

Translate this page into:
In silico screening and validation of KPHS_00890 protein of Klebsiella pneumoniae proteome: An application to bacterial resistance and pathogenesis
⁎Corresponding authors. chandans@jssuni.edu.in (Chandan Shivamallu), assyed@ksu.edu.sa (Asad Syed), shivachemist@gmail.com (Shiva Prasad Kollur)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
Introduction
Nosocomial infections are a notorious subset of infectious diseases, varying between 10% and 20% prevalence worldwide. The infections are concomitant with various treatment complications, multiple-drug resistance, and a high degree of virulence. Klebsiella pneumoniae is a gram-negative bacteria of nosocomial importance.
Objectives
Our current study is gauged to reason and understand why, despite treatment with cutting-edge medicines and technology, the K. pneumoniae remains elusive.
Methods
Using various in silico tools, the KPHS_00890 hypothetical protein of K. pneumoniae subsp. pneumoniae HS11286 was identified and annotated.
Results
A thorough investigation revealed that KPHS_00890 hypothetical protein is a bifunctional 5′-nucleotidase, an enzyme catalyzing the degradation of nucleotides to nucleosides.
Conclusions
Scrutiny and review of the 5′-nucleotidase function across various species ascertained its pertinent role in immune evasion, by suppressing inflammatory responses. Thus, having identified the KPHS_00890 hypothetical protein of K. pneumoniae subsp. pneumoniae HS11286 as a 5′-nucleotidase, we propose that it may be involved in an immune evasion strategy during infection pathogenesis.
Keywords
Molecular docking
Homology modeling
Phylogenetic tree
5′-nucleotidase
Klebsiella pneumoniae
Multiple sequence alignment

1 Introduction
K. pneumoniae is a commensal, Gram-negative Bacillus, encapsulated, and non-motile enteric bacteria. K. pneumoniae can cause a myriad of healthcare-associated infections including pneumonia, bacteremia, meningitis, liver abscess, sepsis, wound or surgical site infections, and urinary tract infections(Martin and Bachman, 2018; Starzyk-Luszcz et al., 2017). Gaining entry into the body is the most pivotal juncture in the K. pneumoniae life-cycle, determining its commensal or pathogenic biological activity. K. pneumoniae accounts for 8% of all the nosocomial infection, wherein pneumonia is the most occurring in the majority of cases. A K. pneumoniae infection occurs only when it can evade and overcome the primary and secondary immune responses of the host, and this is a particularly tangible possibility in hospital settings. The spread of infection happens primarily via person-to-person contact, and although less likely, either through open wounds, unattended cuts, and bruises, or contamination of the environment(Caneiras et al., 2019; Patro and Rathinavelan, 2019; Starzyk-Luszcz et al., 2017; Vading et al., 2018). Upon gaining entry, the K. pneumoniae arsenal against immune response includes: the capsule components (MagA, K2A, RmpA), endotoxins, lipopolysaccharide, siderophores (entB, Ybts), and efflux pumps, to name a few. Each one of these methods of virulence is highly specific and resolute in its pathogenic action. Although K. pneumoniae is a commensal, its strategically guided lifecycle can trump the host system with the slightest of indiscretions(Khaertynov et al., 2018; Remya et al., 2019). It is imperative to fill in the lacunae on the organism’s immune evasion tactics using a quick and efficient approach to assess the microorganism, to better the available medicines, and reduce the prevalence of nosocomial infections. Therefore, this study utilizes the in silico tools for screening and validation of K. pneumoniae functional proteome to decipher novel factors of virulence, antibiotic resistance, and immune evasion. The hypothetical protein KPHS_00890 was screened, annotated, and validated as 5′-nucleotidase (E.C. 3.1.3.5) as a result of this study.
2 Materials and methods
2.1 Strain selection
GenBank database (https://www.ncbi.nlm.nih.gov/genome/browse/#!/prokaryotes/815/) was used to analyse various strains of K. pneumoniae. Strains with completely annotated concerning whole-genome assembly were primarily shortlisted. Among the shortlisted strains only those considered were virulent, drug-resistant, and isolated through clinical samples were considered.
2.2 Proteome screening
The proteome sequence data of the selected K. pneumoniae strain was retrieved from the NCBI database. The proteome was further screened for proteins from the sense strand (+strand). For further studies, the hypothetical protein was selected if it had more than 500 amino acids in its sequence. A hypothetical protein is a protein whose sequence is predicted based on the open-reading frame and deciphered through the whole-genome sequencing of the organism. But the structure and function of such a protein are undeciphered. FASTA sequences of all the proteins matching the above criteria were procured.
2.3 Sequence similarity search
The FASTA sequence of the protein was used to screen the database with identical protein sequences, across the proteome of the organisms in a non-redundant fashion. BLOSUM 62 matrix was used for the scoring parameter with gap costs comprising the existence of 11, an extension of 1.
2.4 Multiple sequence alignment
Sequences selected were identical to the hypothetical protein, and thus, multiple sequence alignment was carried out to analyze the conserved regions across all the sequences. Two independent tools, multiple sequence alignment of NCBI, and MEG X software were used(Kumar et al., 2018). Multiple sequence alignment in NCBI was performed using COBALT, to find the conserved domain and local sequence similarity information among the protein sequences with gap penalties; these were comprised of −11 and gap extension penalty of -l, end gap opening penalty of −5, and end gap extension penalty of -l. (Papadopoulos and Agarwala, 2007). K. pneumoniae hypothetical protein, KPHS_00890 is obtained through BLAST, which was further subjected to multiple sequence alignment using the Clustal W tool of MEGA X software.
2.5 Phylogenetic analysis
The phylogenetic tree was generated through a fast minimum evolution method wherein the maximum sequence difference was set to 0.85. Distance approximation was calculated using the Grishin model, in COBALT. The multiple aligned sequences were subject to phylogenetic analysis through the Maximum likelihood method using MEGA X software.
2.6 Structural elucidation of protein
2.6.1 Homology modelling
The FASTA sequence of the protein was used to predict the structure of the protein using the SWISS-MODEL workspace(Bertoni et al., 2017; Bienert et al., 2017; Guex et al., 2009). ProMod 3 was used as a modelling engine with advanced QMEAND for analysis of QMEAN. The template search for the input sequence was carried out using the Swiss model template library upon a combination of BLAST and HHblits. Global Model Quality Estimate (GMQE) and Quaternary Structure Quality Estimate (QSQE). The top-ranked templates were further used to build the model.
2.6.2 Structural validation
The generated protein structure models were further characterized based on their structural validity. Ramachandran plot of each model was generated using the Rampage tool to check the accuracy of the generated models(Lovell et al., 2003). The model with the highest number of residues in favorable and allowed regions was considered for the downstream analysis. The quality of the protein structures was checked based on their z-score using the ProSA server(Wiederstein and Sippl, 2007).
2.6.3 Protein structure refinement
The selected model was subject to structural refinement to correct the structural errors in the residues found in the disallowed region of the Ramachandran plot. The structural refinement of the selected protein was carried out using the GalaxyRefine tool(Heo et al., 2013).
2.6.4 Validation of refined structure
The rampage tool was used to predict the Ramachandran plot of the previously predicted structure. The percentage of residues in the favorable, allowed, and disallowed regions of the Ramachandran plot were assessed to check if the residues that had disallowed geometry were corrected(Lovell et al., 2003). The quality of the refined protein structure was checked based on the z-score using the ProSA server(Wiederstein and Sippl, 2007).
2.7 Structural comparison
Structural comparison between the refined structure and structure of 3IVD protein was performed using the UCSF chimera tool(Heo et al., 2014). The RMSD between the two structures was calculated after structural alignment, through UCSF chimera(Heo et al., 2014).
2.8 Binding site prediction
Binding site prediction was performed to analyse the probable ligand binding sites in the refined protein structure. GalaxySite tool was used to predict ligand-binding sites in the protein through HH Search(Heo et al., 2014).
2.9 Molecular Docking studies
Molecular interaction studies of the proposed ligands were carried out to check the validity of the proposed function of the protein. The binding affinity and interactions in the binding sites were considered as final functional annotations for the structurally annotated protein. The binding interaction and affinity of the ligands were next compared with those of proteins from another genus in the database that was structural and functional identity. The grid box was set based on the binding site residues, wherein all the binding site residues fit inside the grid box. The molecular interactions in the complexes were analysed in detail using the UCSF chimera tool(Pettersen et al., 2004) and LigPlot+ tool(Laskowski and Swindells, 2011).
2.9.1 Ligand preparation
The 2D structures of the ligand and the inhibitor were sketched using the ACS chem sketch tool. The 2D structures were cleaned for proper geometrical alignment. Subsequently, the 3D structure of the ligands was generated by the addition of the respective 3D coordinates. The geometry of the 3D structure thus generated was cleaned using the Argus lab tool. The refined 3D structure of the ligands was used for molecular docking studies.
The crystal structure of the protein procured from RCSB PDB was further refined by removing water residues, followed by the addition of Gasteiger charges, and was further refined by merging nonpolar hydrogens using AutoDock V.4.0(Morris et al., 2010; Shin et al., 2011).
2.10 Molecular dynamics simulation studies
The MD simulation was carried out for both protein and protein–ligand complexes using GROMACS, forced with CHARMM27 all-atom force field. The parameter files for the ligand were generated using SwissParam online server. TIP3P water model was used to solvate the system and NaCl counter ions were added to neutralize the system. The equilibration was run for 20 (ns) at 300 K temperature and 1 bar pressure.
3 Results and discussion
3.1 Choice of strain
Among 437 completely sequenced K. pneumoniae genome assemblies and annotations, only 54 genome assemblies were found to be classified up to subspecies level as pneumoniae. Out of 54 complete genome assemblies (Supplementary Table 1), 16 were found to be clinical isolates with drug resistance (Supplementary Table 2). Among the 16 genome assemblies, only the strain HS11286, which is also a reference strain showed multi-drug resistance. Additionally, the rest of the 15 genome assemblies were found to have more than 80% identity with this reference strain. Hence, HS11286 was considered for further studies, as the other 15 genome assemblies were neither type strains nor reference strains.
3.2 Proteome assessment
The proteome of K. pneumoniae HS11286 has 5316 proteins in its proteome. The proteome consisted of 1,694 hypothetical proteins. Among them, 65 hypothetical proteins in the proteome had more than 500 amino acid residues in their sequence. Among the 65 hypothetical proteins (Supplementary Table 3), 30 were coded by sense stand. Hence, all 30 hypothetical proteins were considered for further studies (Supplementary Table 4).
3.3 Sequence analogy
Upon sequence similarity search, 29 hypothetical proteins were found to have corresponding identical proteins of known function. Hence, the function of all the 29 hypothetical proteins was consequently established (Supplementary Table 5). Based on the establishment of these functional annotations, the role of these proteins in the bacteria was analyzed. Among the 29 proteins, the hypothetical protein KPHS_00890 was found to have a bifunctional 5′- nucleotidase function. Due to antibiotic overuse, and misuse, and its persistent latent activity, drug resistance is a primary concern in healthcare. Hence, it can be suggested that 5′-nucleotidase of K. pneumoniae proteome has virulent functions particularly involved in evasion of host immune response, and is a probable contributing factor in the rise of multi-drug resistance of K. pneumoniae.
3.4 Sequence congruence
MSA with identical sequences from different genera revealed that 77.41 percentage of the sequence was conserved. MSA with identical sequences from the Klebsiella genus revealed that 84.74 percentage of the sequence was conserved. The results of both COBALT and MEGA X-Clustal W were in agreement. A high percentage of the conserved sequence indicates that the number of mutations in the gene expressing the 5′-nucleotidase protein is extremely low, or implies the presence of silent mutations, thus, not affecting the protein product. The conserved regions also clearly signify that 5′-nucleotidase is vital for bacterial survival and pathogenesis. (Supplementary Fig. 1)
3.5 Evolutionary analysis
Upon phylogenetic analysis of MSA with the Klebsiella genus, the phylogenetic tree with the highest log-likelihood of −2436.71 was obtained (Supplementary Fig. 2). Phylogenetic analysis of MSA with multiple genera resulted in the phylogenetic tree with the highest log-likelihood of −3276.55 (Supplementary Fig. 3). The phylogenetic tree revealed a high degree of relatedness among the sequences, with slight variation, and showed a high degree of evolutionary relatedness.
3.6 Structural annotation
3.6.1 Three dimensional structure of protein
The sequence of KPHS_ 00,890 protein had a sequence identity of 88.35% with proteins PDB ID: 3IVD. Hence, using this protein structure as a template, a model of the prospective protein was built. The model of predicted protein structures had 96.8% of residues in the favored region 2.2% of the residues were found in the Allowed region of the Ramachandran plot, respectively (Fig. 1a and 1b). The structure of model 1, upon refinement with GalaxyWEB, was found to have 98.6% residues in the favorable region, 0.8% of the residues in the allowed region, and 0.6% of the residues in the outlier region (Fig. 1c and 1d).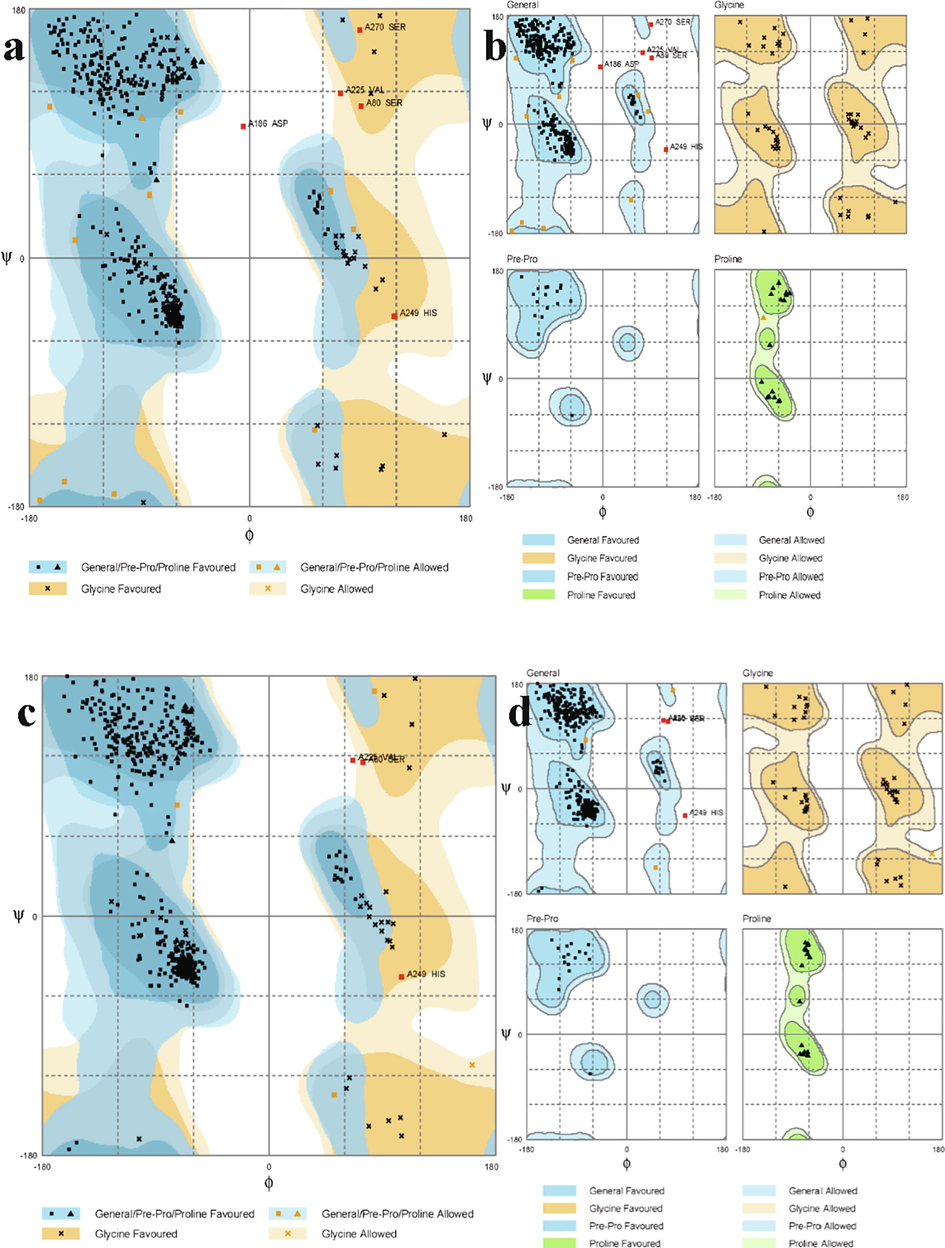
Ramachandran plot exhibiting residues of the model a and b: before refinement and c and d: after refinement.
The overall model quality was predicted through ProSA; it exhibited a Z-score of −10.17, for the model before refinement (Fig. 3a). Improvement in the Z-score of protein structure was observed upon refinement. The Z-score of the refined protein structure was −10.59 (Fig. 2b).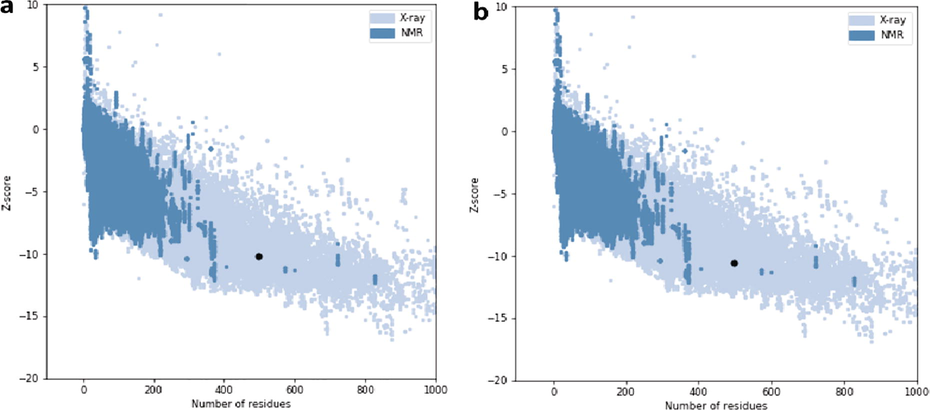
Plot exhibiting z-score of the protein obtained through the ProSA server. a; z-score of the model before refinement. b; z-score of the model after refinement.
3.7 Structural similarity
Structural comparison of the refined protein structure with the structure of 3IVD protein from the RCSB PDB database exhibited a high degree of structural convergence, having an RMSD value of 0.272 Å. The structural alignment was visible upon matching structure through chimera (Fig. 3).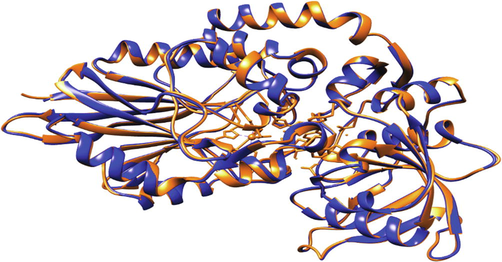
Structure depicting 5′-nucleotidase (golden ribbon) aligned with 3IVD (cyan ribbon) protein structure.
3.8 Binding site evaluation
The binding site of the refined protein was found to be HIS110, ASN113, THR173, PRO214, ASN368, GLY370, GLY371, ARG373, PHE392, ASN394, PHE464, ASP470. The binding site of 3IVD protein comprised of ASN370, ARG375, PHE394, ASN396, PHE466, ASP472 for uridine ligand (Fig. 4). The predicted binding site is precisely the same since it comprises the identical amino acid residues as that of template protein.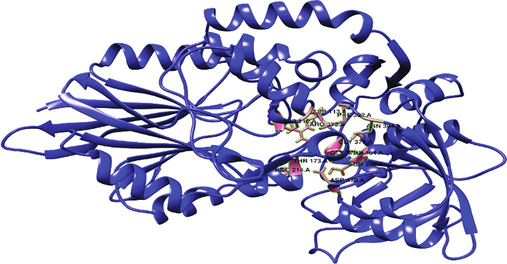
Structure of 5′-nucleotidase with binding site depicted in a pink hue.
3.9 Molecular interaction of predicted 5′-nucleotidase
Uridine interacted with the predicted 5′-nucleotidase enzyme binding site via hydrogen bonds at ARG373, ASP470, and HIS110, and hydrophobic interactions at ASP77, ASN109, THR173, VAL174, PRO214, GLY370, PHE392, and PHE464 (Fig. 5). The binding energy was found to be −6.4 kcal/mol.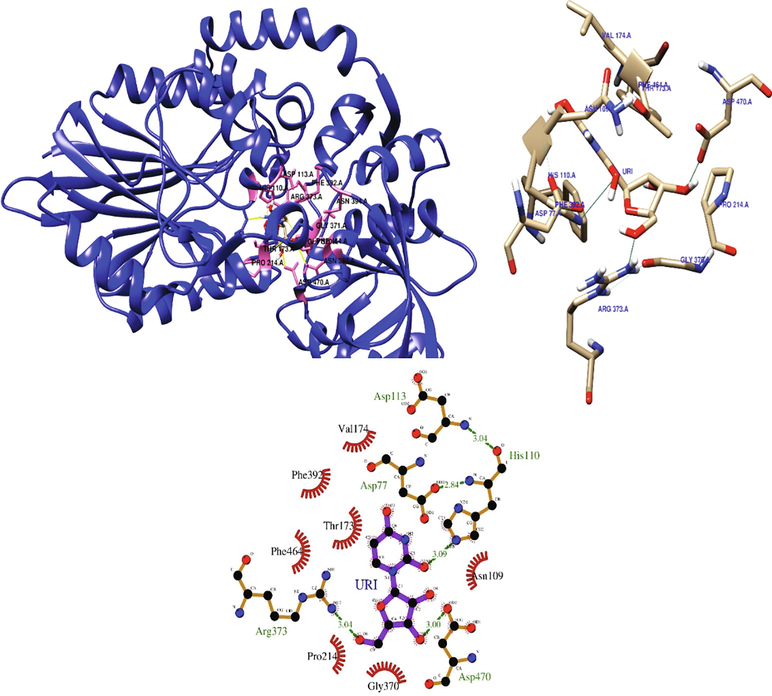
Depicting the interaction of uridine at the binding site of 5′-nucleotidase.
Cytidine interacted with the predicted 5′-nucleotidase enzyme binding site through hydrogen bonds at HIS110, THR173, HIS249, TYR340, ARG373, ASN418, and ASP470. Cytidine also interacted with the residues ASN109, PRO214, ALA215, HIS251, ASN368, GLY370, PHE392, and PHE464 via hydrophobic interactions (Fig. 6). The binding energy of cytidine with 5′-nucleotidase was found to be −6.5 kcal/mol.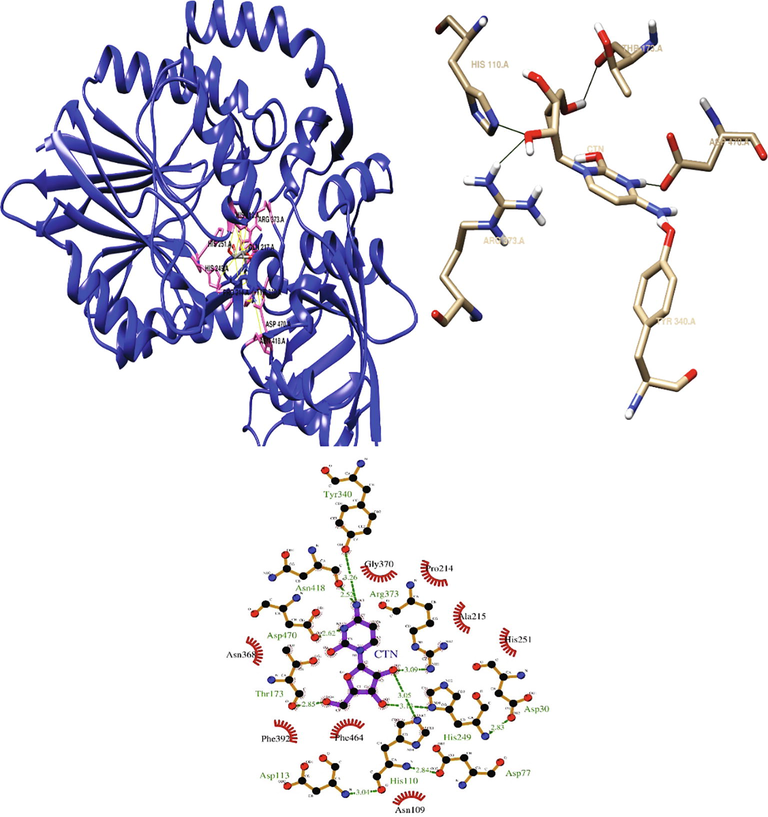
Depicting the interaction of cytidine at the binding site of 5′-nucleotidase.
The inhibitor (chloramphenicol) with potential clinical effects is the widely used antibiotic against several gram-negative bacteria. In the present work, the inhibitor interacted with the predicted 5′-nucleotidase enzyme binding site through hydrogen bonds at TRP42, ARG216, TYR38 and ARG373, and hydrophobic interactions at HIS32, HIS34, VAL40, PRO41, GLY81, GLN217, HIS251, SER270, GLY271, ILE273, ARG338 and GLU375 (Fig. 7) with the binding affinity of −5.7 kcal/mol.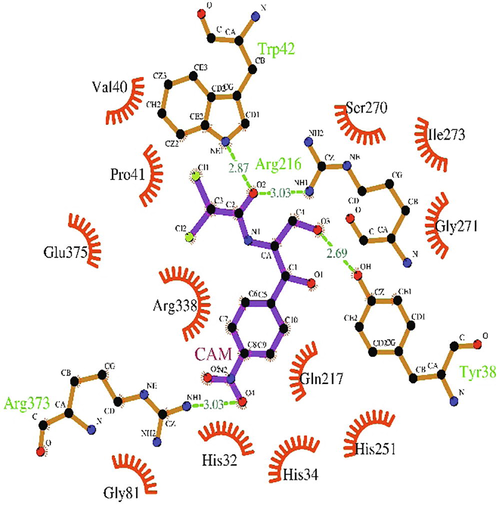
LigPlot + depicting the interaction chloramphenicol (inhibitor) at the binding site of 5′-nucleotidase.
The binding affinity of uridine and cytidine with the predicted 5′nucleotidase structure is comparable with that of the template protein, which was −8.2 kcal/mol and −8.4 kcal/mol, respectively (Supplementary Tables 5-9).
3.10 Molecular dynamics simulation study
The simulation study provides the analysis of root mean square deviation (RMSD), radius of gyration (Rg), solvent accessible surface area (SASA), number of hydrogen bonds maintained throughout the simulation time and variation in protein and their complexes. The simulation was performed at the time duration of 20 ns with the native protein alone and in complex with ligand.
The RMSD plot Fig. 8 (a) of 3IVD alone shows that the protein reached equilibrium approximately at 17–20 ns time and remaining showed stable trajectory with minimal deviation in 0.1–0.15 nm RMSD range, whereas the RMSD plot of 5′-nucleotidase complexed with uridine and cytidine reached equilibrium at 10–15 ns and 5–20 ns (Fig. 9a and Fig. 10a). After the initial fluctuations the ligand bound to the protein reached equilibrium.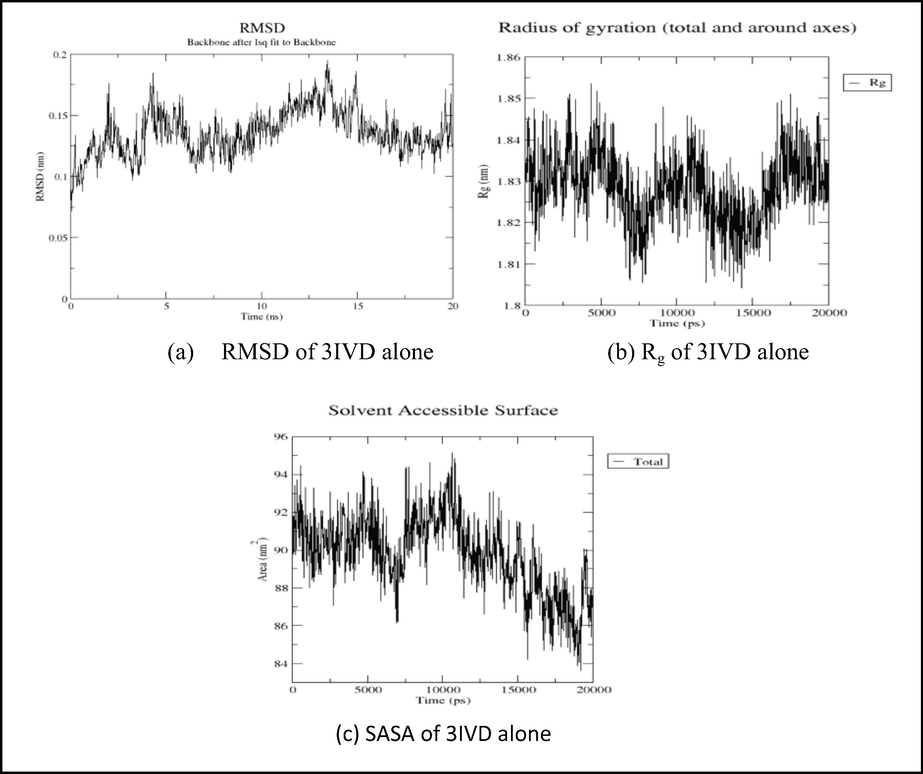
Showing the analysis of RMSD, Rg, and SASA graphs of 3IVD protein alone.
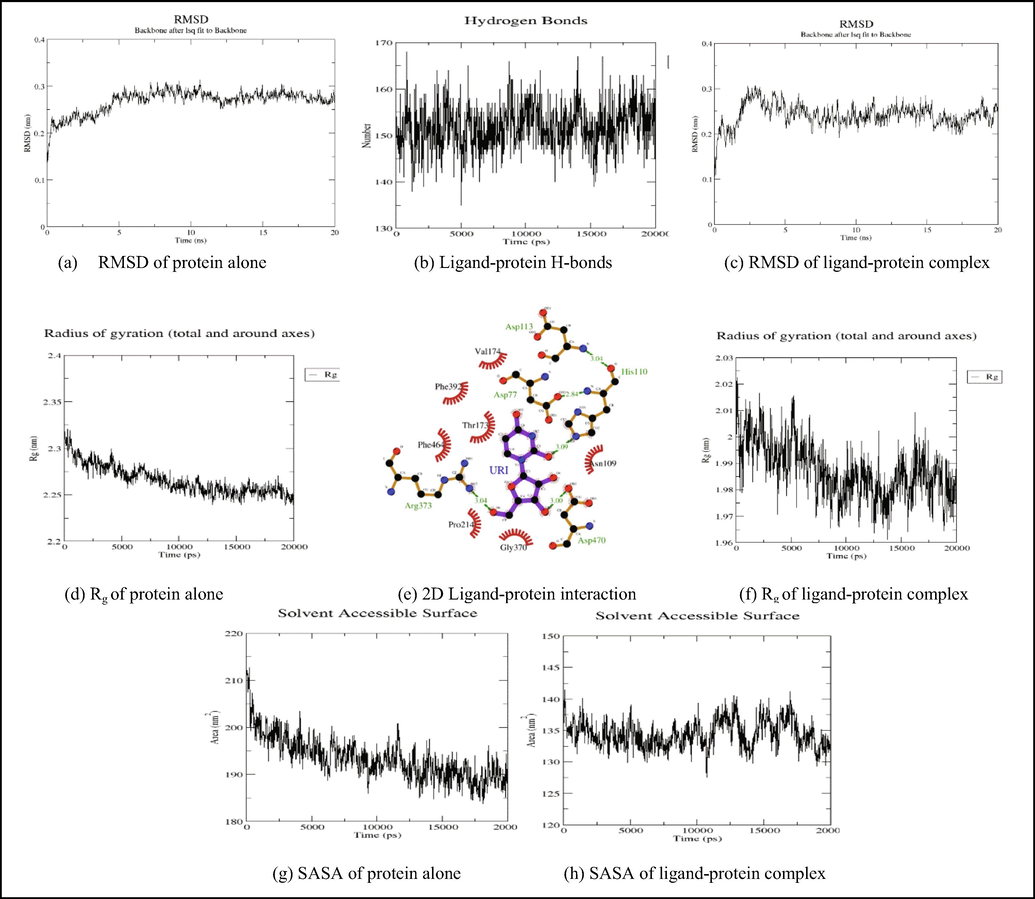
Showing the analysis of RMSD, Rg, SASA graphs and inter hydrogen bonding between 5′nucleotidase-Uridine complex.
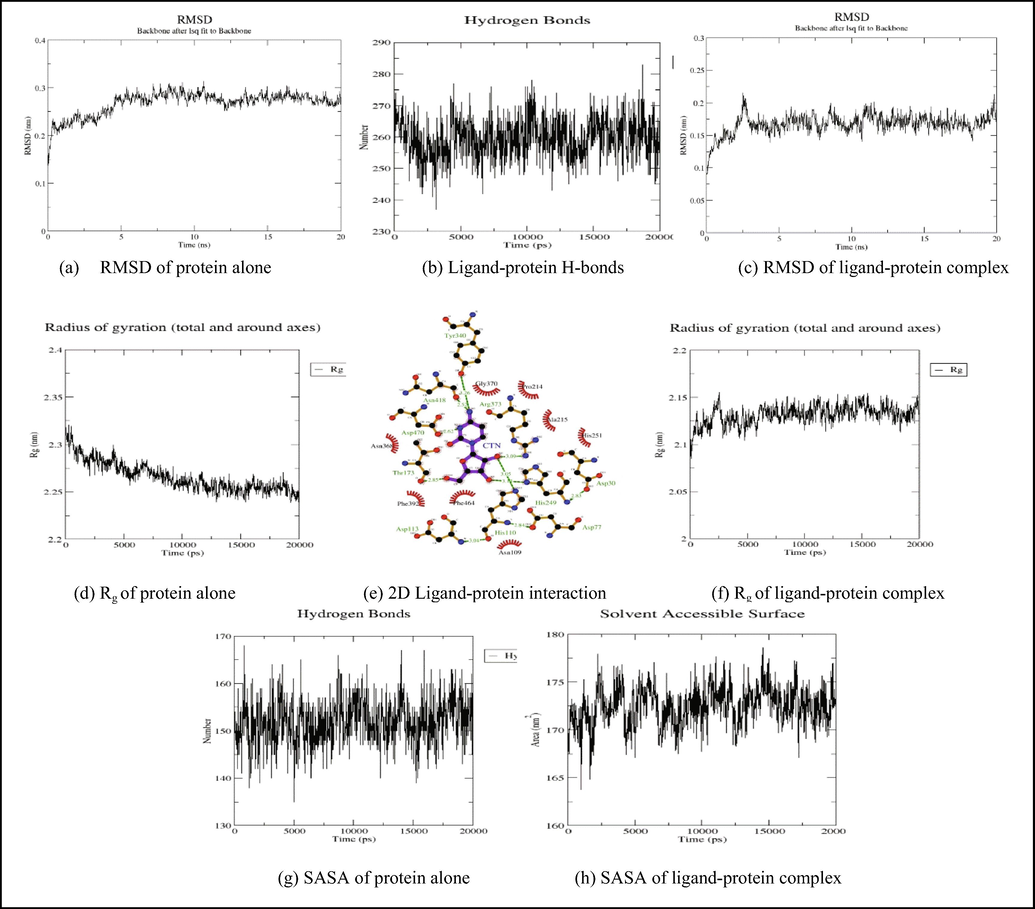
Showing the analysis of RMSD, Rg, SASA graphs and inter hydrogen bonding between 5′nucleotidase- cytidine complex.
The radius of gyration (Rg) considers the varied masses calculated to root mean square distances considering the central axis of rotation. The Rg plot (Fig. 8b, 9 g and 9f) considers the capability, shape and folding during the each time step of the whole trajectory throughout the simulation. The protein and their respective ligand complex exhibited the similar pattern of Rg value with the deviation in the range of 1.81–1.86 nm for 3IVD (Fig. 8b), 0.15–0.31 nm (Fig. 9g) and 0.15–0.25 nm (Fig. 9f).
H-bonds which appear during the Molecular Docking study being analyzed over the total simulation period. All the intermolecular H-bonds among ligands and protein were only considered during the analysis and plotted accordingly (Fig. 9b and 10b). It is evident from the plot that the number of H-bonds during the simulation runs remains consistent with Molecular Docking study, while few bonds were simultaneously broken and rebuilt.
SASA measures the area around hydrophobic core formed between protein–ligand complexes (Fig. 8c, 9 g, 9 h, Fig. 10g and Fig. 10h). Consistent SASA values were observed.
4 Conclusion
In summary, it is evident that the KPHS_00890 codes for protein 5′-nucleotidase. It is also found that the predicted structural and functional protein 5′-nucleotidase is highly identical to E. coli 5′-nucleotidase. The 5′-nucleotidase protein of K. pneumoniae is highly conserved and it reserves a high degree of similarity with nucleotidase proteins of other organisms such as E. coli, Salmonella, etc., thus, indicating a high degree of relatedness, which is also evident from phylogenetic analysis. The binding site for substrates of E. coli and K. pneumoniae nucleotidase was found to overlap, signifying conserved active sites in both the proteins. The substrates cytidine and uridine are natural substrates of 5′-nucleotidase with good biding affinity to K. pneumoniae nucleotidase, comparable to the binding with E. coli nucleotidase.
Author contributions
Conception or design of the work: PHK, CS; The acquisition: CS, GS, MNC, SPK, RV, AMB, AHB; Analysis or interpretation of data: PHK, GS; The creation of new software used in the work: PHK, CS, PK; Have drafted the work or substantively revised it: AS, SPK, AGR, CS.
Acknowledgments
CS, PHK, and MNC acknowledge the support and infrastructure provided by the JSS Academy of Higher Education and Research (JSSAHER), Mysuru, India. SPK thankfully acknowledges the Director, Amrita Vishwa Vidyapeetham, Mysuru campus for infrastructure support. The authors extend their appreciation to the Researchers Supporting Project number (RSP-2021/56), King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep.. 2017;7:1-15.
- [Google Scholar]
- The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res.. 2017;45:D313-D319.
- [Google Scholar]
- Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis. 2009;30(S1):S162-S173.
- [Google Scholar]
- GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res.. 2013;41:384-388.
- [Google Scholar]
- GalaxySite: Ligand-binding-site prediction by using molecular docking. Nucleic Acids Res.. 2014;42:210-214.
- [Google Scholar]
- MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol.. 2018;35:1547-1549.
- [Google Scholar]
- LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model.. 2011;51(10):2778-2786.
- [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem. 2010;30(16):2785-2791.
- [Google Scholar]
- COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics. 2007;23(9):1073-1079.
- [Google Scholar]
- UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem.. 2004;25(13):1605-1612.
- [Google Scholar]
- Characterisation of virulence genes associated with pathogenicity in Klebsiella pneumoniae. Indian J. Med. Microbiol.. 2019;37(2):210-218.
- [Google Scholar]
- LigDockCSA: Protein-ligand docking using conformational space annealing. J. Comput. Chem.. 2011;32(15):3226-3232.
- [Google Scholar]
- ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res.. 2007;35:407-410.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2021.101537.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




