

In search of potent histamine-3 receptor antagonists
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Available online 22 October 2014
Peer review under responsibility of King Saud University.
Abstract
Starting with modafinil, an alertness and wake-promoting agent but inactive in histamine 3 receptor (H3R) binding assay, a series of potent H3 receptor antagonists were developed.
Keywords
Histamine-3
Antagonist
Suzuki reaction
Modafinil

1 Introduction
Due to the beneficial roles played by histamine 3 receptor (H3R) in alertness, wake-promotion and cognition in central nervous system (CNS), the research in this field has become a subject of intense interest in recent years (Singh et al., 2013). Thus the development of potent H3R antagonists has emerged as an attractive target for the treatment of various CNS disorders including narcolepsy, ADHD, and cognitive disorders either as a primary indication or associated with another disease state (Berlin et al., 2011; Vohora and Bhowmik, 2012; Celanire et al., 2005). Our interest in H3R antagonists originated from modafinil (compound 1, Fig. 1) a novel alertness and wake-promoting agent whose mechanism of action at the molecular level remains elusive to date (Saper and Scammell, 2004). Modafinil does not display any significant in vitro binding affinity for the H3 receptor. However, it has been reported that in anesthetized rats, modafinil increased extracellular histamine concentrations (Ishizuka et al., 2003). Thus while designing a series of H3R antagonists, we sought to incorporate some of modafinil’s structural themes, especially its lipophilic bis-aryl moiety attached to a polar (i.e. sulfinyl) moiety. In a previous report, we disclosed results from our initial effort (Dunn et al., 2014). In this Communication, we offer a brief summary from our additional effort.
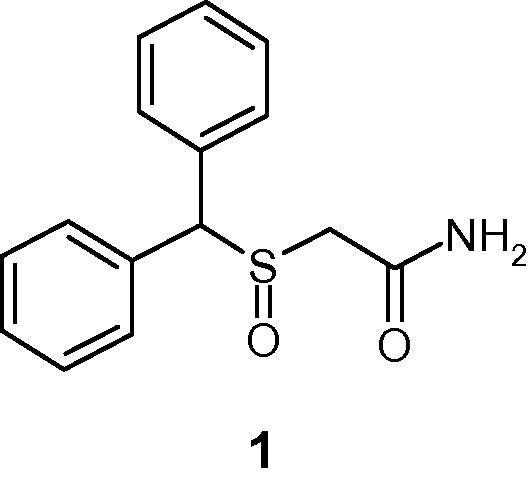
- Chemical structure of compound 1.
2 Chemistry
Scheme 1 depicts the synthesis of target compounds 9–10. Pyridine derivative 2 was coupled with the boronic acid 3 under Suzuki condition to generate compound 4. Separately, enantiomerically pure prolinols (compounds 5–6) underwent reductive amination reactions with cyclobutanone to generate amino alcohols 7–8. O-alkylation of compounds 7–8 with compound 4 generated compounds 9 and 10, respectively.
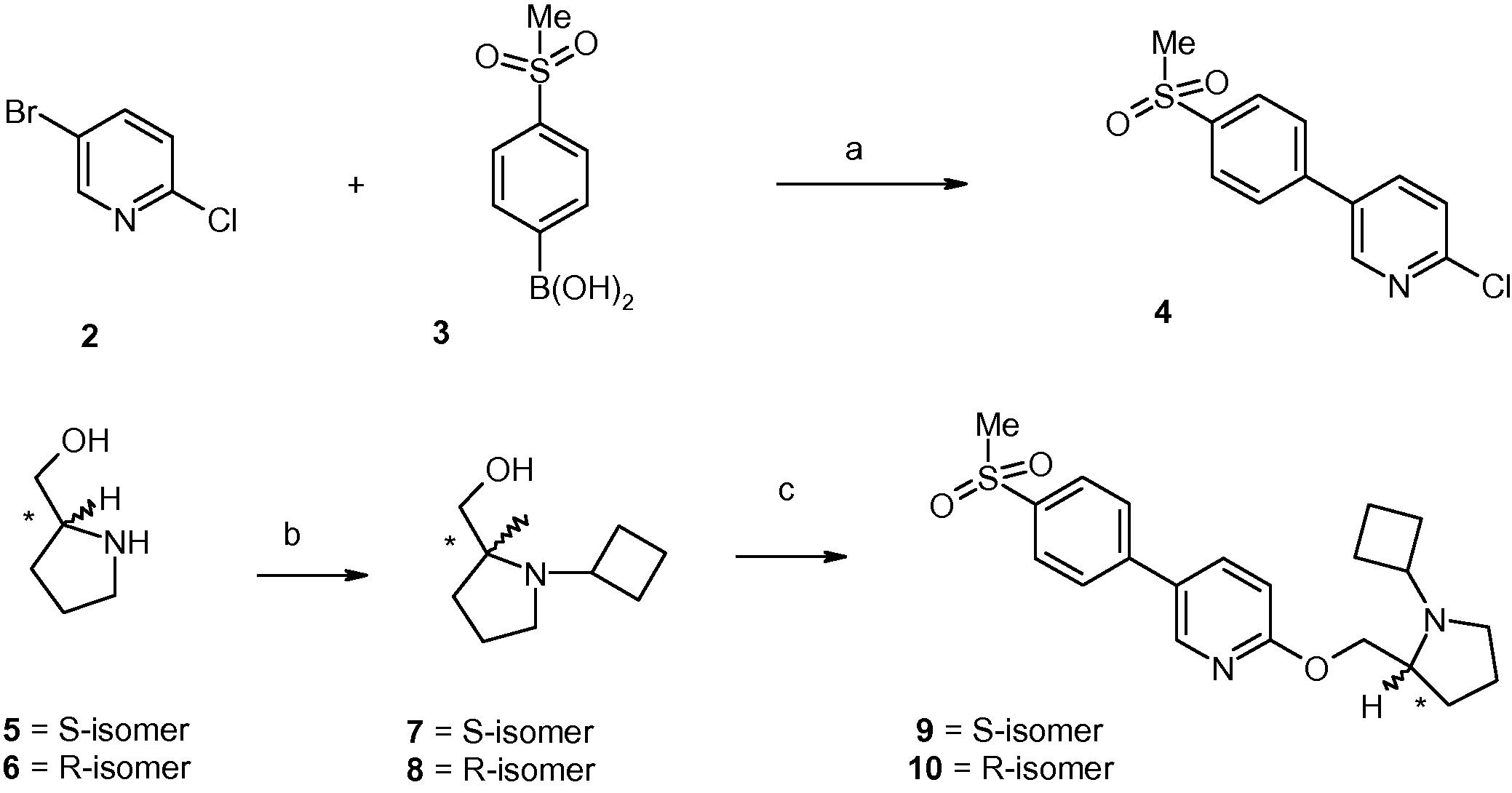
- Reagents and conditions: (a) 4-(Pd(PPh3)4, 2 M aq. Na2CO3, toluene, EtOH, 95–100 °C, 3 h, 70%; (b) (i) cyclobutanone, gl. AcOH (catalytic), CH2Cl2, 0 °C, 20–30 min; (ii) sodium acetoxyborohydride, 0 °C to room temperature, 4–6 h, 60–65%; (c) (i) NaH, DMF, 0 °C to room temperature, 30 min; (ii) compound 4, 100 °C, 2 h, 40–45%.
In Scheme 2, the chloro group of compound 2 was displaced by the alkoxides generated from cyclic alkanols 11–13 to produce a series of compounds of general structure 14. Separately, phenol derivative 15 underwent a Mitsunobu reaction with compound 13 to produce compound of general structure 16. Compounds 14 and 16 underwent separate Suzuki reactions with 4-(methylsulfonyl)benzeneboronic acid to generate compounds of general structure 17 that were N-deprotected to produce free amines of general structure 18. Reductive amination of compounds 18 with a set of cyclic ketones produced target compounds 19–25.
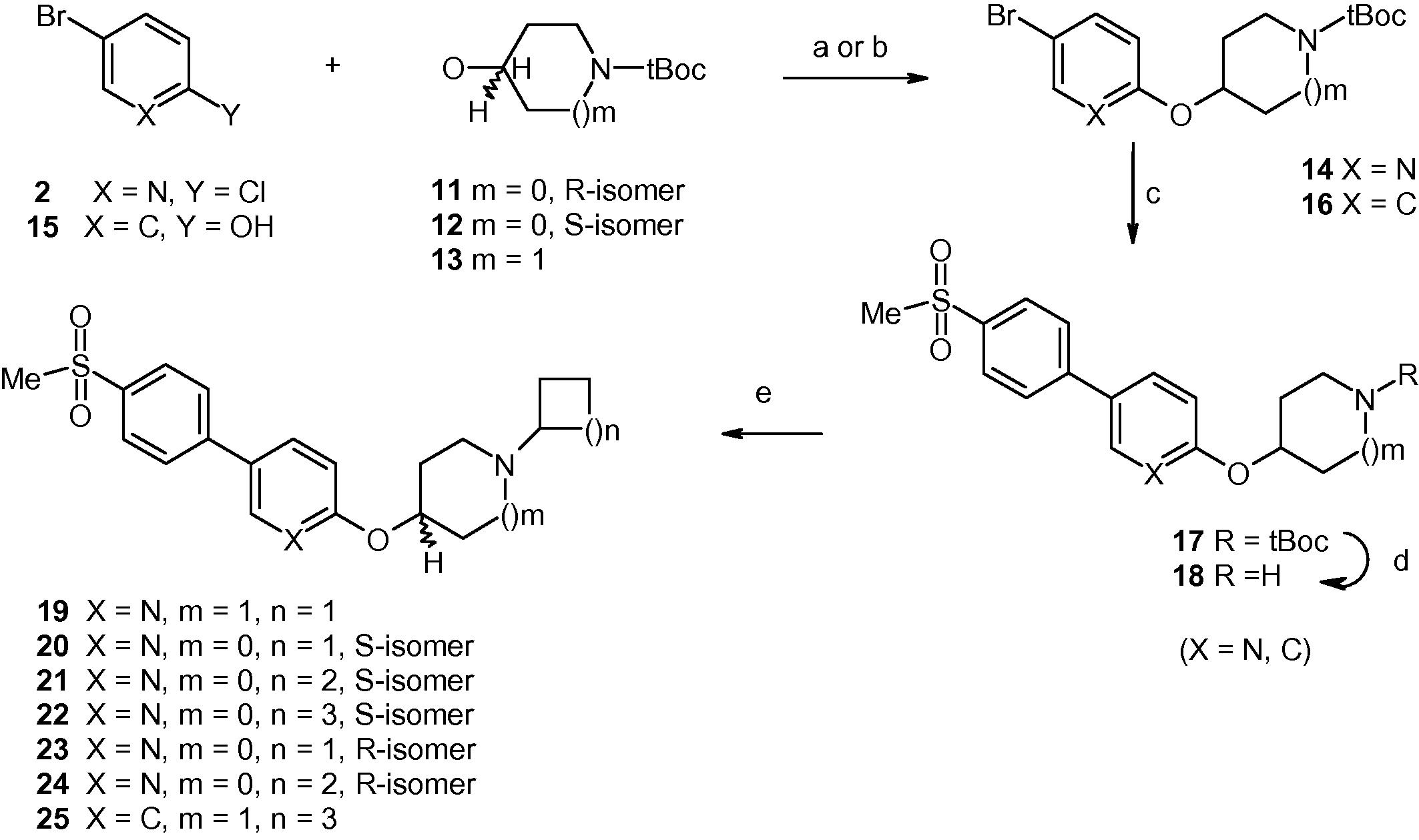
- Reagents and conditions: for compound 14: (a) (i) compounds 11–13, 60%NaH, NMP, 0 °C to room temperature; (ii) compound 2, 90 °C, overnight, 50–60%; for compound 16: (b) PPh3, 40% DEAD in toluene, THF, 0 °C to room temperature, overnight, 60%; (c) 4-(methylsulfonyl)benzeneboronic acid, 4-(Pd(PPh3)4, 2 M aq. Na2CO3, toluene, EtOH, 95–100 °C, 6–8 h, 50–60% (d) 4 N HCl in dioxane, room temperature, 90–95%; (e) (i) cyclic ketone, gl. AcOH (catalytic), CH2Cl2, 0 °C, 30 min; (ii) sodium acetoxyborohydride, 0 °C to room temperature, 4–6 h, 50–60%.
3 Biology
Following a literature procedure, binding properties of the target compounds were assessed against recombinant human H3 (hH3) and rat H3 (rH3) receptors by displacement of [3H]-N-α-methylhistamine and reported as Ki values from an average of three experiments, done in duplicate (US Patent, 2012), while in triplicate for the potent analogs. The results are displayed in Table 1. The table also displays the result for a reference compound Pitolisant that entered into clinical trials (Schwartz, 2011).
| Compound | hH3 Ki (nM)a | rH3 Ki (nM)a |
|---|---|---|
| 1 | b | b |
| 9 | >1000 | >1000 |
| 10 | >1000 | >1000 |
| 19c | 7 ± 2 | 18 ± 3 |
| 20c | 19 ± 3 | 62 ± 4 |
| 21c | 19 ± 4 | 63 ± 4 |
| 22c | 22 ± 2 | 20 ± 3 |
| 23 | 234 | >1000 |
| 24 | 115 | >1000 |
| 25 | 82 | 276 |
| 26 | >1000 | >1000 |
| Pitolisantd | 0.3–1.0 | 17 |
4 Discussion
As mentioned previously, compound 1 displayed no hint of any in vitro binding affinity (even at higher concentrations) for either H3 receptor. Thus, our exploration began by re-orienting the aromatic region into a para-linked bi-aryl system, especially an aryl-heteroaryl system as the central lipophilic region. While a polar sulfonyl group was appended to the aryl ring, the heteroaryl moiety was attached via a methyleneoxy moiety, to a terminal cyclic amine moiety to impart some structural rigidity in that part of the molecule. Initially, structural fragments derived from (S)- and (R)- prolinol were utilized to generate compounds 9 and 10, respectively. The lack of reasonable activity of both compounds 9 and 10 (Table 1) indicated that this initial maneuver was not productive. It was postulated that the basic amine moiety in either compound probably was unable to interact with a critical aspartate residue on the receptor, essential for a productive binding to H3R. Subsequently, the decision was made to incorporate the exocyclic methylene group of either compound as a part of a cyclic moiety, thus generating achiral 2-pyridyl-4-oxypiperidnyl- derivative 19 that imparted immediate potency in both assays (Ki of 7 nM and 18 nM, respectively). To expand the scope of the series, the 4-oxypiperidinyl group in compound 19 was subsequently replaced by chiral 3-oxypyrrolidinyl systems. In the (S)-series of ligands, variation of the size (n = 4 – 5) of the cycloalkyl groups attached to the basic nitrogen generated compounds 20–22 that displayed similar affinity in the hH3 binding assay, but they differed ca. 3-fold in rH3 binding assay (Ki 19–22 nm vs. 62–63 nM). From the (R)-series, the ligands appeared to possess at least one order of magnitude lower affinity than their corresponding counterpart from (S)-series in the hH3 assay (cf. affinities of compound 23 vs. compound 20, and compound 24 vs. compound 21, respectively). Compounds 23–24 appeared to be significantly less active in the rH3 assay also. The reason for this difference in binding activity of compounds 23–24 in two assays is not currently understood. Thus, the chirality of the 3-oxypyrrolidinyl moiety played a role in the binding activity of this class of ligand. Subsequently, the pyridyl group of compound 22 was replaced by a phenyl group generating compound 25 that appeared to have somewhat lower affinity than the parent compound. Finally, the replacement of the entire methylsulfonylphenyl moiety of this class of ligands with a smaller lipophilic group (e.g. bromine as in compound 26, Fig. 2) resulted in significant loss of affinity (cf. compound 26 vs. compound 25) indicating importance of the methylsulfonylphenyl group in the binding affinity of this class of ligands. Representative compounds 19 and 21 were advanced to pharmacokinetic studies in rats. Both compounds were detected in the brain in reasonable level post 1 h ip administration (dose 10 mg/kg). Additional profiling is continuing with several members of the series. After completion of this research, a report of a set of bis-phenyl derivatives appeared in the literature without mentioning any aryl-heteroaryl combination from this current series (Semple et al., 2012).
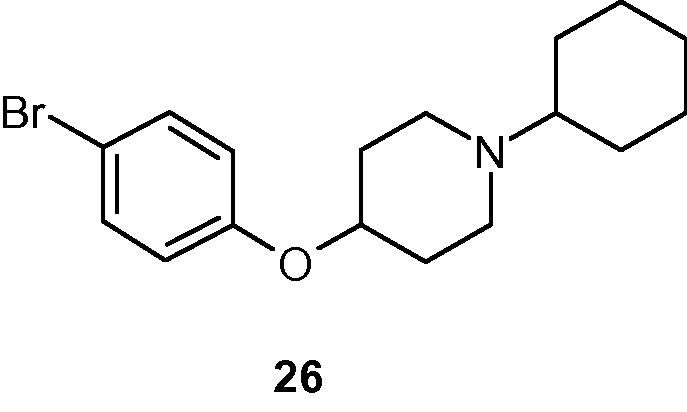
- Chemical structure of compound 26.
5 Conclusions
In this Communication, utilizing modafinil as a launching pad, a series of para phenyl-pyridyl aromatic system compounds that bind potently to both mouse and human recombinant histamine-3 receptors were developed. In PK experiments, representative compounds 19 and 21 displayed brain permeability post 1 h ip administrations.
Acknowledgment
Author wishes to acknowledge the scientific support provided by the members of the biology team.
References
- Keynote review: histamine H3 receptor antagonists reach out for the clinic. Drug Discov. Today. 2005;10:1613-1627.
- [Google Scholar]
- From an atypical wake-promoting agent to potent histamine-3 receptor inverse agonists. Chem. Bio. Drug Des.. 2014;83:149-153.
- [Google Scholar]
- Modafinil increases histamine release in the anterior hypothalamus of rats. Neurosci. Lett.. 2003;339:143-146.
- [Google Scholar]
- The histamine H3 receptor: from discovery to clinical trial. Br. J. Pharmacol.. 2011;163:713-721.
- [Google Scholar]
- Identification of biaryl sulfone derivatives as antagonists of the histamine H3 receptor: Discovery of (R)-1-(2-(4’-(3-methoxypropylsulfonyl)biphenyl-4-yl)ethyl)-2-methylpyrrolidine(APD916) Bioorg. Med. Chem. Lett.. 2012;22:71-75.
- [Google Scholar]
- Histamine H3 receptor function and ligands: recent developments. Mini Rev. Med. Chem.. 2013;13:47-57.
- [Google Scholar]
- US Patent 8,247,414, 2012.
- Histamine H3 receptor antagonists/inverse agonists on cognitive and motor processes: relevance to Alzheimer’s disease, ADHD, schizophrenia, and drug abuse. Front Syst. Neurosci.. 2012;6(72):1-10.
- [Google Scholar]
Appendix
Rat H3 assay
Cell line development and membrane preparation
The rat H3 receptor cDNA was PCR amplified from reverse-transcribed RNA pooled from rat thalamus, hypothalamus, striatum and prefrontal cortex with a sequence corresponding to bp #338–1672 of Genbank file #NM_05306, encoding the entire 445-amino acid rat histamine H3 receptor. This was engineered into the pIRES-neo3 mammalian expression vector, which was stably transfected into the CHO-A3 cell line (Euroscreen, Belgium), followed by clonal selection by limiting dilution. Cells were harvested and cell pellets were frozen (−80 °C). Cell pellets were re-suspended in 5 mM Tris–HCl, pH 7.5 with 5 nM EDTA and a cocktail of protease inhibitors (Complete Protease Inhibitor Tablets, Roche Diagnostics). Cells were disrupted using a polytron cell homogenizer and the suspension was centrifuged at 1000×g for 10 min at 4 °C. The pellet was discarded and the supernatant was centrifuged at 40,000×g for 30 min at 4 °C. The membrane pellet was washed in membrane buffer containing 50 mM Tris–HCl, pH 7.5 with 0.6 mM EDTA, 5 mM MgCl2 and protease inhibitors, re-centrifuged as above and the final pellet re-suspended in membrane buffer plus 250 mM sucrose and frozen at −80 °C.
Radioligand binding
Membranes were re-suspended in 50 mM Tris HCl (pH 7.4), 5 mM MgCl2 and 0.1% BSA. The membrane suspensions (10 μg protein per well) were incubated in a 96 well microtiter plate with [3H]-N-alpha-methylhistamine (approx. 1 nM final concentration), test compounds at various concentrations (0.01 nM–30 μM) and scintillation proximity beads (Perkin Elmer, FlashBlue GPCR Scintillating Beads) in a final volume of 80 μl for 4 h at room temperature, shielded from light. Non-specific binding was determined in the presence of 10 μM clobenpropit. Radioligand bound to receptor, and therefore in proximity to the scintillation beads, was measured using a Microbeta scintillation counter.
Human H3 assay
Cell line development and membrane preparation
CHO cells stably expressing the human H3 receptor (GenBank: NM_007232) were harvested and cell pellets were frozen (−80 °C). Cell pellets were re-suspended in 5 mM Tris–HCl, pH 7.5 with 5 nM EDTA and a cocktail of protease inhibitors (Complete Protease Inhibitor Tablets, Roche Diagnostics). Cells were disrupted using a polytron cell homogenizer and the suspension was centrifuged at 1000×g for 10 min at 4 °C. The pellet was discarded and the supernatant was centrifuged at 40,000×g for 30 min at 4 °C. This membrane pellet was washed in membrane buffer containing 50 mM Tris–HCl, pH 7.5 with 0.6 mM EDTA, 5 mM MgCl2 and protease inhibitors, re-centrifuged as above and the final pellet re-suspended in membrane buffer plus 250 mM sucrose and frozen at −80 °C.
Radioligand binding
Membranes were re-suspended in 50 mM Tris HCl (pH 7.4), 5 mM MgCl2 and 0.1% BSA. The membrane suspensions (10 μg protein per well) were incubated in a 96 well microtiter plate with [3H]-N-alpha-methylhistamine (approx. 1 nM final concentration), test compounds at various concentrations (0.01 nM–30 μM) and scintillation proximity beads (Perkin Elmer, FlashBlueGPCR Scintillating Beads) in a final volume of 80 μl for 4 h at room temperature, shielded from light. Non-specific binding was determined in the presence of 10 μM clobenpropit. Radioligand bound to receptor, and therefore in proximity to the scintillation beads, was measured using a Microbeta scintillation counter.




