phylogeny and evidence of recombination in begomoviruses associated with yellow mosaic disease of moth bean (Vigna aconitifolia) in South India. J. Phytopathol.. 2022;170(5):300-314.
- [Google Scholar]
- Molecular characterization and RSV Co-infection of Nicotiana benthamiana with three distinct begomoviruses. Methods. 2020;183:43-49.
- [Google Scholar]
- Molecular study of geminiviruses with its complex biology, host-vector interactions, and increasing diversity. J. King Saud Univ.-Sci. 2022:102051.
- [Google Scholar]
- Both malvaceous and non-malvaceous betasatellites are associated with two wild cotton species grown under field conditions in Pakistan. Virus Genes. 2010;41(3):417-424.
- [Google Scholar]
- A study of weeds as potential inoculum sources for a tomato-infecting begomovirus in central Brazil. Phytopathology. 2013;103(5):436-444.
- [Google Scholar]
- Cell type–specific expression profiling in plants via cell sorting of protoplasts from fluorescent reporter lines. Nat. Methods. 2005;2(8):615-619.
- [Google Scholar]
- A simple method of DNA isolation from jute (Corchorus olitorius) seed suitable for PCR-based detection of the pathogen Macrophomina phaseolina (Tassi) Goid. Lett. Appl. Microbiol.. 2013;56(2):105-110.
- [Google Scholar]
- Universal primers for the PCR-mediated amplification of DNA β. Mol. Biotechnol.. 2002;20(3):315-318.
- [Google Scholar]
- Recent Advances in Plant Virology. Horizon Scientific Press; 2011.
- Recombination profiles between Tomato yellow leaf curl virus and Tomato yellow leaf curl Sardinia virus in laboratory and field conditions: evolutionary and taxonomic implications. J. Gen. Virol.. 2012;93(12):2712-2717.
- [Google Scholar]
- Rapid amplification of plasmid and phage DNA using phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res.. 2001;11(6):1095-1099.
- [Google Scholar]
- Development of silver nano-based indirect ELISA and Dot-ELISA methods for serological diagnosis of a bacterial fish pathogen Aeromonas veronii. J. Microbiol. Methods. 2023;211:106782
- [Google Scholar]
- Revising the way we conceive and name viruses below the species level: a review of geminivirus taxonomy calls for new standardized isolate descriptors. Arch. Virol. 2005;150(10):2151-2179.
- [Google Scholar]
- Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783-791.
- [Google Scholar]
- Kudzu (Pueraria montana) community responses to herbicides, burning, and high-density loblolly pine. Weed Sci.. 2003;51(6):965-974.
- [Google Scholar]
- Identification and molecular characterization of a novel kudzu-infecting virus of the family Betaflexiviridae. Arch. Virol. 2022;167(8):1707-1711.
- [Google Scholar]
- Characterizing specimens of kudzu and related taxa with RAPD's. Castanea 2003:254-260.
- [Google Scholar]
- Evidence for recombination among the tomato leaf curl virus strains/species from Bangalore, India. Archiv. Virol.. 2002;147(2):255-272.
- [Google Scholar]
- MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol.. 2016;33(7):1870-1874.
- [Google Scholar]
- Avoidance of protein fold disruption in natural virus recombinants. PLoS Pathog.. 2007;3(11):e181.
- [Google Scholar]
- Virus taxonomy: the database of the International Committee on Taxonomy of Viruses (ICTV) Nucleic Acids Res.. 2018;46(D1):D708-D717.
- [Google Scholar]
- Mixed infection of an emaravirus, a crinivirus, and a begomovirus in Pueraria lobata (Willd) Ohwi. Front. Microbiol.. 2022;13:926724
- [Google Scholar]
- Recombination in the TYLCV complex: a mechanism to increase genetic diversity. Implications for plant resistance development. In: Tomato Yellow Leaf Curl Virus Disease. Springer; 2007. p. :119-138.
- [Google Scholar]
- Rapid evolution of the population of begomoviruses associated with the tomato yellow leaf curl disease after invasion of a new ecological niche: a review. Span. J. Agric. Res.. 2008;6(S1):147-159.
- [Google Scholar]
- Characterization of begomovirus components from a weed suggests that begomoviruses may associate with multiple distinct DNA satellites. Virus Genes. 2010;40(3):452-457.
- [Google Scholar]
- Evolution of geminiviruses and their satellites. FEBS Lett.. 2009;583(12):1825-1832.
- [Google Scholar]
- Sequence characterization of tomato leaf curl Sinaloa virus and tomato severe leaf curl virus: phylogeny of New World begomoviruses and detection of recombination. Arch. Virol.. 2005;150(7):1281-1299.
- [Google Scholar]
- Tepovirus, a novel genus in the family Betaflexiviridae. Arch. Virol. 2012;157:1629-1633.
- [Google Scholar]
- Complete nucleotide sequences of cotton leaf curl Rajasthan virus and its associated DNA β molecule infecting tomato. Arch. Virol. 2007;152(11):2131-2134.
- [Google Scholar]
- Additional changes to taxonomy ratified in a special vote by the International Committee on Taxonomy of Viruses (October 2018) Arch. Virol. 2019;164(3):943-946.
- [Google Scholar]
- Differential roles of AC2 and AC4 of cassava geminiviruses in mediating synergism and suppression of posttranscriptional gene silencing. J. Virol.. 2004;78(17):9487-9498.
- [Google Scholar]
- First Report of Kudzu mosaic virus on Pueraria montana (Kudzu) in China. Plant Dis.. 2013;97(1):148.
- [Google Scholar]
- Tomato yellow leaf curl disease in Guangdong is caused by Tomato leaf curl Taiwan virus. Chinese J. Agric. Biotechnol.. 2007;4(2):127-131.
- [Google Scholar]




