

Translate this page into:
Identification of novel mycobacterium tuberculosis leucyl-tRNA synthetase inhibitor using a knowledge-based computational screening approach
⁎Corresponding authors at: Department of Genetic Medicine, Faculty of Medicine, King Abdulaziz University, Jeddah 21589, Saudi Arabia Kingdom of Saudi Arabia. nshaik@kau.edu.sa (Noor Ahmad Shaik), bbabajan@kau.edu.sa (Babajan Banaganapalli)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Objectives
Tuberculosis is a chronic lung disease caused by Mycobacterium tuberculosis (MTB), whose thick cell envelope and drug metabolizing enzymes offering it multidrug resistance. Therefore, there is a need to identify new molecular targets, biologically active as well as clinically safe anti-MTB compounds from natural resources.
Methods
In this study, we performed high throughput computational screening of FDA listed natural bioactive compounds for identifying novel anti-MTB leucyl-tRNA (LeuRS) synthetase inhibitors.
Results
Initial virtual molecular docking of 136 bioactive compounds has identified Docetaxel, Reserpine and Irinotecan as promising lead molecules owing to their structural plasticity and binding affinity with the active site of MTB-LeuRS. Further, deep molecular docking and molecular dynamics (MD) simulation analysis (at 100 ns) of the above three test compounds along with Oxaborole compound (GSK656) has demonstrated the superior binding affinity and stability of Irinotecan in forming molecular complexes with LeuRS protein. Interestingly, it also showed comparable binding residues and affinity parameters (like flexibility, structural divergence) as the GSK656 inhibitor in binding the tRNASyn domain 2 of LeuRS. A good correlation of pharmacokinetic properties (ADME-Tox) like bioavailability, absorption, solubility, and low toxicity between Irinotecan and GSK656 was evident. Competitive binding of Irinotecan to tRNASyn domain 2 of LeuRS is likely to make it unavailable to bind Leucine amino acid, which may negatively impact the protein biosynthesis and eventually inhibit the bacterial growth and attenuate the pathogen’s virulence.
Conclusions
Our findings pave the way for further experimental confirmation of Irinotecan in the quest for a novel anti-LeuRS specific inhibitor to combat drug resistant MTB infection.
Keywords
Mycobacterium tuberculosis
Deep docking
Bioactive compounds
MD simulation
tRNA
Drug resistance

1 Introduction
Mycobacterium tuberculosis (MTB), a rod-shaped aerobic bacterium causes tuberculosis (Furin et al., 2019). It usually remains latent in the host with no symptoms, but 5–10% of patients develop clinical manifestations, which can be intensified with an HIV co-infection (Harries and Dye 2006). When the infection is active, patients may feel fatigued, have night sweats, fever, cough, and hemoptysis (Suárez et al., 2019). As a first line, for the initial 2 months, tuberculosis is treated by a combination therapy of pyrazinamide (PZA), ethambutol (EMB), isoniazid (INH), and rifampicin (RMP), followed by additional therapy of INH and RMP for the next four months (Benaissa et al., 2022) (Ahmed and Saif 2017). Patients infected with INH and RMP drug resistant MTB (MDR-TB) must be treated with fluoroquinolones or other second line injectable drugs such as amikacin, kanamycin, and capreomycin, among others (Suárez et al., 2019). If the patients had an INH, RMP, and second line injectable drug resistance, then the infection is called “extensively drug resistant tuberculosis” (XDR-TB). In such infections, the treatment options are very limited and the chances of mortality is usually very high (Seaworth and Griffith 2017).
The rapid emergence of drug resistance in MTB necessitates us to explore the novel alternative therapeutic candidates, that are more efficient and less toxic than chemically synthesized drug molecules (Alsulaimany et al., 2021). In recent years, naturally occurring bioactive compounds have shown great promise in combating drug resistant strains of MTB (Wang et al., 2019, Pawar et al., 2020). Aminoacyl-tRNA synthetases (aaRSs) owing to their unique solubility profile, stability, and expression profile, they represent as a promising enzyme group to develop anti-MTB drug compounds. These enzymes catalyze the transfer of an amino acid to cognate tRNAs in the protein translation process. Leucyl-tRNA synthetase (LeuRS), belonging to class I aaRSs is a proven therapeutic target for AN2690 (5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole), a bioactive compound in some microorganisms (Ndagi et al., 2021). The distinctive structural features of prokaryotic and eukaryotic Leu-RS catalytic domains make it an ideal therapeutic target by different chemical inhibitors (Gadakh and Van Aerschot 2012). However, the potential of inhibiting MTB leucyl-tRNA synthetase with natural bioactive compounds is not yet well explored.
Laboratory screening of bioactive compounds involving cellular systems and animal models can be an expensive and time-consuming process. On the contrary, bioinformatics methods accelerate drug target identification through rapid drug candidate screening, refinement, and also by predicting pharmacodynamic properties (Shaker et al., 2021). These bioinformatic approaches deploy artificial intelligence methods like machine learning and pattern classification methods (neural networks, support vector machines, and decision trees) to virtually screen bioactive compound libraries for potential antimicrobial molecules (Naik et al., 2020). Throughput computational approaches like molecular dynamics simulations and molecular docking allow us to study the structural plasticity and binding characteristics of inhibitor molecules toward target proteins at a three-dimensional (3D) structural level (Almeleebia et al., 2021). Furthermore, bioinformatics tools are also known to be efficient in predicting the ADME-Tox and other pharmacological properties of potential drug molecules (Aronica et al., 2021; Behl et al., 2021). Because there is a growing need to find new and safe anti-MTB drugs, this study used high-throughput virtual screening of FDA-approved natural bioactive compounds to look for anti-MTB aaRS inhibitory molecules. Deep molecular docking and molecular dynamics simulations were used to look for these molecules.
2 Methodology
2.1 Therapeutic target sequence retrieval and its conservation analysis
The amino acid sequences of LeuRS (MTB: TBMG_00040) were initially retrieved from the KEGG Gene database. Then, the amino acid sequence and physiochemical properties [molecular weight (mw), instability index, pH, and grand average of hydropathy (GRAVY)] of the protein were predicted using the ProtParam tool hosted on the ExPASy server (https://web.expasy.org/protparam) (Gasteiger et al., 2003). The LeuRS protein sequences of MTB, Escherichia coli, Helicobacter pylori, Salmonella bongori, Haemophilus influenza, and Plasmodium falciparum were aligned using the Clustal Omega sequence alignment webserver (https://multalin.toulouse.inra.fr/multalin) for identifying conserved regions, insertions, and deletions across different species (Sievers and Higgins 2018). Clustal Omega is a unique multiple sequence alignment program that generates alignments between three or more sequences using seeded guide trees and HMM profile-profile algorithms.
2.2 Protein model preparation
At first, the amino acid sequence of MTB LeuRS (KEGG ID: T00940) was provided as input to PSI-BLAST tool for searching the experiment sourced structures deposited in the Protein Data Bank (PDB) database. Since a fully solved LeuRS structure was not available, a SWISSMODEL based homology modelling approach, followed by energy minimization (Steepest descent) using steps with SwissPdbViewer 3.5, was adopted (Waterhouse et al., 2018) (Guex and Peitsch 1997). The structural evaluation and stereochemical quality of the predicted LeuRS model were checked with the PROCHECK webserver ( http://services.mbi.ucla.edu). The PyMOL molecular visualization tool was used for visual exploration of the LeuRS molecular structures ( https://pymol.org).
2.3 Bioactive compounds collection and energy refinement
A total of 136 Food and Drug Administration (FDA) listed natural bioactive compounds were downloaded from the ZINC database ( https://zinc.docking.org), using the following option, “substances > filters > FDA approved compounds”. All these compounds were downloaded in compressed structure data format (SDF). Energy refinement of all the downloaded compounds was done using Open Babel in the PyRx 0.8 tool ( https://pyrx.sourceforge.io).
2.4 Receptor-Based virtual screening
The potential anti-MTB LeuRS therapeutic molecules from the bioactive compound library were identified through virtual docking by PyRx, a Graphical User Interface (GUI) software. Both the target protein and bioactive compounds were initially prepared in PDBQT file formats. The physical parameters of the docking grid box around the ligand molecules were defined using the ‘Maximize’ [Center X: 22.4884, Y: 11.0007, Z: 24.4633; Dimensions (Å) X: 108.6, Y: 97.8, Z: 85.53] option available in PyRx software for ensuring the availability and accessibility of protein surface areas during the docking procedure. Using the default exhaustiveness value of 8, molecular docking was performed, by keeping the protein receptor rigid and the ligand molecule in a flexible conformation.
2.5 Deep molecular docking
The virtual docking of top-ranked bioactive compounds was further explored by deep molecular docking analysis with MTB-LeuRS by the DockThor program (Santos et al., 2020, Guedes et al., 2021). The receptor and ligand files were prepared by adding polar hydrogen atoms and partial charges by applying the MMFF94S force field. A grid box configuration is determined by the redocking of each complex according to its reference ligand. The grid box was defined using the center (average of the coordinates) and size (20 Å in each dimension). The discretization was maintained at 0.25 Å. The redocking was verified using the root mean square of atomic position deviation of ≤ 2 Å.
2.6 Drug-Likeness analysis
By using SwissADME, the top-scoring molecules were further evaluated for toxicity, drug-likeness, and pharmacokinetics (Pires et al., 2015; Daina et al., 2017). SwissADME was used to evaluate the drug-likeness of molecules retrieved from the ZINC database. Lipinski’s rule assesses various parameters needed for drug design. As per this rule, any drug should fulfill the following drug-likeness parameters: (a) molecular mass < 500 Daltons (Da), (b) < 5H bond donors, (c) < 10H bond acceptors, and (d) octanol/water partition coefficient logP is < 5, to be considered a biologically active drug molecule. The biological toxicity and pharmacokinetic properties of the compounds were validated using the SwissADME tool.
2.7 Molecular dynamics (MD) simulations
MD simulation was performed using Desmond41 software (https://www.deshawresearch.com/index.html) to study the protein–ligand complex interaction and binding energy analysis. The proteins and their ligand systems were created with the “System Builder” node in Desmond workflow. The hydration model was created by the top ranked energy minimized docking complex that was placed in the orthorhombic box (10 Å buffer distance), with SCP water mode l42 as water model implementing default cut-off metrics for Van der Waal forces (9 Å), time step (2.0 fs), initial temperature (300 K) and pressure (1.013 Pa) bar of the system. Desmond software calculates electrostatic force by fractionating near term and far term values with a 9 Å internal boundary. Furthermore, the sampling interval during simulation was set to 100 ps (picoseconds). Lastly, MD simulations were performed under the NPT ensemble (isothermal-isobaric ensemble) for 100 ns (nanoseconds). The structures with the largest populations were included in the analysis (ligand binding sites, H-bonds, root-mean-square fluctuation (RMSF), root-mean-square deviation (RMSD), and torsion angels etc.,).
3 Results
3.1 LeuRS protein sequence analysis
The physiochemical characterization results of MTB LeuRS protein (969 aa) are as follows; molecular weight of 107594.77 Da, pH of 5.08 (acidic), stability index of 30.90 (stable), and a grand average of hydropathicity (GRAVY) index of −0.317 (hydrophobic). Amino acid sequence alignment of MTB LeuRS proteins with different bacteria has suggested a 40% similarity with Salmonella bongori, 38% with E. coli, 37.9% with Haemophilus, and 30% with Helicobacter pylori (Fig. 1).
Multiple sequence alignment of LeuRS protein across 6 bacterial species. Highly conserved regions in star (∗), semi conserved regions in colon (:), mutated region in dot (.) and deleted regions in dashes (-).
3.2 Computational 3D protein modeling
Homology modelling is a suitable approach to study 3D structures of proteins, which are not yet characterized through the conventional x-ray crystallography method (24). Therefore, we modelled the 3D structure of LeuRS using the SWISS-MODEL webserver. Based on the PSI-BLAST search results, homologous sequence of Leucyl-tRNA synthetase protein from Thermus thermophilus was identified to have 40.02 % overall identity with MTB and had the highest query coverage of 96 %. Initially, five MTB-LeuRS models were assembled using the SWISS-MODEL tool utilizing Thermus thermophilus Leucyl-tRNA (PDB ID: 1H3N) as a template. Quality assessment of predicted models was done with GMQE and QMEAN scoring tools. The best model (Fig. 2A) was selected based on the template model score, root mean square deviation, and confidence score metrics (Table 1). The disordered regions in the predicted models were illustrated with the help of the Local Quality Plot (Fig. 2B). The residues in these disordered regions have low structural reliability as their individual QMEAN scores are below the 0.6 threshold value. A PROCHECK-based quality check with a Ramachandran plot has confirmed that out of the 969 amino acids, 97.2 % fell into allowed regions and 2.8 % fell into disallowed regions (Fig. 2C). The secondary structure of 1H3N is aligned with the sequence of MTB LeuRS (Fig. 2D).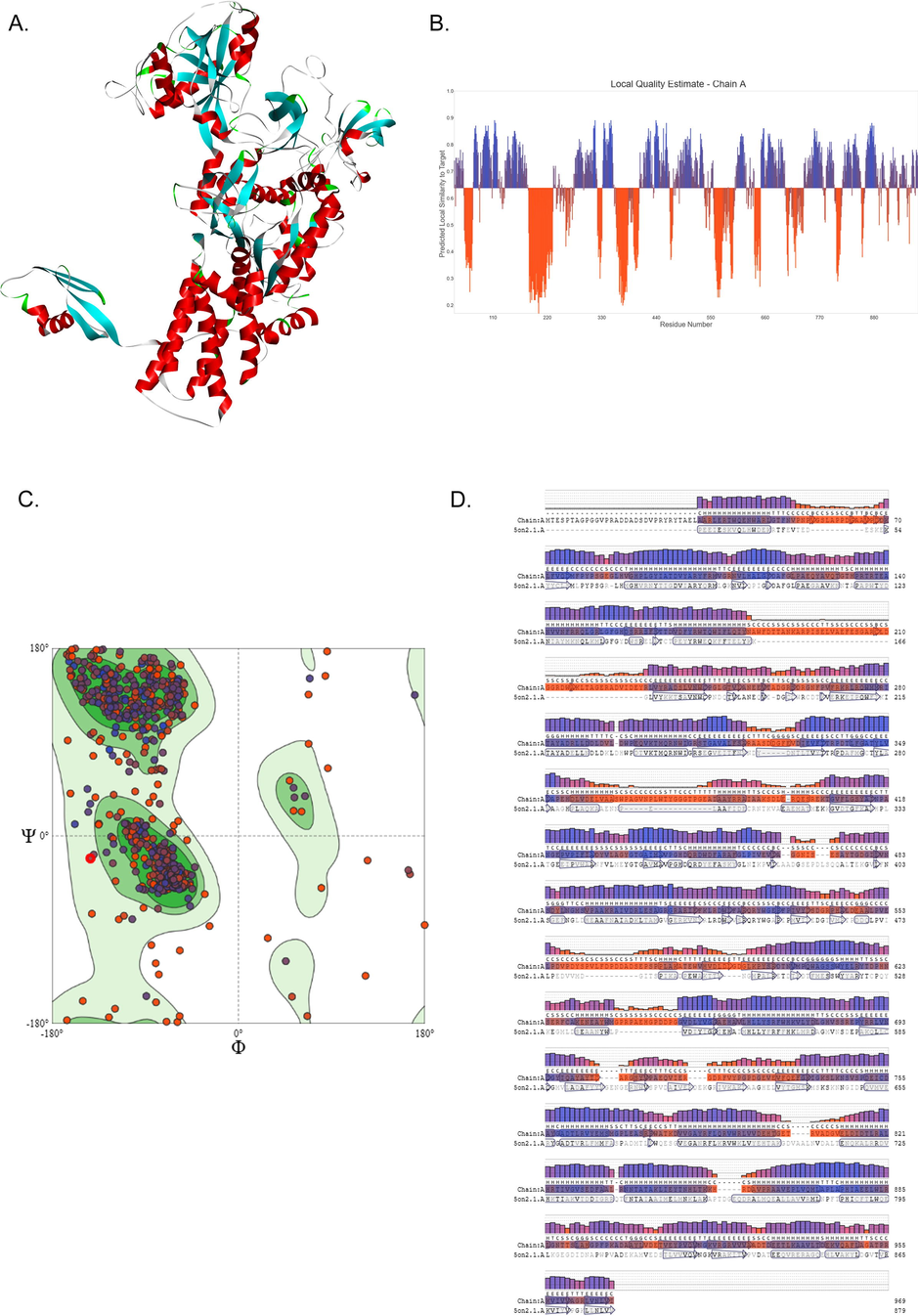
Homology modeling of the MTB-LeuRS. (A) 3D structure of MTB-LeuRS B) Residue local quality plot of MTB-LeuRS, generated from Swiss-Model server C). MTB-LeuRS structural residue distribution in Ramachandran plot D). Secondary structure elements of MTB-LeuRS via Swiss model DSSP method.
S.No
Compound Name
Zinc ID
Energy Minimize
Binding
Affinity
Interacting Amino Acids
H-bonds
1
Docetaxel
ZINC000085537053
E = 1580.93
−10.2
Phe77, His89, Asp117, His661
4
2
Reserpine
ZINC000003938746
E = 824.92
−9.8
Tyr79, Arg526, Gln694
3
3
Irinotecan
ZINC000001612996
E = 787.89
−9.7
Arg389
2
4
Toposar
ZINC000003938684
E = 9257.10
−9.7
Phe77, His661
2
5
Idarubicin
ZINC000003920266
E = 393.58
−9.7
Asp97
1
6
Piperacillin
ZINC000003913937
E = 1217.81
−9.4
Tyr79, Arg526
3
7
Methotrexate
ZINC000001529323
E = 352.96
−9.2
Arg617, Ser611
2
8
Synribo
ZINC000043450326
E = 603.92
−9.2
Gln694
1
9
Synribo
ZINC000043450324
E = 723.50
−9.1
Tyr79, Gln694
3
3.3 Virtual docking of bioactive compounds
Virtual screening of 136 bioactive compound libraries of natural bioactive compounds by molecular docking analysis has revealed that, 10 compounds showed > -9.1 highest binding affinity with MTB LeuRS protein. Based on the binding affinity cut off value of > -9.7 Kcal/Mol., we selected three bioactive compounds, i.e., Docetaxel, Reserpine, and Irinotecan, for targeting the LeuRS molecule (Table 1).
3.4 ADME-Tox of natural bioactive compounds
All the ADME-tox analyses of the three test compounds, along with the known LeuRS inhibitor (GSK656), have shown good pharmacokinetic properties owing to the bioavailability score of < 0.55. Also, the two compounds, Reserpine, and Irinotecan, show good absorption that is almost 80 % identical to the known inhibitor. The absorption percentage is a functional variable that demonstrates drug transport properties. The compounds that possess significant permeability of the plasma membrane have TPSA values < 224 Å. Out of the three compounds, Irinotecan has shown a good correlation with the known inhibitor in the physio-chemical properties. The octanol–water partition coefficient (log P = 4.95) indicates a decent absorbency, while log S = − 5.83 demonstrates good solubility in the body. Fig. 3 presents the bioavailability radars of the three test compounds in a graphical format. The drug-likeness snapshots are hexagons consisting of vertices, each indicating a bioavailability parameter of the prospective drug. Pink sections represent size (MW between 150 and 500 g/mol), lipophilicity (logP value of a reference compound between 0.7 and + 5.0), solubility (log S between 0.7 and + 5.0), polarity (TPSA between 20 and 130), flexibility (no>9 rotatable bonds), and saturation (fraction of carbons in the sp3 hybridization at no<0.25).Red distorted hexagons represent drug-likeness properties within the pink background (Table 2). This graph indicates that Irinotecan is marginally outside of the pink region on one side. In other test compounds, Reserpine and Docetaxel, an offshoot of one of the vertices to the polar regions is shown in Fig. 3. The target prediction has identified that all compounds can possibly inhibit multiple target proteins such as receptors, kinases, proteases, transporters, and structural proteins.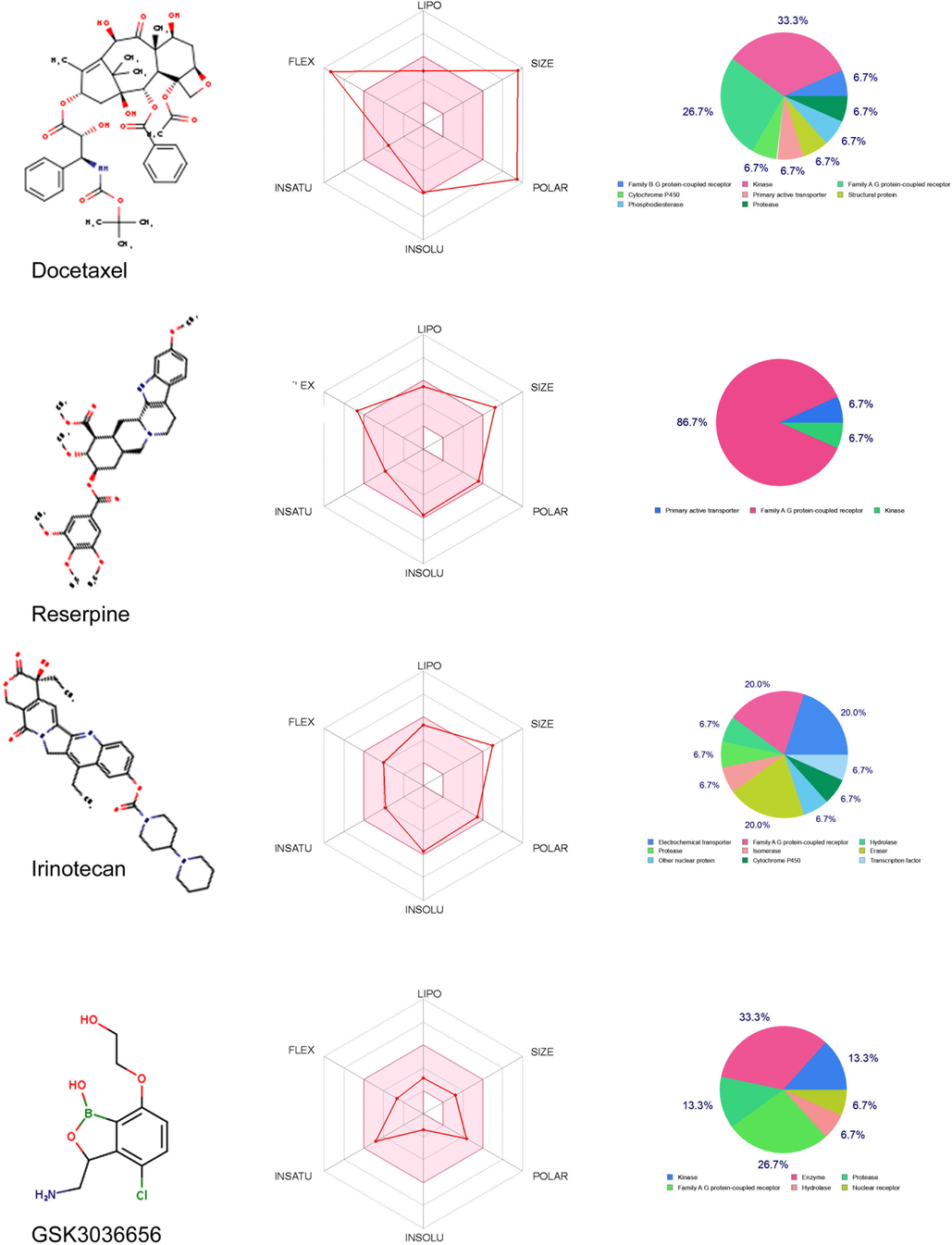
SwissADME bioavailability and Target prediction plots of Docetaxel, Reserpine, Irinotecan and GS3036656. The pink area on the bioavailability radar represents the optimal range for each property: Molecular Weight (SIZE) (between 150 and 500 gmol −1), lipophilicity (LIPO) (XLOGP3 between 0.7 + 5.0), Solubility (INSOLU) (log S > 6), polarity (POLAR) (TPSA 20–130), saturation (INSATU) (<9 rotatable bonds). The pie chart denotes the predicted molecular targets of 4 compounds.
Compound
Formula
MW
Rotatable bonds
H-bond acceptors
H2 bond donors
TPSA
iLOGP
Ali solubility class
GI absorption
CYP1A2 inhibitor
CYP2C19 inhibitor
Bioavailability Score
MRTD (mg/day)
Docetaxel
C43H53NO14
807.88
14
14
5
224.45
4.1
Poorly soluble
Low
No
No
0.17
268
Reserpine
C33H40N2O9
608.68
10
10
1
117.78
5.21
Poorly soluble
High
No
No
0.17
1.1
Irinotecan
C33H38N4O6
586.68
6
8
1
114.2
4.95
Moderately soluble
High
No
No
0.55
188
GSK3036656
C10H13BClNO4
257.48
4
5
3
84.94
0
Very soluble
High
No
No
0.55
2515
3.5 Deep docking of natural bioactive compounds with LeuRS
The three test bioactive compounds and known inhibitor GSK656 were docked with MTB LeuRS protein using the DockThor server, which is a grid-based method that implements the steady-state genetic algorithm for evaluating the docking poses using the MMFF94S force field. Deep docking results suggest that all the test compounds show the highest binding affinity value of > -7.389 Kcal/Mol with the LeuRS protein (Table 3) (Fig. 4). Interestingly, the Irinotecan is seen to be forming a strong H-bond with Glu367 of LeuRS, and its binding region is identical to that of the GSK656 inhibitor (Supplementary Figure S1). Note: Cintra/Inter molecular Energy.
Name
DockT Scorea
T. Energyb
I. Energyc
vdW Energyd
Elec. Energye
Interacting AA
H2 Bonds and Distance
Docetaxel
−7.389
257.417
−41.916
−11.14
−30.776
Glu367, Glu705
3 (<1.8 Å)
Irinotecan
−7.956
69.03
−40.392
−20.246
−20.146
Glu367
1 (2.3 Å)
Reserpine
−7.793
505.355
−37.686
−12.544
−25.142
Asn221, Trp577, Arg493, Lys708
5 (<2.4 Å)
GSK3036656
−7.798
−17.608
−34.976
−17.743
−17.233
Glu367,Lys708
2 (<1.9 Å)
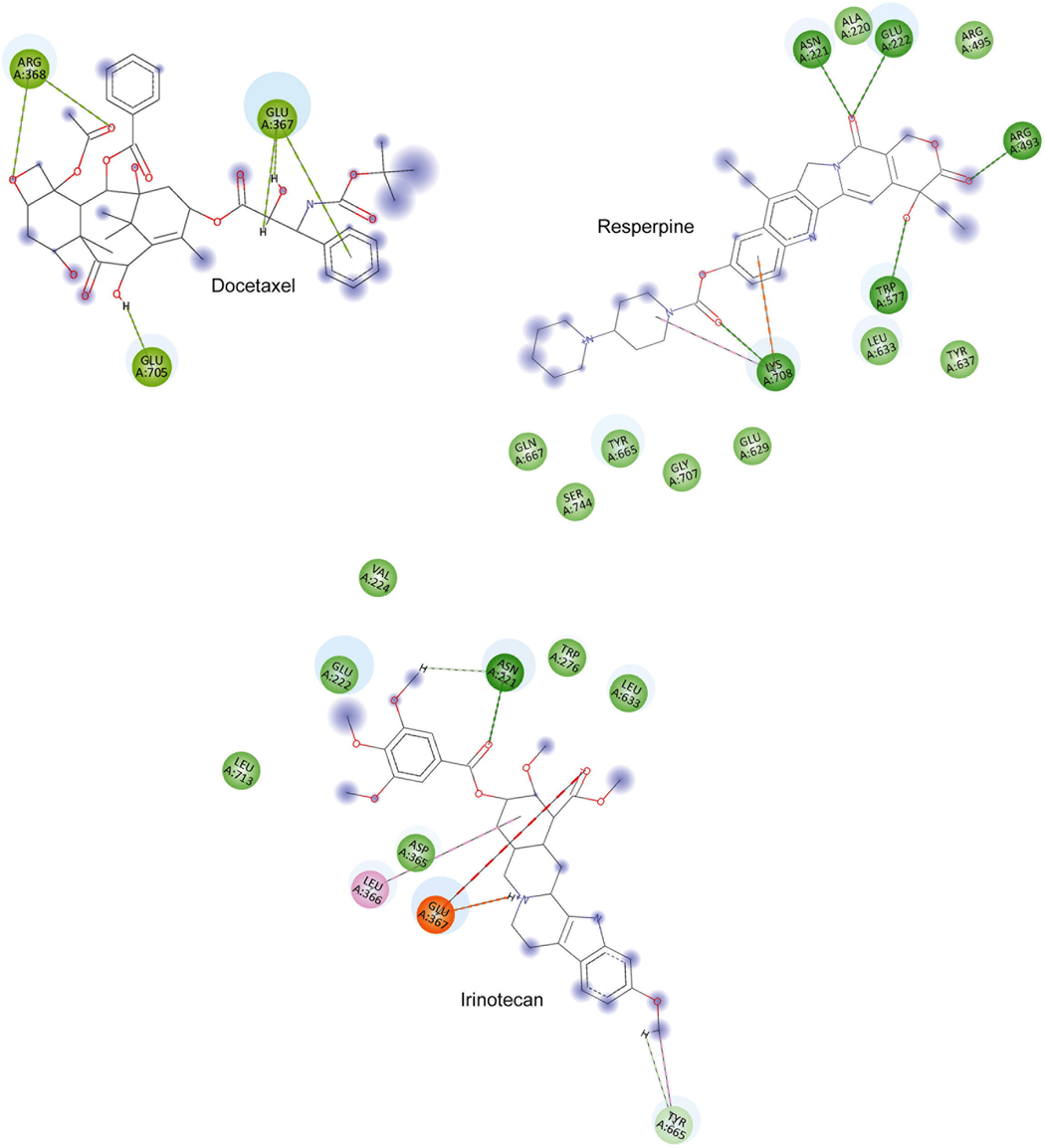
Docetaxel, Reserpine and Irinotecan in molecular docking with MTB-LeuRS. The 2D-residual interaction diagram created with Discovery Studio (according to convention, interaction types are distinguished by colored circles (residues) and dashed lines (directed to the ligand moiety).
3.6 Complex molecular dynamics simulations
The relative binding affinity and stability of the LeuRS and bioactive compound complexes were explored by studying the corresponding RMSD, RMSF, H-bond, and ligand torsion profiles predicted by MD simulation. During the 100 ns timeframe of MD simulation, after 20 ns, RMSD curves of Irinotecan-LeuRS were seen to become uniform with average values of 6.5 Å and 5.5 Å, respectively. On the other hand, during the entire simulation period, the RMSD curve for LeuRS-Docetaxel and LeuRS- Reserpine complexes converged at 60 ns with values ranging from 5.5 Å to 7.0 Å, while for the LeuRS- GSK656 complex, it was on the lower side (3Å– 6Å). Despite the RMSD curve of Irinotecan-LeuRS complex being on the top side compared to GSK656, the relative deviations were very low (2Å) throughout the RMSD curve analysis. This information shows that Irinotecan has a stable binding profile when compared to that of GSK656 against LeuRS. Both ligands showed minimum deviations while interacting with the amino acid residues in LeuRS protein. Interestingly, the flexibility of LeuRS increased to > 3 Å, while interacting with all the 3 bioactive compounds compared to GSK656 (Fig. 5). Protein-ligand interactions such as hydrogen bonds, hydrophobic interactions, ionic and water bridges were also monitored throughout the 100 ns period. Fig. 6 illustrate a stacked bar chart of the Docetaxel-LeuRS complex, revealing that G676, Y678, Q704, and P708 residues showed interaction values of > 0.75, and these residues formed > 70% of the interactions with LeuRS throughout the 100 ns simulation period. The Reserpine-LeuRS complex showed that Y665 and K708 residues formed 60% of the interactions throughout the simulation period. However, the Irinotecan compounds showed the highest interaction with five amino acid residues (N221, E222, L490, W517, Y637) and these residues (>65%) were shown to constantly interact with protein throughout the simulation period (Fig. 6) The GSK656 showed constant interaction of three residues (L208, W276 and R487) with the LeuRS protein over a 100 ns simulation period. Flexibility in the proteins might have allowed Irinotecan to interact with LeuRS protein more strongly compared to the GSK656 compound. Fig. 6 illustrate the ligand torsion plot of rotatable bonds in Docetaxel, Irinotecan, Reserpine, and GSK656 at a 100 ns simulation period. The Irinotecan shows the lowest of seven rotatable bonds, and the bonds' rotations are constantly seen throughout the simulation period. Other test compounds like Docetaxel, Reserpine, and GSK656 demonstrated 18, 9, and 15 degrees of freedom, respectively (Supplementary Figure S2). However, these bonds were uncertain during the simulation period. Overall, our results showed the conformational diversity of Docetaxel, Reserpine, and Irinotecan test compounds, but Irinotecan has demonstrated a superior binding affinity and stability in forming molecular complexes with LeuRS protein.Fig. 7..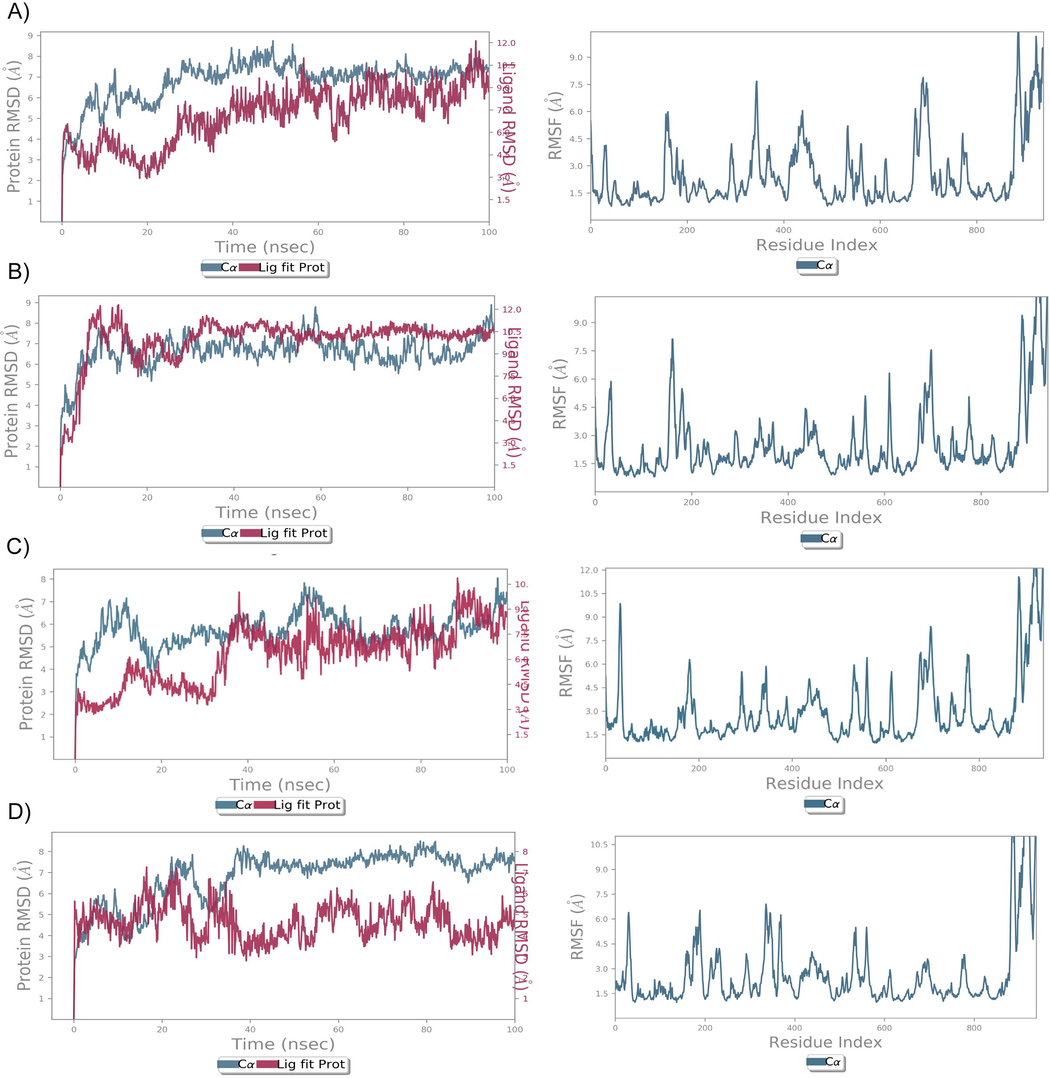
RMSD and RMSF analysis of MTB-LeuRS lead compounds using MD simulation. The RMSD plot shows the C α backbone (blue) of MTB-LeuS and lead compound fluctuations (red) at 100 ns. The RMF plot shows the fluctuation of the MtbLeuS protein while interacting with lead compounds at a 100 ns simulation period.
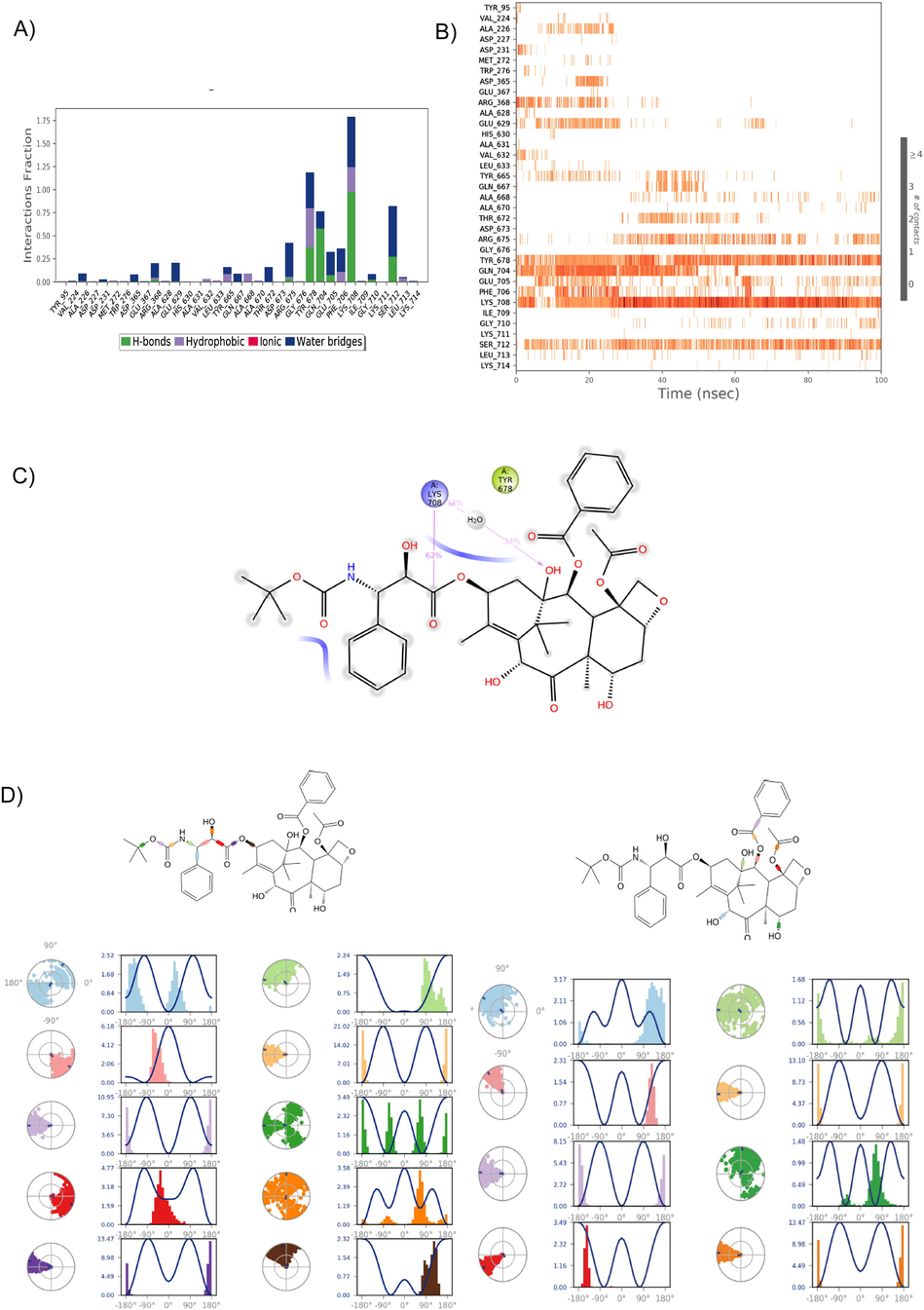
Analysis of the Docetaxel H-bond and torsional angel over a 100 ns simulation period. A) The 35 ionic interactions formed during the simulation by natural Docetaxel compound with the protein. B) Heatmap of the LueS-Docetaxel H-bonds over 100 ns. C) the 2D structure of Docetaxel stable interaction with protein during simulation. D) The rotational energy barrier as a function of bond rotation angle is typically represented in simulation graphs. The radial plots denote the 18 torsional angels’ conformation space of Docetaxel during the simulation period, and the histogram represents the torsional energy profile of the LeuRS- Docetaxel complex during the 100 ns simulation period.
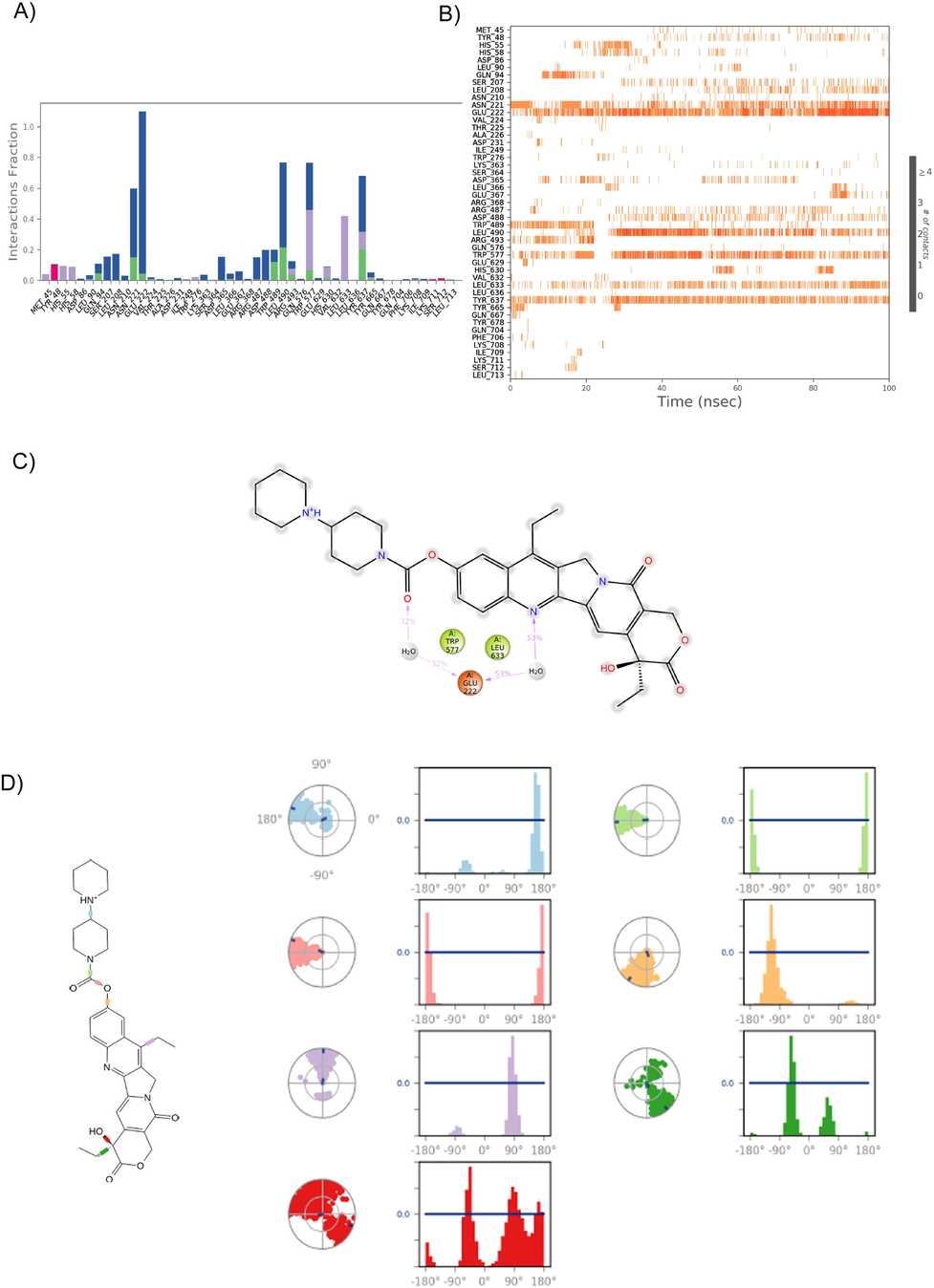
Analysis of the LeuRS-Irinotecan H-bond and torsion angle over a 100 ns simulation period. A) The 49 ionic interactions formed during the simulation by natural Irinotecan compound with the LeuRS protein. B) Heatmap of the LueRS- Irinotecan H-bonds over 100 ns. C) The 2D structure of Irinotecan shows stable interaction with the protein during the entire simulation. D) The radial plots denote the 7 torsion angels’ conformation space of Irinotecan during the simulation period, and the histogram represents the torsional energy profile of the LeuRS- Irinotecan complex during the 100 ns simulation period.
4 Discussion
Drug-resistant MTB is posing a significant challenge in reducing global TB infection-related morbidity and mortality. So, there has been a continuous search for new drugs to improve the treatment of TB infection and patient recovery. Those drugs can efficiently target DNA replication, energy metabolism, folate metabolism, RNA synthesis, and bacterial cell wall synthesis of MTB (Bahuguna and Rawat 2020). Natural compounds extracted from multicellular or unicellular organisms, either in crude mixtures or pure compounds, present a potential opportunity to discover new therapeutic candidates due to their enormous chemical diversity and biological safety (Rizwan et al., 2014) (Fatima et al., 2019). In MTB, aaRSs mediate the transfer of a specific amino acid to cognate tRNA, forming a charged aminoacyly-tRNA (aaRS) (Ndagi et al., 2021). Because of their role in protein translation, these enzymes are thought to represent new therapeutic targets for antimicrobial agents (Bouz and Zitko 2021). Computational molecular docking approaches have proven to be an efficient method for discovering novel compounds in drug discovery (Shaik et al., 2019). Inhibitors of aaRS have the potential to target multiple regions, including the editing domain, ATP binding, tRNA recognition, amino acid binding, and the allosteric sites. Mupirocin, a clinically approved aaRS, binds to the catalytic domain of the bacterial isoleucyl-tRNA synthetase and does the competitive inhibition of the isoleucine- adenylate complex.
As mentioned above, the present study has explored the anti-microbial activity of 136 FDA approved natural products (secondary metabolites) as a potential alternate to conventional anti-MTB drugs. Out of all the tested products, molecular docking results predict that Docetaxel, Irinotecan, Reserpine compounds have the highest binding affinity with the MTB-aaRS. In recent years, Deep Learning (DL) approaches have been proposed to replace classical scoring systems in the context of molecular docking (Gentile et al., 2020). DL applications have become popular because they can predict the properties of drug compounds, design new drugs, predict reactions, and make new compounds (Jiménez-Luna et al., 2020). Findings from our Deep Docking method have confirmed that Irinotecan has the highest binding affinity with aaRS and is comparable to the GSK656 compound.
Irinotecan [(S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo1H-pyrano[3′,4′:6,7]-indolizino[1,2-b]quinolin-9-yl-[1,4′bipiperidine]-1′-carboxylate] belongs to pyranoindolizinoquinolines. The hydrochloride salt trihydrate of Irinotecan, in combination with fluorouracil and leucovorin, are used to treat patients with metastatic adenocarcinoma of the pancreas treated with gemcitabine-based therapy (Fujita et al., 2015). Historically, Irinotecan is known to inhibit the action of the topoisomerase I enzyme, which regulates DNA supercoiling and topology, and enables nuclear activities such as DNA replication, transcription, and repair. Irinotecan prevents re-ligation of the DNA strand, binds to the topoisomerase I-DNA complex, and forms a ternary complex that restricts the replication fork from moving (de Man et al., 2018). Correspondingly, double-stranded breaks and replication arrest in the DNA can occur. As a result, apoptosis (programmed cell death) occurs due to the inefficient repair of the developed DNA damage (Bailly 2019).
The interfering role of Irinotecan on the aaRS activity in MTB infections is not yet well explored. The LeuRS of MTB belongs to the Class I category of aaRS, which has a distinct structure in the catalytic domain (Fujita et al., 2015). It is made up of three functional domains: the tRNA synthetase class I (M) domain (71–192), the Leucyl tRNA synthetase domain 2 (305-504aa), and the anti-codon binding domain (814–938aa). Irinotecan inhibits LeuRS activity by binding to L490 amino acid residues in domain 2 via the Oxaborole tRNA-trapping (OBORT) mechanism, where it forms a covalent adduct that links Oxaborole and the terminal ribose of the tRNA. It is likely that when aaRS is inhibited in bacteria, it leads to a buildup of uncharged tRNA. That will lead to the induction of the relA gene, exerting a negative feedback on RNA polymerase. The relA gene is accountable for guanosine tetrapeptides and pentapeptide biosynthesis. High energy activities like macromolecule biosynthesis will be downregulated. Ultimately, the bacterial growth will be inhibited and the pathogen’s virulence will be attenuated in vivo (Hurdle et al., 2005). In drug-likeness assessment, Irinotecan has shown better physiological and pharmacological properties in comparison to Lipinski’s principles. Interestingly, Irinotecan showed a good absorption score (0.55) as equivalent to GSK656. The absorption property defines drug transport properties. Besides, Irinotecan has also shown ideal molecular bonding characteristics like rotatable bonds (6), H-bond acceptors (8), bond donors (1), and iLogP (4.95).
One of the anti-tuberculosis drugs under consideration is GSK070, a protein synthesis inhibitor. It inhibits MTB LeuRS with an MIC90 = 0.08 μM (GSK656) and is currently undergoing preclinical studies (Shetye et al., 2020). Gudzera et al. (2016) discovered MTB-LeuRS inhibitors derived from 5-phenylamino-2H–[1,2,4] triazin-3-one (Gudzera et al., 2016).The inhibitory activity of those inhibitors against pathogenic MTB LeuRS is 10-fold more selective than against the human enzyme (Gudzera et al., 2016). In 2016, (Palencia et al., 2016) synthesized a 3- aminomethyl-4-halogenbenzoxaborole exhibiting potent in vitro MTB-LeuRS inhibition. It can also kill the H37Rv strain of MTB in vitro. Further structural modifications to improve pharmacokinetics properties and oral bioavailability resulted in the development of GSK656 (GSK3036656), which has submicromolar MTB-LeuRS inhibitory activity with excellent in vitro growth inhibition activity, and an effective anti-MTB in vivo in a mouse MTB model (ED99 = 0.4 mg/kg) (Li et al., 2017). Similar to first line anti-MTB drugs, GSK656 has an ideal molecular weight (257.48 g/mol), low PSA (53.71 A2), and clogD7.4 (-0.4) values. GSK656 is the first aaRS inhibitor in phase IIa clinical trials to be used systemically against MTB (ClinicalTrials.gov Identifier: NCT03557281) (Tenero et al., 2019). It should be noted that the anti-Leu-RS activity results of Irinotecan obtained through computational molecular screening can only show the effective binding and inhibitor stability. However, further laboratory studies are vital in validating and comparing the biological activities, toxicities, and therapeutic indexes of GSK656 and Irinotecan to establish their anti MTB inhibitors.
In summary, we screened 136 FDA approved natural secondary metabolite compounds against the M.tuberculosis infection. The best inhibitory compounds (Docetaxel, Reserpine, and Irinotecan) demonstrated robust binding with anti-Leu RS. Deep molecular docking has shown that Irinotecan interacts with leucyl-tRNA synthetase through an oxaborole tRNA-trapping (OBORT) mechanism, downregulating high energy demanding biological mechanisms including biosynthesis of nucleic acids, proteins, and lipids, eventually inhibiting the bacterial growth and attenuating the pathogen’s virulence. The multidirectional computational screening methods used in this study once again establish the potential of knowledge-based screening approaches in the repurposing of naturally occurring natural compounds for treating TB infection. This study also recommends implementing highly efficient computational protocols for discovering novel anti-MTB compounds. However, we sincerely admit that our findings are unable to elucidate complex drug metabolism and toxicity reactions occurring inside the human body. Our findings pave the way to testing novel MTB proteasome inhibitors in experimental conditions in the quest of finding novel inhibitors to combat pathogenic TB infections.
Acknowledgements
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number IFPRC-063-247-2020 and King Abdulaziz University, DSR, Jeddah, Saudi Arabia. The authors also extend their sincere thanks to Saudi Ajal for Health Services for their help in data analysis and presentation of our study results.
Data Availability Statement
All datasets analyzed for this study are included in the article/Supplementary Material.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Pulmonary Tuberculosis Versus Recurrent Chemotherapy-Induced Pneumonitis: A Clinical Dilemma. Cureus.. 2017;9(10):e1742
- [CrossRef] [Google Scholar]
- Identification of New Mycobacterium tuberculosis Proteasome Inhibitors Using a Knowledge-Based Computational Screening Approach. Molecules (Basel. Switzerland). 2021;26(8):2326.
- [Google Scholar]
- Transcriptome-Based Molecular Networks Uncovered Interplay Between Druggable Genes of CD8(+) T Cells and Changes in Immune Cell Landscape in Patients With Pulmonary Tuberculosis. Front Med (Lausanne).. 2021;8:812857
- [CrossRef] [Google Scholar]
- Computational Methods and Tools in Antimicrobial Peptide Research. J Chem Inf Model.. 2021;61(7):3172-3196.
- [Google Scholar]
- An overview of new antitubercular drugs, drug candidates, and their targets. Medicinal research reviews.. 2020;40(1):263-292.
- [CrossRef] [Google Scholar]
- Irinotecan: 25 years of cancer treatment. Pharmacol Res.. 2019;148:104398
- [CrossRef] [Google Scholar]
- Bioinformatics Accelerates the Major Tetrad: A Real Boost for the Pharmaceutical Industry. Int J Mol Sci.. 2021;22(12):6184.
- [Google Scholar]
- Evaluation of a Molecular Test for Detection of Mycobacterium tuberculosis Isolates Resistant to Rifampicin and Isoniazid. Clinical laboratory.. 2022;68(03/2022)
- [CrossRef] [Google Scholar]
- Inhibitors of aminoacyl-tRNA synthetases as antimycobacterial compounds: An up-to-date review. Bioorganic Chemistry.. 2021;110
- [CrossRef] [Google Scholar]
- Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics. Clinical pharmacokinetics.. 2018;57(10):1229-1254.
- [Google Scholar]
- L-asparaginase produced from soil isolates of Pseudomonas aeruginosa shows potent anti-cancer activity on HeLa cells. Saudi J Biol Sci.. 2019;26(6):1146-1153.
- [CrossRef] [Google Scholar]
- Irinotecan, a key chemotherapeutic drug for metastatic colorectal cancer. World journal of gastroenterology.. 2015;21(43):12234-12248.
- [CrossRef] [Google Scholar]
- Aminoacyl-tRNA synthetase inhibitors as antimicrobial agents: a patent review from 2006 till present. Expert opinion on therapeutic patents.. 2012;22(12):1453-1465.
- [CrossRef] [Google Scholar]
- ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res.. 2003;31(13):3784-3788.
- [CrossRef] [Google Scholar]
- Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Central Science.. 2020;6(6):939-949.
- [Google Scholar]
- Identification of Mycobacterium tuberculosis leucyl-tRNA synthetase (LeuRS) inhibitors among the derivatives of 5-phenylamino-2H-[1,2,4]triazin-3-one. J Enzyme Inhib Med Chem.. 2016;31(sup2):201-207.
- [Google Scholar]
- New machine learning and physics-based scoring functions for drug discovery. Scientific reports.. 2021;11(1)
- [CrossRef] [Google Scholar]
- SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis.. 1997;18(15):2714-2723.
- [CrossRef] [Google Scholar]
- Tuberculosis. Annals of tropical medicine and parasitology.. 2006;100(5–6):415-431.
- [CrossRef] [Google Scholar]
- Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrobial agents and chemotherapy.. 2005;49(12):4821-4833.
- [CrossRef] [Google Scholar]
- A Deep-Learning Approach toward Rational Molecular Docking Protocol Selection. Molecules (Basel, Switzerland). 2020;25(11):2487.
- [Google Scholar]
- Discovery of a Potent and Specific M. tuberculosis Leucyl-tRNA Synthetase Inhibitor: (S)-3-(Aminomethyl)-4-chloro-7-(2-hydroxyethoxy)benzo[c][1,2]oxaborol-1(3H)-ol (GSK656) J Med Chem.. 2017;60(19):8011-8026.
- [Google Scholar]
- High throughput virtual screening reveals SARS-CoV-2 multi-target binding natural compounds to lead instant therapy for COVID-19 treatment. International journal of biological macromolecules.. 2020;160:1-17.
- [Google Scholar]
- A consequence of drug targeting of aminoacyl-tRNA synthetases in Mycobacteriumtuberculosis. Chemical biology & drug design.. 2021;98(3):421-434.
- [CrossRef] [Google Scholar]
- Discovery of Novel Oral Protein Synthesis Inhibitors of Mycobacterium tuberculosis That Target Leucyl-tRNA Synthetase. Antimicrobial agents and chemotherapy.. 2016;60(10):6271-6280.
- [Google Scholar]
- Screening of natural compounds that targets glutamate racemase of Mycobacterium tuberculosis reveals the anti-tubercular potential of flavonoids. Scientific reports.. 2020;10(1)
- [CrossRef] [Google Scholar]
- Epidemiology and pattern of antibiotic resistance in Helicobacter pylori: scenario from Saudi Arabia. Saudi J Gastroenterol.. 2014;20(4):212-218.
- [CrossRef] [Google Scholar]
- Highly Flexible Ligand Docking: Benchmarking of the DockThor Program on the LEADS-PEP Protein-Peptide Data Set. Journal of chemical information and modeling.. 2020;60(2):667-683.
- [Google Scholar]
- Molecular designing, virtual screening and docking study of novel curcumin analogue as mutation (S769L and K846R) selective inhibitor for EGFR. Saudi J. Biol. Sci.. 2019;26(3):439-448.
- [Google Scholar]
- In silico methods and tools for drug discovery. Comput Biol Med.. 2021;137
- [CrossRef] [Google Scholar]
- New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Translational Research.. 2020;220:68-97.
- [CrossRef] [Google Scholar]
- Clustal Omega for making accurate alignments of many protein sequences. Protein Sci.. 2018;27(1):135-145.
- [CrossRef] [Google Scholar]
- The Diagnosis and Treatment of Tuberculosis. Deutsches Arzteblatt international.. 2019;116(43):729-735.
- [CrossRef] [Google Scholar]
- First-Time-in-Human Study and Prediction of Early Bactericidal Activity for GSK3036656, a Potent Leucyl-tRNA Synthetase Inhibitor for Tuberculosis Treatment. Antimicrobial agents and chemotherapy.. 2019;63(8)
- [CrossRef] [Google Scholar]
- Enniatin A1, A Natural Compound with Bactericidal Activity against Mycobacterium tuberculosis In Vitro. Molecules (Basel. Switzerland). 2019;25(1):38.
- [Google Scholar]
- SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic acids research.. 2018;46(W1):W296-W303.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102032.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




