

Hydrogen bonds interactions in biuret-water clusters: FTIR, X-ray diffraction, AIM, DFT, RDG, ELF, NLO analysis
⁎Corresponding authors at: Siberian Federal University, pr. Svobodny, 79, Krasnoyarsk 660041, Russia. leo_lion_leo@mail.ru (Aleksandr S. Kazachenko), kazachenko.as@icct.krasn.ru (Aleksandr S. Kazachenko), omar@ksu.edu.sa (Omar Al-Dossary)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
-
Biuret-(H2O)n (n = 1–10) clusters was calculated using DFT method.
-
Intermolecular interactions of Biuret with water discovered using AIM, ELF and RDG.
-
Biuret-(H2O) was investigated by FTIR and XRD.
-
Chemical reactivity was simulated via HOMO-LUMO, MEP.
Abstract
In this work, we studied intermolecular aqueous clusters of biuret, an important urea derivative. FTIR showed an increase in the intensity of absorption bands when water molecules are introduced into the biuret. X-ray diffraction analysis showed that the introduction of water molecules into the biuret structure significantly increases the intensity of the bands on the diffraction patterns in the range from 14 to 65 2˚⊖. Aqueous biuret clusters have also been studied in the gas phase by theoretical methods: density functional theory and Atoms in Molecules (AIM) using the DFT level B3LYP/6–31 + G (d, p). The nature of molecular interactions between water and biuret through hydrogen bonds was also investigated using the electron localization function (ELF) and non-covalent reduced density gradient (NC-RDG). The thermodynamic and Non-linear optical properties of biuret-water clusters were performed also.
Keywords
Biuret
Clusters
Water
DFT
QTAIM
RDG

1 Introduction
Urea is recognized as the first artificially synthesized organic molecule (Yokoya et al., 2021). This discovery provided an opportunity for the development of the field of synthetic organic chemistry, and the synthesis of very complex and / or strained molecules became possible (Nicolaou et al., 2005).
Urea derivatives using in engineering and chemical-catalytic processes (Bernhard et al., 2012), due to the increased use of urea as a reducing agent for the selective catalytic reduction of nitrogen oxides in the after treatment of diesel engine exhaust gases (Koebel et al., 2000), and new materials (Dong et al., 2011), due to recent studies of urea derivatives as precursors of carbon nitride materials (Liu et al., 2011).
Biuret (carbamylurea) is a chemical compound with the chemical formula [H2NC(O)]2NH, formed as a result of the condensation of two equivalents of urea (Hughes et al., 1961). Biuret is also used as a non-protein nitrogen source in ruminant feed (Kunkle et al., 2013), where it is converted to protein by intestinal microorganisms. (Oltjen et al., 1969) It is less preferred than urea due to its higher cost and lower digestibility, but the latter characteristic also slows down its digestion and thus reduces the risk of ammonia poisoning (Fonnesbeck et al., 1975).
In recent studies (Kazachenko et al., 2021), biuret has been investigated as a catalyst for the sulfation of polysaccharides with sulfamic acid, as an alternative to urea.
In aqueous solutions, the phenomenon of clustering of some organic and inorganic molecules is encountered, which has recently been actively studied, both theoretically and experimentally (Akman et al., 2020; Kazachenko et al., 2021). Water clusters are discrete hydrogen-bonded assemblies or clusters of molecules in water (Ludwig, 2001).
Water clusters of various classes of substances have been actively studied in recent years. Thus, water clusters of ozone in various water/ozone ratios were studied in (Yadav et al., 2017), and the maximum binding capacity of water clusters of various sizes with respect to ozone molecules was shown. In (Wang et al., 2015), the interaction of new polymers with a water cluster was studied. Water clusters in crystalline hydrates were studied in (Supriya and Das, 2003). In (Akman et al., 2020), aqueous thiourea clusters were studied by both experimental and theoretical methods. A similar work was presented for ammonium sulfamate (Kazachenko et al., 2021) and sulfamic acid (Kazachenko et al., 2022). It is shown that despite the similarity of the structure of ammonium sulfamate and sulfamic acid, their clustering with water occurs differently, which is affected by the presence of the ammonium cation in the sulfamate. The influence of urea on water, including in clusters, was actively studied by experimental and theoretical methods in (Lovrinčević et al., 2020; Carr et al., 2013) and others.
Based on all of the above, the importance of studying water clusters of various substances is obvious. This work is a systematic continuation of the previously started topic of the study of water clusters of some acids, salts and urea derivatives. In this regard, the combined use of experimental and theoretical methods allows us to obtain the most reliable picture.
In this work, we studied aqueous clusters of biuret by FTIR, X-ray diffraction, QTAIM, DFT, RDG, ELF, NLO.
2 Experimental
2.1 FTIR and XRD analysis
Samples of the Biuret-water cluster were obtained by dissolving biuret in distilled water at 70˚C, followed by precipitation of the cluster at room temperature.
Shimadzu IR Tracer-100 spectrometer (Japan) within the wavelength range of 400–4000 cm−1 was used for registration FTIR spectra of biuret and biuret-water cluster. OPUS program (version 5.0) was used for analysis spectral data. Solid samples for analysis were prepared in the form of pills in a KBr matrix (2 mg sample/1000 mg KBr).
DRON-3 X-ray diffractometer (CuKα monochromatized radiation (λ = 0.154 nm), voltage 30 kV, current 25 mA) was used for X-ray diffraction (XRD) phase analysis. The scanning step is 0.02 deg; intervals for 1 s per data point. The measurement was carried out in the interval of the Bragg angles 2Θ from 5.00 to 70.00 deg.
2.2 Computational details
Molecular stability of each biuret-Water cluster has been investigated by calculating the SCF energy of these different structures. The calculation was performed using B3LYP/6–31 + G(d,p) method via Gaussian 09 software (Frisch et al., 2013). The output of various theories shows similar results but DFT theory results was more accurate compared with experimental data, so we are mentioning the results of DFT theory only. DFT theory is an effective tool for performing chemical calculations of compounds, better representation of polar bonds and accretion of lower basis set can also be done (Petersson and Al‐Laham, 1991; Petersson et al., 1988). The initial configuration searches for the biuret-water clusters were based on two steps. Firstly, the isomer component of the ABCluster software (Zhang and Dolg, 2015) was used to generate the initial structures of biuret water clusters. Both 2D and 3D initial guesses have been considered to make sure we get the true global minima of each structure. Secondly, Each of the generated structure was then fully optimized by DFT using B3LYP functional with the polarized basis sets (6–31 + G(d,p)) by Gaussian 09 package to obtain the respective total energy and locate the most stable geometry for each cluster. The optimized geometry of the considered clusters was confirmed to be located at the local true minima on the potential energy surface, as indicated by the lack of imaginary frequencies in the vibrational mode calculation. In addition, molecular electrostatic potential (MEP) surface was plotted for the considered clusters by using B3LYP/6–31 + G(d,p) level of theory. The MEP and all the output files were visualized by means of GaussView software (Dennington et al., 2010). The important parameters was the energy gap which considered the differences between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO). This gap gives the stability and tells about hard and soft nature of cluster. Then, topological analysis was carried out to discover the non covalent interactions. The wavefunctions obtained at the B3LYP/6–31 + G(d,p) level were used to determine the electron density ρc and the Laplacien electron density (∇2ρc) at the bond critical points (BCPs). The AIM, RDG and ELF visual representations were generated throughout Multiwfn 3.8 program (Lu and Chen, 2012).
3 Results and discussions
3.1 FTIR and XRD analysis
The initial biuret and its water clusters were analyzed by FTIR spectroscopy (Fig. 1).
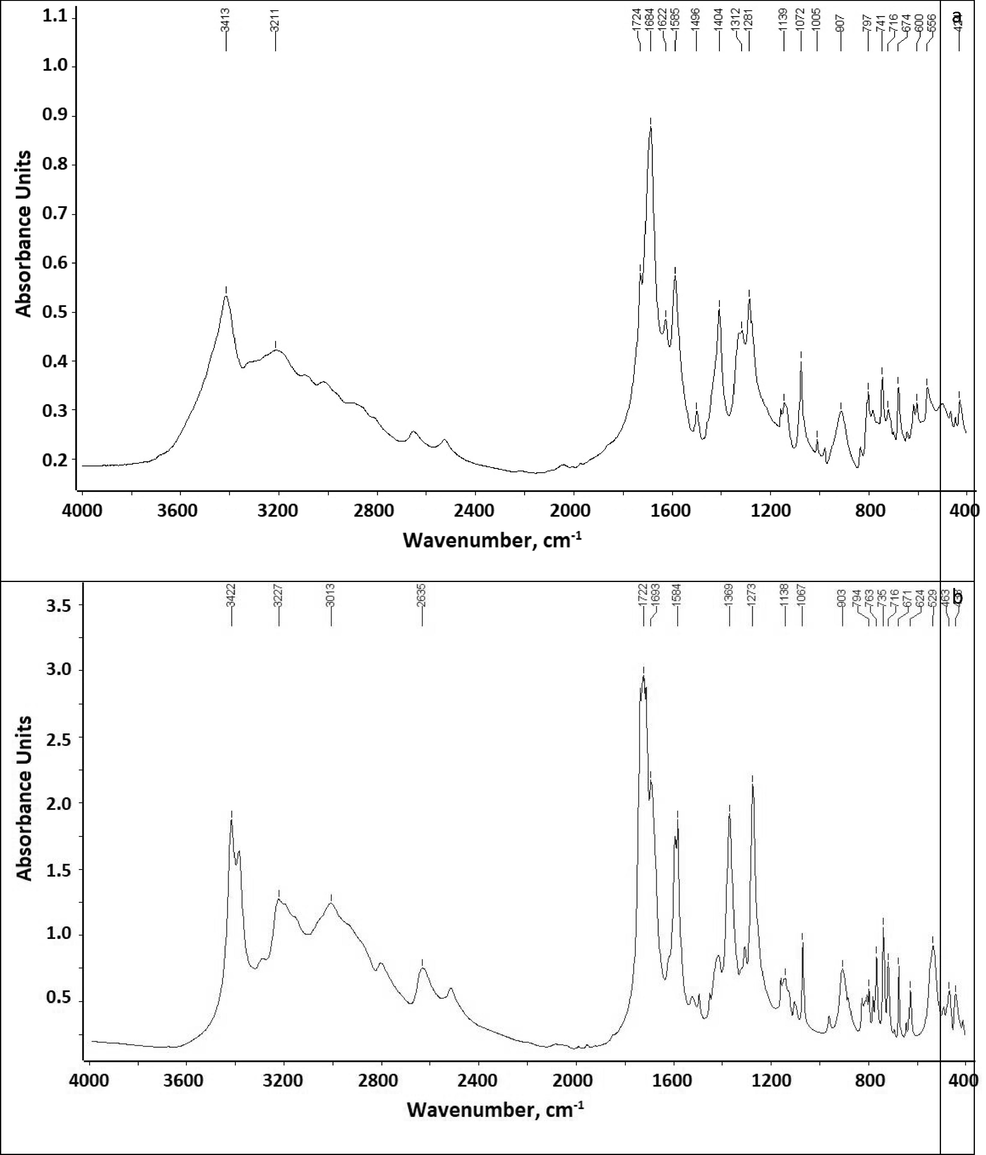
- FTIR spectra: a - Biuret, b - Biuret water cluster.
Fig. 1 shows the spectra of biuret and water biuret cluster. Basic infrared spectral data of biuret and its water cluster are presented in Table 1.
| Wavenumber, cm−1 | Vibration type | |
|---|---|---|
| Biuret | Biuret-water | |
| 3413 | 3422 | νas (NH2) + ν(H2O)/νas(NH2) |
| 3211 | 3227 | νas(NH2) |
| 1724 | 1722 | ν(C⚌O) |
| 1585 | 1584 | δ(NH2) |
| 1496, 1404 | 1369 | ν(C—N) + ν(C − NH2) |
| 1281 | 1273 | δ(N—H) |
| 1139 | 1138 | ν(C—N) + δ(N—H) |
| 1072 | 1067 | ν(C—N) |
| 907 | 903 | ν(C—N) + ν(C − NH2) |
| 797 | 794 | δ(C − NH2) |
| 716 | 716 | δ(C⚌O) |
In the spectra of biuret and its water cluster, two bands are observed (3200–3420 cm -1) in the region of stretching vibrations of N—H. The former is alternatively referred to as bridging hydroxide, while the band of water hydration appears at about 3420 cm –1 (Wang et al., 2016). Bending N — H vibrations are observed at 1585 cm – 1 with significant intensity. It is reported that the frequencies of carbonyl stretching vibrations in compounds containing the CO-NH-CO group give two bands (Uno and Machida, 1962); the peak of the asymmetric stretching vibration appears above 1720 cm -1, and the peak of the symmetric vibration appears at about 1680–1695 cm -1 (Hajji et al., 2021; Hajji et al., 2021).
When coordination occurs, it determines the degree of electron delocalization in the N-CO-N system; thus, coordination through the oxygen atom will lead to a decrease in the nature of the double bond of the C⚌O bonds and will lead to a shift in the carbonyl extension mode to the region of lower frequencies (Udupa and Indira, 1975).
The peaks of stretching vibrations of C⚌O bonds are found at 1684, 1693, 1622 cm -1, respectively.
The peaks of bending vibrations of C⚌O bonds, found in the region of 624–671 cm -1, may indicate some coordination between water models and oxygen atoms in biuret.
In addition, for the aqueous biuret cluster, there is a significant increase in the signal intensity of almost all absorption bands in comparison with the initial biuret, which is consistent with the data (Akman et al., 2020; Kazachenko et al., 2021). The peak of intermolecular hydrogen bonds of OH groups is observed at 3500–3200 cm−1, and its intensity increases by about 5 times when passing from biuret to an aqueous biuret cluster.
According to Fig. 2, the background of the X-ray diffraction patterns is low, and the diffraction intensity is high, which indicates that the complex has a fine-crystalline state. The X-ray diffraction pattern of the original biuret has high intensity bands at 11,15,17,19,21,23,29,39 2⊖ (deg), which corresponds to the data given in (Udachin et al., 2011).
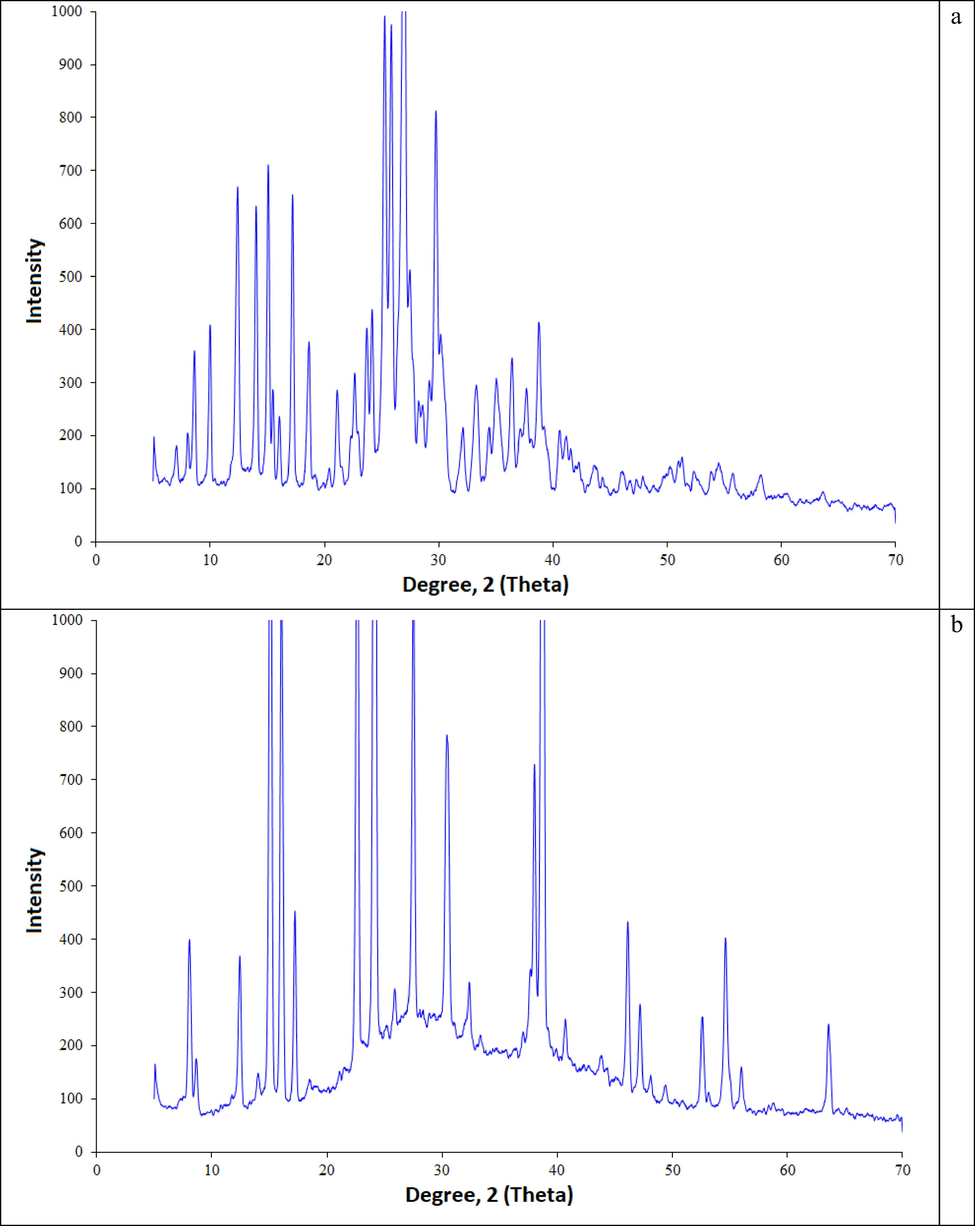
- XRD data: a - Biuret, b - Biuret water cluster.
The inclusion of water molecules in the biuret crystal leads to a significant increase in the intensity of most peaks in the range from 14 to 65 2⊖ (deg). A similar phenomenon was observed for thiourea (Akman et al., 2020), ammonium sulfamate (Kazachenko et al., 2021), which can also be explained by the specific effect of water molecules on the crystal structure of compounds (Volz and Clayden, 2011).
3.2 Structural analysis of Biuret -(H2O)n clusters (n = 1–10)
Urea and its derivatives, which have both acceptor and donor parts of the hydrogen bond, are an ideal structure for the formation of various intermolecular clusters (Yokoya et al., 2021), including water clusters (Akman et al., 2020). Recently, urea derivatives have attracted particular attention due to the fact that they contain important functional groups (Hammami et al., 2015).
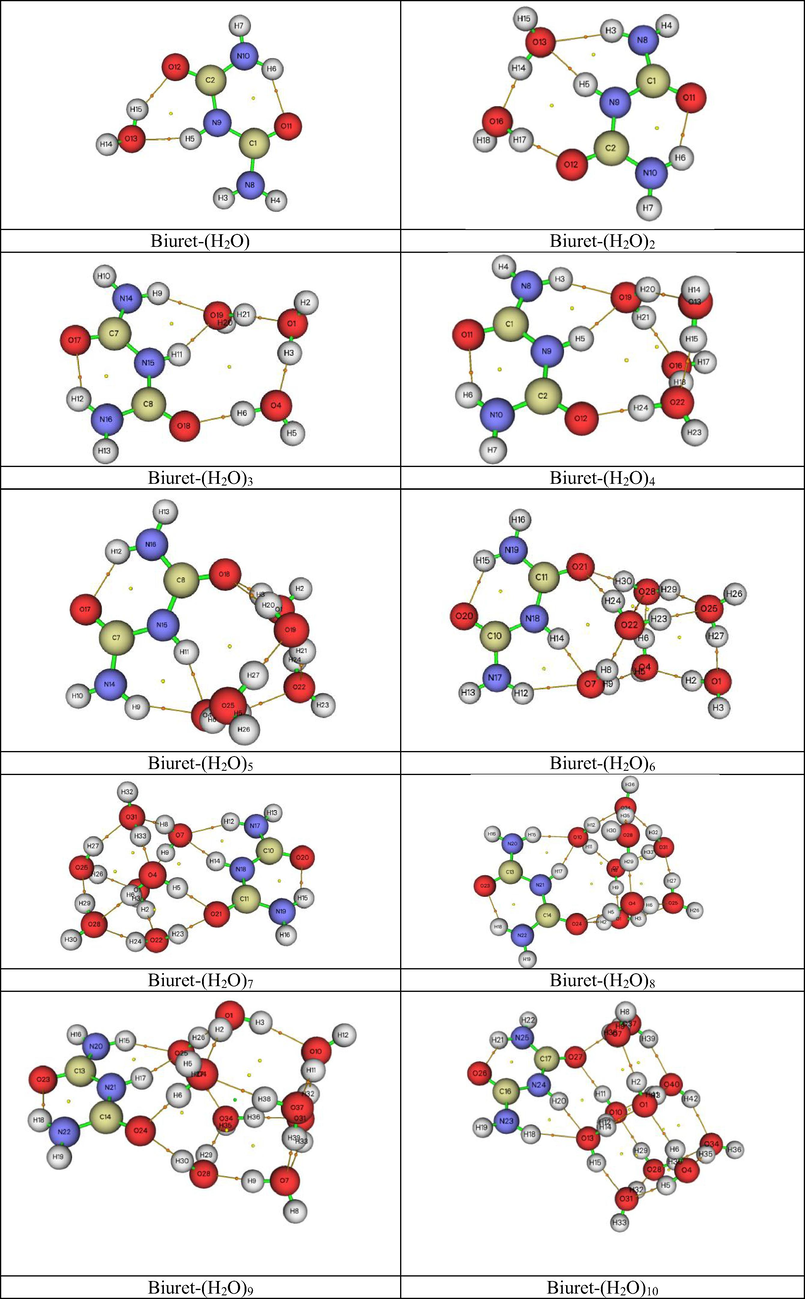
To determine the interactions of the biuret molecule in water, the most stable clusters of biuret with water were identified. The most stable clusters are possible, such as Biuret -(H2O), Biuret -(H2O)2, Biuret -(H2O)3, Biuret -(H2O)4, Biuret -(H2O)5, Biuret -(H2O)6, Biuret -(H2O)7, Biuret -(H2O)8, Biuret-(H2O)9, Biuret-(H2O)10, have been optimized at the level of B3LYP theory with a 6–31 + G (d, p) basis set and are represented by atomic numbers in Fig. 3 (a-l).
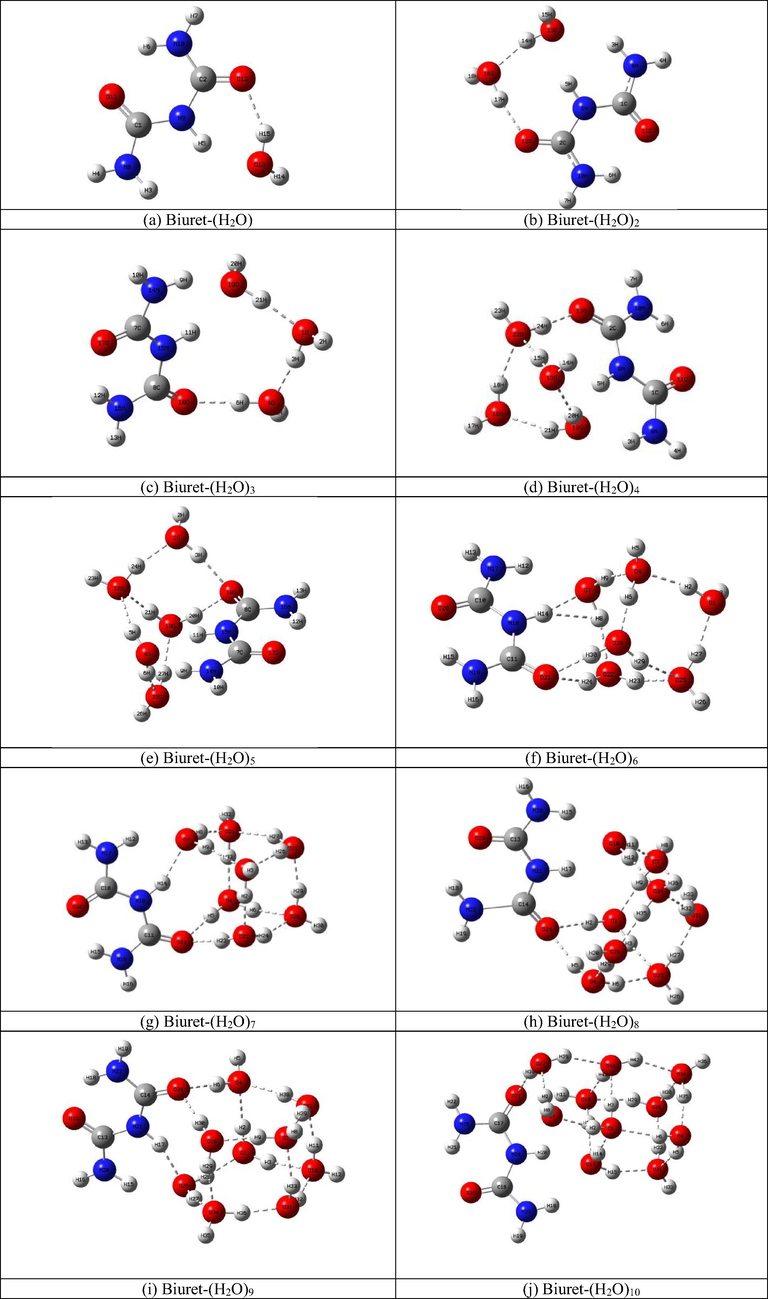
- The Optimized structures of Biuret-water clusters.
The optimized parameter such as bond lengths of Biuret-(H2O)(1−10) clusters were computed using the B3LYP theory with a 6–31 + G (d, p) method and compared with each other. The optimized bond parameters of the biuret water clusters (Biuret-(H2O)(1−10)) are shown in Table 2 and 3.
| Biuret-(H2O) | Biuret-(H2O)2 | Biuret-(H2O)3 | Biuret-(H2O)4 | Biuret-(H2O)5 | |||||
|---|---|---|---|---|---|---|---|---|---|
| C1-N8 | 1.3701 | C1-N8 | 1.3632 | O1-H2 | 0.9613 | C1-N8 | 1.3567 | O1-H2 | 0.9644 |
| C1-N9 | 1.389 | C1-N9 | 1.3958 | O1-H3 | 0.9868 | C1-N9 | 1.4013 | O1-H3 | 0.9859 |
| C1-O11 | 1.2319 | C1-O11 | 1.2327 | O1-H21 | 1.716 | C1-O11 | 1.2348 | O1-H24 | 1.7355 |
| C2-N9 | 1.4036 | C2-N9 | 1.3934 | H3-O4 | 1.7221 | C2-N9 | 1.3857 | H3-O18 | 1.7639 |
| C2-O10 | 1.3476 | C2-O10 | 1.3474 | O4-H5 | 0.9612 | C2-N10 | 1.3463 | O4-H5 | 0.9757 |
| C2-O12 | 1.2385 | C2-O12 | 1.2425 | O4-H6 | 0.9836 | C2-O12 | 1.2475 | O4-H6 | 0.9846 |
| H3-N8 | 1.0091 | H3-N8 | 1.0134 | H6-O18 | 1.7445 | H3-N8 | 1.0166 | O5-H22 | 1.9572 |
| H4-N8 | 1.0087 | H4-N8 | 1.0077 | C7-N14 | 1.3579 | H4-N8 | 1.0071 | H6-O25 | 1.7929 |
| H5-N9 | 1.0217 | H5-N9 | 1.0201 | C7-N15 | 1.3997 | H5-N9 | 1.0259 | C7-N14 | 1.3558 |
| H6-N10 | 1.0157 | H6-N10 | 1.0165 | C7-O17 | 1.2264 | H6-N10 | 1.0175 | C7-N15 | 1.4022 |
| H7-N10 | 1.0074 | H7-N10 | 1.0074 | C8-N15 | 1.393 | H7-N10 | 1.0071 | C7-O17 | 1.2343 |
| O12-H15 | 1.8677 | O13-H14 | 0.9891 | C8-N16 | 1.3465 | O12-H24 | 1.6408 | C8-N15 | 1.3801 |
| O13-H14 | 0.9642 | O13-H15 | 0.965 | C8-O18 | 1.2348 | O13-H14 | 0.9642 | C8-N16 | 1.3429 |
| O13-H15 | 0.9828 | O12-H17 | 1.7547 | H9-N14 | 1.014 | O13-H15 | 0.9805 | C8-O18 | 1.2554 |
| H14-O16 | 1.7497 | H10-N14 | 1.0058 | O13-H20 | 1.884 | H9-N14 | 1.0157 | ||
| O16-H17 | 0.9893 | H11-N15 | 1.0188 | H15-O22 | 1.8528 | H10-N14 | 1.0071 | ||
| O16-H18 | 0.964 | H12-N16 | 1.0152 | O16-H17 | 0.9643 | H11-N15 | 1.0276 | ||
| H13-N16 | 1.0058 | O16-H18 | 0.98 | H12-N16 | 1.0182 | ||||
| O19-H20 | 0.9621 | O16-H21 | 1.8522 | H13-N16 | 1.0074 | ||||
| O19-H21 | 0.9883 | H18-O22 | 1.8754 | O18-H20 | 1.9672 | ||||
| O19-H20 | 0.9793 | O19-H20 | 0.9763 | ||||||
| O19-H21 | 0.981 | O19-H21 | 0.9749 | ||||||
| O22-H23 | 0.9651 | O19-H27 | 1.755 | ||||||
| O22-H24 | 1.001 | H21-O22 | 1.9877 | ||||||
| O22-H23 | 0.9645 | ||||||||
| O22-H24 | 0.9903 | ||||||||
| O25-H26 | 0.9636 | ||||||||
| O25-H27 | 0.9893 | ||||||||
| Biuret-(H2O)6 | Biuret-(H2O)7 | Biuret-(H2O)8 | Biuret-(H2O)9 | Biuret-(H2O)10 | |||||
|---|---|---|---|---|---|---|---|---|---|
| O1-H2 | 0.9853 | O1-H2 | 1.0029 | O1-H2 | 0.979 | O1-H2 | 0.9755 | O1-H2 | 0.9853 |
| O1-H3 | 0.9648 | O1-H3 | 0.9648 | O1-H3 | 0.9784 | O1-H3 | 0.9773 | O1-H3 | 0.9874 |
| O1-H27 | 1.7327 | O1-H9 | 1.8579 | O1-H9 | 1.6438 | O1-H26 | 1.7459 | O1-H6 | 1.9219 |
| H2-O4 | 1.7939 | O1-H26 | 1.9227 | H2-H24 | 1.8715 | H2-O4 | 1.9493 | O1-H14 | 1.7329 |
| O4-H5 | 0.9651 | H2-H22 | 1.6563 | H3-H25 | 1.8994 | H3-O10 | 1.9145 | H2-O7 | 1.8013 |
| O4-H6 | 1.0059 | O4-H5 | 0.9787 | O4-H5 | 0.977 | O4-H5 | 0.9653 | H3-O40 | 1.7952 |
| O4-H9 | 1.9351 | O4-H6 | 0.9779 | O4-H6 | 0.9789 | O4-H6 | 0.9891 | O4-H5 | 0.9764 |
| H6-O28 | 1.6406 | O4-H33 | 1.6563 | O4-H29 | 1.6593 | O4-H38 | 1.8858 | O4-H6 | 0.9781 |
| O7-H8 | 0.9862 | H5-O21 | 1.8863 | H5-O24 | 1.8893 | H6-O24 | 1.7409 | O4-H35 | 1.6905 |
| O7-H9 | 0.9763 | H6-O28 | 1.9058 | H6-O25 | 1.8707 | O7-H8 | 0.9647 | H5-O31 | 1.9595 |
| O7-H14 | 1.8288 | O7-H8 | 0.9802 | O7-H8 | 0.9647 | O7-H9 | 1.0018 | O7-H8 | 0.9639 |
| H8-H14 | 2.0819 | O7-H9 | 0.9802 | O7-H9 | 1.0045 | O7-H33 | 1.9216 | O7-H9 | 0.9898 |
| H8-O22 | 1.8322 | O7-H14 | 1.8744 | O7-H11 | 1.8428 | O7-H39 | 1.8559 | H9-O37 | 1.7538 |
| C10-N17 | 1.361 | H8-O31 | 1.8579 | O7-H33 | 1.9138 | H9-O28 | 1.6533 | O10-H11 | 0.98 |
| C10-N18 | 1.3973 | C10-N17 | 1.3572 | O10-H11 | 0.9805 | O10-H11 | 1.0019 | O10-H12 | 0.9807 |
| C10-O20 | 1.2333 | C10-N18 | 1.4011 | O10-H12 | 0.9798 | O10-H12 | 0.9649 | O10-H29 | 1.999 |
| C11-N18 | 1.3845 | C10-O20 | 1.2341 | H12-O34 | 1.853 | O10-H32 | 1.8747 | O10-H41 | 1.7914 |
| C11-N19 | 1.3447 | C11-N18 | 1.3836 | C13-N20 | 1.3563 | H11-O37 | 1.6594 | H11-O27 | 1.9157 |
| C11-O21 | 1.2504 | C11-N19 | 1.3435 | C13-N21 | 1.4021 | C13-N20 | 1.3548 | H12-O13 | 1.8985 |
| H12-N17 | 1.0143 | C11-O21 | 1.2518 | C13-O23 | 1.2346 | C13-N21 | 1.404 | O13-H14 | 0.9946 |
| H13-N17 | 1.0075 | H12-N17 | 1.0158 | C14-N21 | 1.385 | C13-O23 | 1.2346 | O13-H15 | 0.9814 |
| H14-N18 | 1.0285 | H13-N17 | 1.0072 | C14-N22 | 1.3441 | C14-N21 | 1.3765 | H15-O31 | 1.8648 |
| H15-N19 | 1.0173 | H14-N18 | 1.0271 | C14-O24 | 1.2505 | C14-N22 | 1.3425 | C16-N23 | 1.359 |
| H16-N19 | 1.0075 | H15-N19 | 1.0177 | H15-N20 | 1.0171 | C14-O24 | 1.2583 | C16-N24 | 1.4007 |
| O21-H24 | 2.0134 | H16 -N19 | 1.0075 | H16-N20 | 1.0072 | H15-N20 | 1.0174 | C16-O26 | 1.232 |
| O21-H30 | 1.8644 | O21-H23 | 1.8863 | H17-N21 | 1.0256 | H16-N20 | 1.0072 | C17-N24 | 1.388 |
| O22-H23 | 0.9753 | O22-H23 | 0.9787 | H18-N22 | 1.0179 | H17-N21 | 1.0329 | C17-N25 | 1.343 |
| O22-H24 | 0.9756 | O22-H24 | 0.9779 | H19-N22 | 1.0076 | H17-O25 | 1.8091 | C17-O27 | 1.2499 |
| H23-O25 | 1.9372 | H24-O28 | 1.9061 | O25-H26 | 0.9647 | H18-N22 | 1.0185 | H18-N23 | 1.0146 |
| O25-H26 | 0.9647 | O25-H26 | 0.9779 | O25-H27 | 1.0003 | H19-N22 | 1.0075 | H19-N23 | 1.0076 |
| O25-H27 | 0.9907 | O25-H27 | 0.9779 | H27-O31 | 1.6716 | O24-H30 | 1.8866 | H20-N24 | 1.0175 |
| O25-H29 | 1.9415 | O25-H29 | 1.6852 | O28-H29 | 0.9998 | O25-H26 | 0.9903 | H21-N25 | 1.0174 |
| O28-H29 | 0.9768 | H27-O31 | 1.9219 | O28-H30 | 0.9637 | O25-H27 | 0.9792 | H22-N25 | 1.0076 |
| O28-H30 | 0.9799 | O28-H29 | 0.9993 | O28-H35 | 1.647 | H27-O34 | 1.8597 | O27-H38 | 1.9624 |
| O28-H30 | 0.9645 | O31-H32 | 0.979 | O28-H29 | 0.9791 | O28-H29 | 0.9744 | ||
| O31-H32 | 0.9648 | O31-H33 | 0.9777 | O28-H30 | 0.9777 | O28-H30 | 0.9795 | ||
| O31-H33 | 1.0029 | H32-O34 | 1.8825 | H29-O34 | 1.8917 | O28-H32 | 1.6752 | ||
| O34-H35 | 1.0004 | O31-H32 | 0.9799 | H30-O34 | 1.9012 | ||||
| O34-H36 | 0.9649 | O31-H33 | 0.9779 | O31-H32 | 1.0007 | ||||
| O31-H36 | 1.6489 | O31-H33 | 0.965 | ||||||
| O34-H35 | 0.9651 | O34-H35 | 0.9992 | ||||||
| O34-H36 | 1.0033 | O34-H36 | 0.9649 | ||||||
| O37-H38 | 0.978 | O34-H42 | 1.9657 | ||||||
| O37-H39 | 0.9799 | O37-H38 | 0.9757 | ||||||
| H39-O40 | 2.0021 | ||||||||
| O40-H41 | 0.9855 | ||||||||
| O40-H42 | 0.9761 | ||||||||
According to the data given in Tables 2 and 3, the bond lengths C1-N8, C2-O10, H4-N8, H5-N9, O13-H15 in a cluster of water biuret with one water molecule (Biuret- (H2O)) are slightly longer than in other water clusters, while the lengths of the C2-N9, C2-O12, C1-O11, and H3-N8 bonds in the Biuret- (H2O) cluster are shorter than in other clusters, that these bonds may be associated with greater sensitivity to hydration (Akman et al., 2020; Kazachenko et al., 2021).
The shortest bond length with a value of 0.9613 Å is observed for O1-H2 in the Biuret- (H2O)3 cluster. It should also be noted that a group of OH bonds with rather low values is observed for the Biuret- (H2O) 5 cluster: O22-H23 (0.9645 Å), O22-H24 (0.9903 Å), O25-H26 (0.9636 Å), and O25-H27 (0.9893 Å). The longest bond lengths H8-H14 (2.0819 Å) and O21-H24 (2.0134 Å) are observed for the Biuret cluster - (H2O) 6, H39-O40 (2.0021 Å) for the Biuret - (H2O) 10 cluster.
In the Biuret-(H2O)cluster (Fig. 3a, Table 4), the bonding energy is −58.45 kJ/mol (Table 5), and its is formed by one hydrogen bonds, such as, O13-H15…O12 with values X-H··· X 1.867. In the Biuret-(H2O)2cluster (Fig. 3b), the bonding energy is −111.45 kJ/mol, and its structure is formed by two hydrogen bonds, such as, O16- H17…O12, O13-H14…O16with values X-H··· X:1.750, 1.740, respectively. In the Biuret-(H2O)3 cluster (Fig. 3c), the bonding energy is −636.45 kJ/mol, and its structure is formed by three hydrogen bonds, such as, O4-H14…O16, O1-H3…O4, O19-H21…O1with values X-H··· X:1.74, 1.72, 1.71, respectively. It should be noted that the ring structure for aqueous biuret complexes is formed only for clusters with four water molecules and higher. In the Biuret-(H2O)4 cluster (Fig. 3d), the bonding energy is −242.45 kJ/mol, and its structure is formed by five hydrogen bonds, such as, O22-H24…O12, O16-H18…O22, O19-H21…O16, O13-H15…O22, O19-H20…O13 with values X-H··· X:1.64, 1.87, 1.85, 1.85, 1.88, respectively. In the Biuret-(H2O)5cluster (Fig. 3e), the bonding energy is −294.45 kJ/mol, and its ring structure is formed by six hydrogen bonds, such as, O1-H3…O18, O19-H20…O18, O22-H24…O1, O4-H5…O22, O4-H6…O25, O25-H27…O19with values X-H··· X:1.76, 1.96, 1.73, 1.95, 1.79, 1.75, respectively. In the Biuret-(H2O)6 cluster (Fig. 3f), the bonding energy is −347.45 kJ/mol, and its ring structure is formed by elevenhydrogen bonds, such as, N18-H14…O7, N18-H14…O8, O7-H9…O4, O1-H2…O4, O25-H27…O1, O28-H29…O25, O22-H23…O25, O4-H6…O28, O28-H30…O21, O22-H24…O21, O7-H8…O22 with values X-H··· X:1.82, 2.01, 1.93, 1.79, 1.73, 1.94, 1.93, 1.64, 1.86, 2.01, 1.83, respectively. In the Biuret-(H2O)7 cluster (Fig. 3g), the bonding energy is −426.45 kJ/mol, and its ring structure is formed by twelve hydrogen bonds, such as, N18-H14…O7, O7-H8…O31, O25-H27…O31, O4-H5…O21, O22-H23…O21, O31-H33…O4, O4-H6…O28, O22-H24…O28, O28-H29…O25, O25-H27…O31, O25-H26…O1, O1-H2…O22 with values X-H··· X:1.87, 1.85, 1.92, 1.88, 1.88, 1.65, 1.90, 1.96, 1.68, 1.92, 1.92, 1.65, respectively. In the Biuret-(H2O)8 cluster (Fig. 3h), the bonding energy is −478.45 kJ/mol, and its ring structure is formed by twelve hydrogen bonds, such as, O1-H2…O24, O4-H5…O24, O4-H6…O25, O25-H27…O31, O31-H32…O34, O10-H11…O7, O31-H33…O7, O10-H12…O34, O7-H9…O1, O1-H3…O25, O34-H35…O28, O28-H29…O4with values X-H··· X:1.87, 1.88, 1.87, 1.67, 1.88, 1.84, 1.91, 1.85, 1.64, 1.89, 1.64, 1.65, respectively. In the Biuret-(H2O)9 cluster (Fig. 3i), the bonding energy is −556.45 kJ/mol, and its ring structure is formed by fourteen hydrogen bonds, such as, O4-H6…O24, O1-H2…O4, O28-H30…O24, N21-H17…O25, O25-H27…O34, O28-H29…O34, O7-H9…O28, O34-H36…O31, O1-H3…O10, O31-H32…O10, O10-H11…O37, O37-H38…O4, O37-H39…O7, O25-H26…O1with values X-H··· X:1.74, 1.94, 1.88, 1.80, 1.85, 1.89, 1.65, 1.64, 1.91, 1.87, 1.65, 1.88, 1.85, 1.74, respectively. In the Biuret-(H2O)10cluster (Fig. 3j), the bonding energy is −583.45 kJ/mol, and its ring structure is formed by fifteen hydrogen bonds, such as, O37-H38…O27, O37-H39…O40, O7-H9…O37, O1-H2…O7, O1-H3…O40, O13-H14…O1, O28-H29…O10, O28-H30…O34, O40-H42…O34, O34-H35…O4, O4-H5…O31, O13-H15…O31, O31-H32…O28, O10-H12…O13, O4-H6…O1 with values X-H··· X:1.96, 2.00, 1.75, 1.80, 1.79, 1.73, 1.99, 1.90, 1.96, 1.69, 1.95, 1.86, 1.67, 1.89, 1.92, respectively.
Hydrogen bonds O ⋯ O between water molecules are also observed and, as was found, are shorter than hydrogen bonds NH … O, which indicates that the cyclic parts of aqueous biuret clusters with 6 (or more) water molecules are especially stabilized by OH … O hydrogen bonds, which is consistent with (Zumdahl, 2000). DFT calculations for aqueous biuret clusters with 6 (or more) water molecules show that a probable cluster is stabilized due to the formation of a ring structure around some parts of the central biuret molecule.
Intermolecular interaction energies of with hydrogen bonds calculated by the B3LYP / 6–31 + G (d, p) method (Table 4). The energies of intermolecular interaction with hydrogen bonds in the biuret water clusters calculated by the formula: ΔE = E(cluster)-[E(Biuret) + n*E(H2O)] (1).
| H-Bond | X-H…X | X…X | |
|---|---|---|---|
| Biuret-(H2O) | O13-H15…O12 | 1.867 | 2.75 |
| Biuret-(H2O)2 | O16- H17…O12 | 1.75 | 2.73 |
| O13-H14…O16 | 1.74 | 2.69 | |
| Biuret-(H2O)3 | O4-H14…O16 | 1.74 | 2.72 |
| O1-H3…O4 | 1.72 | 2.70 | |
| O19-H21…O1 | 1.71 | 2.69 | |
| Biuret-(H2O)4 | O22-H24…O12 | 1.64 | 2.64 |
| O16-H18…O22 | 1.87 | 2.82 | |
| O19-H21…O16 | 1.85 | 2.78 | |
| O13-H15…O22 | 1.85 | 2.79 | |
| O19-H20…O13 | 1.88 | 2.08 | |
| Biuret-(H2O)5 | O1-H3…O18 | 1.76 | 2.74 |
| O19-H20…O18 | 1.96 | 2.91 | |
| O22-H24…O1 | 1.73 | 2.69 | |
| O4-H5…O22 | 1.95 | 2.88 | |
| O4-H6…O25 | 1.79 | 2.74 | |
| O25-H27…O19 | 1.75 | 2.71 | |
| Biuret-(H2O)6 | N18-H14…O7 | 1.82 | 2.83 |
| N18-H14…O8 | 2.01 | 3.04 | |
| O7-H9…O4 | 1.93 | 2.87 | |
| O1-H2…O4 | 1.79 | 2.75 | |
| O25-H27…O1 | 1.73 | 2.70 | |
| O28-H29…O25 | 1.94 | 2.86 | |
| O22-H23…O25 | 1.93 | 2.87 | |
| O4-H6…O28 | 1.64 | 2.63 | |
| O28-H30…O21 | 1.86 | 2.83 | |
| O22-H24…O21 | 2.01 | 2.95 | |
| O7-H8…O22 | 1.83 | 2.77 | |
| Biuret-(H2O)7 | N18-H14…O7 | 1.87 | 2.85 |
| O7-H8…O31 | 1.85 | 2.80 | |
| O25-H27…O31 | 1.92 | 2.85 | |
| O4-H5…O21 | 1.88 | 2.85 | |
| O22-H23…O21 | 1.88 | 2.85 | |
| O31-H33…O4 | 1.65 | 2.65 | |
| O4-H6…O28 | 1.90 | 2.83 | |
| O22-H24…O28 | 1.96 | 2.83 | |
| O28-H29…O25 | 1.68 | 2.66 | |
| O25-H27…O31 | 1.92 | 2.85 | |
| O25-H26…O1 | 1.92 | 2.85 | |
| O1-H2…O22 | 1.65 | 2.65 | |
| Biuret-(H2O)8 | O1-H2…O24 | 1.87 | 2.83 |
| O4-H5…O24 | 1.88 | 2.85 | |
| O4-H6…O25 | 1.87 | 2.83 | |
| O25-H27…O31 | 1.67 | 2.66 | |
| O31-H32…O34 | 1.88 | 2.84 | |
| O10-H11…O7 | 1.84 | 2.79 | |
| O31-H33…O7 | 1.91 | 2.84 | |
| O10-H12…O34 | 1.85 | 2.82 | |
| O7-H9…O1 | 1.64 | 2.63 | |
| O1-H3…O25 | 1.89 | 2.83 | |
| O34-H35…O28 | 1.64 | 2.64 | |
| O28-H29…O4 | 1.65 | 2.65 | |
| Biuret-(H2O)9 | O4-H6…O24 | 1.74 | 2.72 |
| O1-H2…O4 | 1.94 | 2.90 | |
| O28-H30…O24 | 1.88 | 2.86 | |
| N21-H17…O25 | 1.80 | 2.79 | |
| O25-H27…O34 | 1.85 | 2.82 | |
| O28-H29…O34 | 1.89 | 2.84 | |
| O7-H9…O28 | 1.65 | 2.64 | |
| O34-H36…O31 | 1.64 | 2.64 | |
| O1-H3…O10 | 1.91 | 2.86 | |
| O31-H32…O10 | 1.87 | 2.82 | |
| O10-H11…O37 | 1.65 | 2.64 | |
| O37-H38…O4 | 1.88 | 2.84 | |
| O37-H39…O7 | 1.85 | 2.81 | |
| O25-H26…O1 | 1.74 | 2.72 | |
| Biuret-(H2O)10 | O37-H38…O27 | 1.96 | 2.92 |
| O37-H39…O40 | 2.00 | 2.89 | |
| O7-H9…O37 | 1.75 | 2.72 | |
| O1-H2…O7 | 1.80 | 2.73 | |
| O1-H3…O40 | 1.79 | 2.77 | |
| O13-H14…O1 | 1.73 | 2.71 | |
| O28-H29…O10 | 1.99 | 2.86 | |
| O28-H30…O34 | 1.90 | 2.84 | |
| O40-H42…O34 | 1.96 | 2.84 | |
| O34-H35…O4 | 1.69 | 2.67 | |
| O4-H5…O31 | 1.95 | 2.88 | |
| O13-H15…O31 | 1.86 | 2.80 | |
| O31-H32…O28 | 1.67 | 2.66 | |
| O10-H12…O13 | 1.89 | 2.87 | |
| O4-H6…O1 | 1.92 | 2.82 |
According to Table 5, the minimum value of Bonding energy (-58.45 kJ / mol) is observed for an aqueous biuret cluster with one water molecule. It should be noted that the maximum value of Bonding energy (-636.45 kJ / mol) is observed for the Biuret- (H2O) 3 cluster, which may be associated with some energy intensity of this structure. The ring structure is observed in Biuret- (H2O)n (n = 4–10). Bonding energy values for these clusters change almost linearly from R2 = 0.9924 (Fig. 4).
| Number of water molecules | Energy (kJ/mol) | Bonding energy ΔE (kJ/mol) |
|---|---|---|
| 1 | −1235219 | −58.45 |
| 2 | −1435939 | −111.45 |
| 3 | −1637131 | −636.45 |
| 4 | −1837404 | −242.45 |
| 5 | −2038123 | −294.45 |
| 6 | −2238843 | −347.45 |
| 7 | −2439589 | −426.45 |
| 8 | −2640308 | −478.45 |
| 9 | −2841053 | −556.45 |
| 10 | −3041747 | −583.45 |
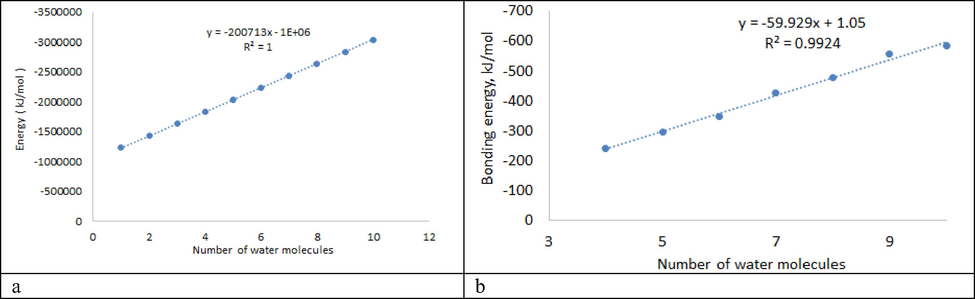
- Influence of the number of water molecules in water clusters on: (a) - Energy, (б) - Bonding energy.
For all water clusters of biuret, starting from Biuret- (H2O), with an increase in the number of molecules in the cluster, there is a linear change in the Energy value from −1235219 to −3041747 kJ / mol (Fig. 4, Table 5).
3.3 NLO and thermodynamic properties of biuret-water clusters
NLO properties are important for frequency shift, optical modulation, switching, laser, fiber, optical material logic, and optical memory for emerging technologies in areas such as telecommunications, signal processing, and optical interface. compounds (Noureddine et al., 2021).
Non-linear optical properties (NLO), such as dipole moment, hyperpolarizability and polarizability for biuret-water clusters were calculated using the DFT method (Table 6). The effect on polarizability (α0) of the water molecules numbers in biuret-water clusters is shown in Fig. 5.
| Number of water molecule | Dipole moment | Polarisability | Hyperpolarisability |
|---|---|---|---|
| 1 | 1.758 | 9.429 | 6.139 |
| 2 | 2.068 | 10.080 | 7.460 |
| 3 | 1.796 | 12.376 | 9.667 |
| 4 | 3.112 | 13.614 | 9.609 |
| 5 | 2.896 | 14.664 | 12.771 |
| 6 | 2.500 | 15.931 | 8.501 |
| 7 | 1.212 | 17.281 | 10.868 |
| 8 | 2.215 | 19.004 | 10.405 |
| 9 | 1.567 | 20.659 | 6.170 |
| 10 | 2.031 | 21.462 | 7.787 |
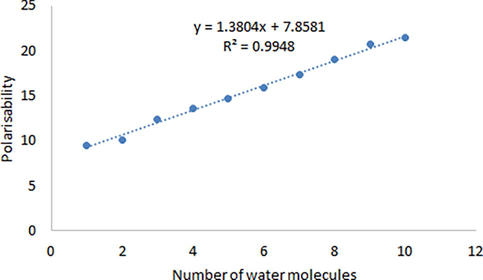
- The effect on polarizability of the number of water molecules in biuret-water clusters.
According to the data shown in Table 6, the values of the dipole moment change nonlinearly with an increase in the number of molecules in aqueous biuret clusters. The maximum value (3.112 Debye) of the dipole moment is observed for the Biuret- (H2O) 4 cluster, and the minimum value (1.212 Debye) is observed for the Biuret- (H2O) 7 cluster. Hyperpolarisability values change in the same way. Maximum value (12.771*10-31 e.s.u)Hyperpolarisability is observed for the Biuret- (H2O) 5 cluster, and the minimum value (6.139*10-31 e.s.u) - observed for the Biuret- (H2O) cluster.
At the same time, the polarisability values increase almost linearly with an increase in the number of water molecules in aqueous biuret clusters with an R2 value (0.9948). A similar phenomenon was observed in the case of thiourea (Akman et al., 2020) and ammonium sulfamate (Kazachenko et al., 2021).
Various thermodynamic functions are used to predict the reactivity of chemicals and to determine the likelihood of different reaction routes (Fazilath Basha et al., 2021).Standard thermodynamic functions such as zero-point energy correction, heat capacity, entropy (S) electronic energy (EE), thermal correction to energy, thermal correction to enthalpy, thermal correction to free energy, and other parameters were calculated using B3LYP methods with a basis set of 6–31 + G (d, p) (Table 7).
| Parameters | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| E(RB3LYP) | −470.4703 | −546.9272 | −623.5516 | −699.8305 | −776.2813 | −852.7348 | −929.1959 | −1005.6473 | −1082.1018 | −1158.5479 |
| Electronic Energy (EE) | −470.4703 | −546.9272 | −623.5516 | −699.8305 | −766.2818 | −852.7348 | −929.1959 | 1005.6473 | −1082.1018 | −1158.5479 |
| Zero-point Energy Correction | 0.1159 | 0.1418 | 0.1664 | 0.19301 | 0.2187 | 0.2455 | 0.2722 | 0.2972 | 0.3237 | 0.3503 |
| Thermal Correction to Energy | 0.1260 | 0.1543 | 0.1819 | 0.2106 | 0.2388 | 0.2676 | 0.2960 | 0.3238 | 0.3532 | 0.3814 |
| Thermal Correction to Enthalpy | 0.1270 | 0.1553 | 0.1829 | 0.2116 | 0.2398 | 0.2685 | 0.2970 | 0.3247 | 0.3532 | 0.3823 |
| Thermal Correction to Free Energy | 0.0807 | 0.1031 | 0.1231 | 0.1474 | 0.1701 | 0.1948 | 0.2200 | 0.2412 | 0.2656 | 0.2890 |
| EE + Zero-point Energy | −470.3544 | −546.7854 | −623.3851 | −699.6374 | −776.0638 | −852.4892 | −928.9237 | −1005.3499 | −1081.7780 | −1158.1976 |
| EE + Thermal Energy Correction | −470.3442 | −546.7728 | −623.3697 | −699.6198 | −766.0429 | −852.4671 | −928.8999 | −1005.3234 | −1081.7494 | −1158.1665 |
| EE + Thermal Enthalpy Correction | −470.3433 | −546.7719 | −623.3687 | −699.6188 | −766.0419 | −852.4662 | −928.9888 | −1005.3225 | −1081.7485 | −1158.1655 |
| EE + Thermal Free Energy Correction | −470.3896 | −546.8241 | −623.4285 | −699.6830 | −766.1117 | −852.5399 | −928.9758 | −1005.4060 | −108.8361 | −1158.2588 |
| E (Thermal) | 79.12 | 96.881 | 114.186 | 132.201 | 149.91 | 167.933 | 185.779 | 203.209 | 221.080 | 239.348 |
| Heat Capacity (Cv) | 35.33 | 43.661 | 52.922 | 61.305 | 69.603 | 76.988 | 84.144 | 72.880 | 100.659 | 109.245 |
| Entropy (S) | 97.39 | 109.75 | 125.876 | 135.098 | 146.736 | 155.039 | 161.861 | 175.904 | 184.415 | 196.433 |
According to the data given in Table 7 and Fig. 6, the values of E (Thermal), Heat Capacity (Cv), Entropy (S) increase linearly as water molecules in the biuret cluster increase. For the change in E (Thermal) R2 = 1, which is high. For the dependence of the change in Heat Capacity (Cv) on the content of water molecules in the water cluster of biuret, R2 = 0.9939 is observed. For the dependence of the change in Entropy (S) on the content of water molecules in the aqueous biuret cluster, R2 = 0.9325 is observed.
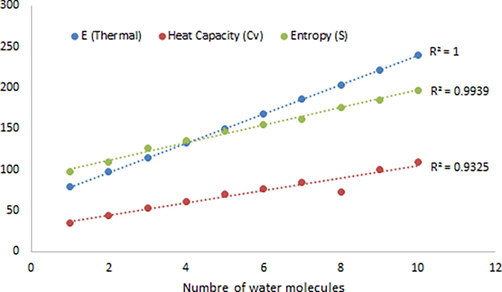
- The variation of thermodynamic parameters as a function of the water molecules numbers in the biuret-water clusters.
It should be noted that other thermodynamic characteristics also change almost linearly with a regular change in water molecules in the biuret cluster. A similar phenomenon was also observed in (Kazachenko et al., 2021).
3.4 HOMO-LUMO analysis and electronic parameters of Biuret-(H2O)n clusters
Frontier molecular orbitals (FMO) are a valuable practical model for describing chemical reactivity. This theory permit the identification of electrophilic and nucleophilic attacks which responsible to the formation of hydrogen bonding interactions. An important aspect of the theory of boundary electrons is the emphasis on the busiest and lowest unoccupied molecular orbitals (HOMO and LUMO). So, according to this theory, attention is paid to the localization of the HOMO orbital, because electrons from this orbital are most free to participate in the reaction. Likewise, the boundary orbit theory predicts that the location of the lowest unoccupied orbital (LUMO) is a good electrophilic site (Gatfaoui et al., 2019).
Using the energy gap between HOMO-LUMO, ionization potential (IP), electronegativity (χ), softness(ς),electron affinity (EA), electrophilicity index (ɷ),chemical potential (μ) (Table 8) were calculated (at B3LYP/6–31 + G(d,p) basis set, according (Fleming, 1976) by the following equations:
| EHOMO | ELUMO | Gap | Ionisation energy | Electron affinity | Chemical potential | Chemical hardness (η) | Softness | Electronegativity | Electrophilicity | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −7,33 | −0,33 | −7 | 7,33 | 0,33 | −3,83 | 3,5 | 0,14 | 25,67 | 3,83 |
| 2 | −7,76 | −0,16 | −7,6 | 7,76 | 0,16 | −3,96 | 3,8 | 0,13 | 29,79 | 3,96 |
| 3 | −7,76 | −0,66 | −7,1 | 7,76 | 0,66 | −4,21 | 3,55 | 0,14 | 31,46 | 4,21 |
| 4 | −7,58 | −0,19 | −7,39 | 7,58 | 0,19 | −3,88 | 3,695 | 0,13 | 27,88 | 3,885 |
| 5 | −7,68 | −0,27 | −7,41 | 7,68 | 0,27 | −3,97 | 3,705 | 0,13 | 29,27 | 3,975 |
| 6 | −7,75 | −0,3 | −7,45 | 7,75 | 0,3 | −4,02 | 3,725 | 0,13 | 30,17 | 4,025 |
| 7 | −7,76 | −0,3 | −7,46 | 7,76 | 0,3 | −4,03 | 3,73 | 0,13 | 30,28 | 4,03 |
| 8 | −7,73 | −0,32 | −7,41 | 7,73 | 0,32 | −4,02 | 3,705 | 0,13 | 30,01 | 4,025 |
| 9 | −7,77 | −0,82 | −6,95 | 7,77 | 0,82 | −4,29 | 3,475 | 0,14 | 32,05 | 4,295 |
| 10 | −7,93 | −0,44 | −7,49 | 7,93 | 0,44 | −4,18 | 3,745 | 0,13 | 32,79 | 4,185 |
A small band gap indicates that the molecule has high polarization, chemical reactivity and biological activity, and low kinetic stability (Bader, 1990). Based on the lower energy gap, it should be noted that the biuret cluster with nine water molecules has a higher chemical stability than other clusters.
According to the data presented in Table 8, the Softness values practically do not change with an increase in the number of water molecules in the biuret cluster and are 0.13–0.14. The Electronegativity values change nonlinearly with an increase in the number of water molecules in the biuret cluster, while the minimum Electronegativity (25.67) corresponds to the minimum number of water molecules in the biuret cluster, and the maximum Electronegativity (32.79) corresponds to the maximum number of water molecules (in the us spaces (1–10)). Electrophilicity values also change non-linearly. The minimum Electrophilicity value (3.83) corresponds to the Biuret- (H2O) cluster, and the maximum Electrophilicity value (4.295) corresponds to the Biuret- (H2O) 9 cluster. Other characteristics, such as Ionisation energy, Electron affinity, Chemical potential and Chemical hardness, change nonlinearly with an increase in the amount of water in the water clusters of biuret.
3.5 AIM, RDG and ELF topological analysis
The theory of atoms in molecules (AIM) is actively used to determine the types of interactions in various molecular systems (Johnson et al., 2010). Topological parameters such as electron density ρ (r), the Laplacian of electron density ∇2ρ (r), potential energy density V (r), Lagrangian kinetic energy G (r), Kinetic energy of Hamiltonian H (r) = G (r) + V (r) and the binding energy Eint = V (r) / 2 can help to understand the properties of hydrogen bonds between compounds (Akman et al., 2020). The Molecular diagram of the Biuret-water clusters is shown in Fig. 7.
- AIM graphical visualization of Biuret-water clusters.
According to (Johnson et al., 2010), the interactions of hydrogen bonds can be defined as follows:
-
(1)
∇2ρ (r) > 0 and H (r) > 0 = Weak hydrogen bonds;
-
(2)
∇2ρ (r) > 0 and H (r) < 0 = Moderate hydrogen bonds;
-
(3)
∇2ρ (r) < 0 and H (r) < 0 = Strong hydrogen bonds.
The electron density ρ (r) and its Laplacian ∇2ρ(r) help determine the nature of interactions. On the whole, large values of the electron density ρ (r) and its Laplacian ∇2ρ(r) show the power of hydrogen interactions. Negative values of the Laplacian ∇2ρ(r) indicate a strong covalent character, while positive values indicate a decrease in the charge in the internuclear region (Akman et al., 2020; Kazachenko et al., 2021).
In the Biuret-(H2O) cluster, three types of interactions were observed: O13-H15…O12, N9-H5…O13, N10-H6…O11, where the electron density values are 0.0312, 0.0256, 0.0290a. u. and the Laplacian values are 0.0887, 0.0775, 0.0912a. u. respectively. As clearly seen from Table 9, the first hydrogen bond (O13-H15…O12) was matched to the biggest binding energy with value equal to 62.16 kcal/mol. In the Biuret-(H2O)2 cluster, five H-bonds interactions were observed: N8-H3…O13, N9-H6…O13, O16-H17…O12, O13-H14…O16, N10-H6…O11, where the electron density values are 0.0139, 0.0267, 0.0380, 0.0390, 0.0288a. u. and the Laplacian values are 0.0445, 0.0742, 0.1122, 0.1171, 0.0898a. u. respectively. The major interaction energy value corresponds to the fourth hydrogen bond with 76.85 kcal/mol. In the Biuret-(H2O)3 cluster, three types of interactions were observed: N14-H9…O19, N15-H11…O19, N16-H12…O17, O19-H21…O1, O1-H3…O4, O4-H6…O18, where the electron density values are 0.0211, 0.0226, 0.0290, 0.0421, 0.0411, 0.0367a. u. and the Laplacian values are 0.0807, 0.0792, 0.1103, 0.1347, 0.1347, 0.1308a. u. respectively. In the Biuret-(H2O)4 cluster, three types of interactions were observed: N18-H3…O19, O19-H20…O13, N9-H5…O19, O19-H21…O16, O13-H15…O22, O22-H24…O12, N10-H6…O11, where the electron density values are 0.0216, 0.0289, 0.0300, 0.0313, 0.0308, 0.0487, 0.0308a. u. and the Laplacian values are 0.0654, 0.0847, 0.0822, 0.0913, 0.0903, 0.1459, 0.0960a. u. respectively. Concerning Biuret-(H2O)5 cluster, three types of interactions were observed: N16-H12…O17, N15-H11…O4, N14-H9…O4, O4-H5…O22, O25-H27…O19, O1-H3…O18, O19-H20…O18, O22-H24…O1, O4-H6…O25, where the electron density values are 0.0307, 0.0321, 0.0195, 0.2451, 0.0398, 0.0361, 0.0232, 0.0402, 0.0349a. u. and the Laplacian values are 0.0956, 0.0857, 0.0597, 0.0710, 0.1139, 0.1113, 0.0677, 0.1195, 0.1053a. u. respectively. The EH…O energy value of these interactions were ranging from 40 to 77 kcal/mol. Regarding Biuret-(H2O)6 cluster, three types of interactions were observed: N17-H12…O7, N18-H14…O7, O28-H30…O21, O22-H24…O21, O1-H2…O4, O4-H6…O28, O25-H27…O1, O7-H8…O22, O7-H9…O4, O28-H29…O25, O22-H23…O25, O22-H23…O21, where the electron density values are 0.0140, 0.0358, 0.0284, 0.0220, 0.0355, 0.0523, 0.0408, 0.0337, 0.0259, 0.0256, 0.0254, 0.0220a. u. and the Laplacian values are0.0447, 0.0693, 0.0864, 0.0608, 0.0141, 0.1407, 0.1197, 0.0941, 0.0472, 0.0736, 0.0740, 0.0608a. u. respectively. In the Biuret-(H2O)7 cluster, twelve hydrogen bonding interactions were identified: N17-H12…O7, N18-H14…O7, N19-H15…O20, O4-H5…O21, O22-H23…O21, O31-H33…O4, O4-H6…O28, O22-H24…O28, O25-H27…O31, O1-H2…O22, O25-H26…O1, O7-H9…O1,where the electron density values are 0.0200, 0.0322, 0.0302, 0.0272, 0.0272, 0.0501, 0.0275, 0.0275, 0.0269, 0.0501, 0.0268, 0.0306a. u. and the Laplacian values are0.0607, 0.0877, 0.0940, 0.0817, 0.0818, 0.1372, 0.0800, 0.0799, 0.0768, 0.1372, 0.0767, 0.0893a. u. respectively. In the Biuret-(H2O)8 cluster, three types of interactions were observed: N20-H15…O10, N21-H17…O10, N22-H18…O23, O1-H2…O24, O4-H5…O24, O10-H12…O34, O10-H11…O7, O31-H32…O34, O31-H33…O7, O25-H27…O31, O1-H3…O25, O4-H6…O25, O34-H35…O28, O28-H29…O4, O7-H9…O1,where the electron density values are0.0228, 0.0290, 0.0309, 0.0279, 0.0263, 0.0305, 0.0315, 0.0292, 0.0273, 0.0483, 0.0280, 0.0296, 0.0494, 0.0492, 0.0518a. u. and the Laplacian values are0.0688, 0.0786, 0.0963, 0.0853, 0.0816, 0.0897, 0.0928, 0.0830, 0.0782, 0.1337, 0.0810, 0.0863, 0.1429, 0.1378, 0.1404a. u. respectively. In the Biuret-(H2O)9 cluster, three types of interactions were observed: N20-H15…O25, N21-H17…O25, N22-H18…O23, O4-H6…O24, O28-H30…O24, O28-H29…O34, O25-H27…O34, O37-H38…O4, O1-H3…O10, O10-H11…O37, O31-H32…O10, O31-H33…O7, O37-H39…O7, O7-H9…O28, O34-H36…O31, O25-H26…O1,where the electron density values are 0.0214, 0.0372, 0.0309, 0.0379, 0.0273, 0.0285, 0.0303, 0.0286, 0.0272, 0.0502, 0.0296, 0.0268, 0.0306, 0.0502, 0.0511, 0.0402a. u. and the Laplacian values are 0.0641, 0.1023, 0.0959, 0.1168, 0.0809, 0.0812, 0.0887, 0.0829, 0.0774, 0.1374, 0.0851, 0.0765, 0.0899, 0.1394, 0.1396, 0.1153a. u. respectively.In the Biuret-(H2O)10 cluster, three types of interactions were observed: N25-H21…O26, N24-H20…O13, N23-H18…O13, O10-H11…O27, O37-H38…O27, O37-H39…O40, O40-H42…O34, O4-H6…O1, O28-H29…O10, O13-H15…O31, O4-H5…O31, O28-H30…O34, O40-H41…O10, O13-H14…O1, O1-H2…O7, O7-H9…O37, O34-H35…O4,where the electron density values are 0.0294, 0.0243, 0.0157, 0.0229, 0.0270, 0.0213, 0.0246, 0.0261, 0.0216, 0.0302, 0.0248, 0.3371, 0.0372, 0.0423, 0.0347, 0.0394, 0.0464a. u. and the Laplacian values are 0.0917, 0.0718, 0.0455, 0.0675, 0.0779, 0.0658, 0.0717, 0.00786, 0.0679, 0.0879, 0.0700, 1.8245, 0.1045, 0.1175, 0.1032, 0.1133, 0.1282a. u. respectively. As it is shown in Table 9, the O28-H30…O34 bond was associated to the bigger interaction energy which is in the range 1449.22 kcal/mol.
| H-bonds | ρ | Δρ | H | VBCP | EH…O | |
|---|---|---|---|---|---|---|
| 1 | O13-H15…O12 | 0.0312 | 0.0887 | −0.7661 0.10-3 | −0.0237 | 62.16 |
| N9-H5…O13 | 0.0256 | 0.0775 | −0.1145 0.10-3 | −0.0196 | 51.41 | |
| N10-H6…O11 | 0.0290 | 0.0912 | −0.1751 0.10-3 | −0.0231 | 60.59 | |
| 2 | N8-H3…O13 | 0.0139 | 0.0445 | 0.0002 | −0.0107 | 28.06 |
| N9-H6…O13 | 0.0267 | 0.0742 | −0.8114. 10-3 | −0.0201 | 52.72 | |
| O16-H17…O12 | 0.0380 | 0.1122 | 0.1598. 10-3 | −0.0278 | 72.92 | |
| O13-H14…O16 | 0.0390 | 0.1171 | −0.1390. 10-4 | −0.0293 | 76.85 | |
| N10-H6…O11 | 0.0288 | 0.0898 | −0.2422. 10-3 | −0.0229 | 60.06 | |
| 3 | N14-H9…O19 | 0.0211 | 0.0807 | 0.0025 | −0.0151 | 39.6 |
| N15-H11…O19 | 0.0226 | 0.0792 | 0.0019 | −0.0159 | 41.7 | |
| N16-H12…O17 | 0.0290 | 0.1103 | 0.0021. 10-2 | −0.0233 | 61.11 | |
| O19-H21…O1 | 0.0421 | 0.1347 | −0.0026 | −0.0388 | 101.77 | |
| O1-H3…O4 | 0.0411 | 0.1347 | −0.0020 | −0.0376 | 98.62 | |
| O4-H6…O18 | 0.0367 | 0.1308 | −0.1208. 10-3 | −0.0329 | 86.29 | |
| 4 | N18-H3…O19 | 0.0216 | 0.0654 | −0.2954. 10-3 | −0.0169 | 44.32 |
| O19-H20…O13 | 0.0289 | 0.0847 | −0.3305. 10-3 | −0.0218 | 57.18 | |
| N9-H5…O19 | 0.0300 | 0.0822 | −0.0012 | −0.0220 | 57.7 | |
| O19-H21…O16 | 0.0313 | 0.0913 | −0.3356. 10-3 | −0.0230 | 60.32 | |
| O13-H15…O22 | 0.0308 | 0.0903 | −0.1315. 10-3 | −0.0220 | 57.7 | |
| O22-H24…O12 | 0.0487 | 0.1459 | −0.8494. 10-3 | −0.0381 | 99.9 | |
| N10-H6…O11 | 0.0308 | 0.0960 | −0.2685. 10-3 | −0.0240 | 62.95 | |
| 5 | N16-H12…O17 | 0.0307 | 0.0956 | −0.2765. 10-3 | −0.0244 | 64 |
| N15-H11…O4 | 0.0321 | 0.0857 | −0.0012 | −0.0238 | 62.42 | |
| N14-H9…O4 | 0.0195 | 0.0597 | −0.1891. 10-3 | −0.0153 | 40.13 | |
| O4-H5…O22 | 0.2451 | 0.0710 | −0.4564. 10-3 | −0.0186 | 48.78 | |
| O25-H27…O19 | 0.0398 | 0.1139 | −0.5233. 10-3 | −0.0295 | 77.37 | |
| O1-H3…O18 | 0.0361 | 0.1113 | 0.4838. 10-3 | −0.0268 | 70.29 | |
| O19-H20…O18 | 0.0232 | 0.0677 | −0.0360. 10-3 | −0.0176 | 46.16 | |
| O22-H24…O1 | 0.0402 | 0.1195 | −0.1286. 10-3 | −0.0299 | 78.42 | |
| O4-H6…O25 | 0.0349 | 0.1053 | 0.1354. 10-3 | −0.0260 | 68.19 | |
| 6 | N17-H12…O7 | 0.0140 | 0.0447 | 0.1762. 10-3 | −0.0108 | 28.32 |
| N18-H14…O7 | 0.0358 | 0.0693 | −0.0011. 10-3 | −0.0265 | 69.51 | |
| O28-H30…O21 | 0.0284 | 0.0864 | 0.2467. 10-3 | −0.0211 | 55.34 | |
| O22-H24…O21 | 0.0220 | 0.0608 | −0.7184. 10-3 | −0.0166 | 43.54 | |
| O1-H2…O4 | 0.0355 | 0.0141 | −0.1166. 10-3 | −0.0262 | 68.72 | |
| O4-H6…O28 | 0.0523 | 0.1407 | −0.0029 | −0.0411 | 107.80 | |
| O25-H27…O1 | 0.0408 | 0.1197 | −0.1788. 10-3 | −0.0302 | 79.21 | |
| O7-H8…O22 | 0.0337 | 0.0941 | −0.7059. 10-3 | −0.0249 | 65.31 | |
| O7-H9…O4 | 0.0259 | 0.0472 | −0.5059. 10-3 | −0.0195 | 51.14 | |
| O28-H29…O25 | 0.0256 | 0.0736 | −0.5354. 10-3 | −0.0194 | 50.88 | |
| O22-H23…O25 | 0.0254 | 0.0740 | −0.4054. 10-3 | −0.0193 | 50.62 | |
| O22-H23…O21 | 0.0220 | 0.0608 | −0.0718. 10-3 | −0.0166 | 43.54 | |
| 7 | N17-H12…O7 | 0.0200 | 0.0607 | −0.2761. 10-3 | −0.0157 | 41.18 |
| N18-H14…O7 | 0.0322 | 0.0877 | −0.0010 | −0.0204 | 53.50 | |
| N19-H15…O20 | 0.0302 | 0.0940 | −0.2657. 10-3 | −0.0240 | 62.95 | |
| O4-H5…O21 | 0.0272 | 0.0817 | 0.7739. 10-3 | −0.0202 | 52.98 | |
| O22-H23…O21 | 0.0272 | 0.0818 | 0.7847. 10-3 | −0.0202 | 52.98 | |
| O31-H33…O4 | 0.0501 | 0.1372 | −0.0022 | −0.0388 | 101.77 | |
| O4-H6…O28 | 0.0275 | 0.0800 | −0.3986. 10-3 | −0.0208 | 54.55 | |
| O22-H24…O28 | 0.0275 | 0.0799 | −0.3995. 10-3 | −0.0207 | 54.29 | |
| O25-H27…O31 | 0.0269 | 0.0768 | −0.5550. 10-3 | −0.0203 | 53.24 | |
| O1-H2…O22 | 0.0501 | 0.1372 | −0.0022 | −0.0388 | 101.77 | |
| O25-H26…O1 | 0.0268 | 0.0767 | −0.5565. 10-3 | −0.0202 | 52.98 | |
| O7-H9…O1 | 0.0306 | 0.0893 | −0.2426. 10-3 | −0.0228 | 59.80 | |
| 8 | N20-H15…O10 | 0.0228 | 0.0688 | −0.3425. 10-3 | −0.0178 | 46.68 |
| N21-H17…O10 | 0.0290 | 0.0786 | −0.0010 | −0.0217 | 56.91 | |
| N22-H18…O23 | 0.0309 | 0.0963 | −0.2652. 10-3 | −0.0246 | 64.52 | |
| O1-H2…O24 | 0.0279 | 0.0853 | 0.2338. 10-3 | −0.0208 | 54.55 | |
| O4-H5…O24 | 0.0263 | 0.0816 | 0.2973. 10-3 | −0.0198 | 51.93 | |
| O10-H12…O34 | 0.0305 | 0.0897 | −0.8836. 10-4 | −0.0226 | 59.28 | |
| O10-H11…O7 | 0.0315 | 0.0928 | −0.1378. 10-3 | −0.0234 | 61.37 | |
| O31-H32…O34 | 0.0292 | 0.0830 | −0.4410. 10-3 | −0.0216 | 56.65 | |
| O31-H33…O7 | 0.0273 | 0.0782 | −0.5124. 10-3 | −0.0205 | 53.77 | |
| O25-H27…O31 | 0.0483 | 0.1337 | −0.0017 | −0.0369 | 96.78 | |
| O1-H3…O25 | 0.0280 | 0.0810 | −0.4246. 10-3 | −0.0211 | 55.34 | |
| O4-H6…O25 | 0.0296 | 0.0863 | −0.2494. 10-3 | −0.0220 | 57.70 | |
| O34-H35…O28 | 0.0494 | 0.1429 | −0.0014 | −0.0385 | 100.98 | |
| O28-H29…O4 | 0.0492 | 0.1378 | −0.0017 | −0.0380 | 99.67 | |
| O7-H9…O1 | 0.0518 | 0.1404 | −0.0027 | −0.0406 | 106.49 | |
| 9 | N20-H15…O25 | 0.0214 | 0.0641 | −0.3824. 10-3 | −0.0168 | 44.06 |
| N21-H17…O25 | 0.0372 | 0.1023 | −0.0010 | −0.0277 | 72.65 | |
| N22-H18…O23 | 0.0309 | 0.0959 | −0.3016. 10-3 | −0.0245 | 64.26 | |
| O4-H6…O24 | 0.0379 | 0.1168 | 0.5208. 10-3 | −0.0281 | 73.70 | |
| O28-H30…O24 | 0.0273 | 0.0809 | −0.3776. 10-3 | −0.0203 | 53.24 | |
| O28-H29…O34 | 0.0285 | 0.0812 | −0.4496. 10-3 | −0.0212 | 55.60 | |
| O25-H27…O34 | 0.0303 | 0.0887 | −0.1957. 10-3 | −0.0225 | 59.01 | |
| O37-H38…O4 | 0.0286 | 0.0829 | −0.2831. 10-3 | −0.0213 | 55.87 | |
| O1-H3…O10 | 0.0272 | 0.0774 | −0.5441. 10-3 | −0.0204 | 53.50 | |
| O10-H11…O37 | 0.0502 | 0.1374 | −0.0023 | −0.0389 | 102.03 | |
| O31-H32…O10 | 0.0296 | 0.0851 | −0.3996. 10-3 | −0.0220 | 57.70 | |
| O31-H33…O7 | 0.0268 | 0.0765 | −0.5232. 10-3 | −0.0201 | 52.72 | |
| O37-H39…O7 | 0.0306 | 0.0899 | −0.1787. 10-3 | −0.0228 | 59.80 | |
| O7-H9…O28 | 0.0502 | 0.1394 | −0.0021 | −0.0391 | 102.56 | |
| O34-H36…O31 | 0.0511 | 0.1396 | −0.0025 | −0.0399 | 104.65 | |
| O25-H26…O1 | 0.0402 | 0.1153 | −0.5262. 10-3 | −0.0298 | 78.16 | |
| 10 | N25-H21…O26 | 0.0294 | 0.0917 | −0.2402. 10-3 | −0.0234 | 61.37 |
| N24-H20…O13 | 0.0243 | 0.0718 | −0.4222. 10-3 | −0.0158 | 41.44 | |
| N23-H18…O13 | 0.0157 | 0.0455 | −0.2542. 10-3 | −0.0118 | 30.95 | |
| O10-H11…O27 | 0.0229 | 0.0675 | −0.1948. 10-3 | −0.0172 | 45.11 | |
| O37-H38…O27 | 0.0270 | 0.0779 | −0.5162. 10-3 | −0.0205 | 53.77 | |
| O37-H39…O40 | 0.0213 | 0.0658 | −0.1644. 10-3 | −0.0167 | 43.80 | |
| O40-H42…O34 | 0.0246 | 0.0717 | −0.5626. 10-3 | −0.0190 | 49.83 | |
| O4-H6…O1 | 0.0261 | 0.00786 | −0.2814. 10-3 | −0.0202 | 52.98 | |
| O28-H29…O10 | 0.0216 | 0.0679 | −0.1086. 10-3 | −0.0172 | 45.11 | |
| O13-H15…O31 | 0.0302 | 0.0879 | −0.3318. 10-3 | −0.0226 | 59.28 | |
| O4-H5…O31 | 0.0248 | 0.0700 | −0.6309. 10-3 | −0.0187 | 49.05 | |
| O28-H30…O34 | 0.3371 | 1.8245 | −0.5043. 10-3 | −0.5525 | 1449.22 | |
| O40-H41…O10 | 0.0372 | 0.1045 | −0.7301. 10-3 | −0.0275 | 72.13 | |
| O13-H14…O1 | 0.0423 | 0.1175 | −0.0010 | −0.0315 | 82.62 | |
| O1-H2…O7 | 0.0347 | 0.1032 | −0.1245. 10-3 | −0.0260 | 68.19 | |
| O7-H9…O37 | 0.0394 | 0.1133 | −0.3466. 10-3 | −0.0203 | 53.24 | |
| O34-H35…O4 | 0.0464 | 0.1282 | −0.0014 | −0.0350 | 91.80 |
The Reduced Density Gradient (RDG) function is used to understand non-covalent interactions as a wide range of real space in a molecule. Its value is determined by the electron density ρ (r) and the first derivative (Contreras Aguilar et al., 2019):
The peaks of the two-dimensional plots of the dependence of the reduced gradient on the sign of (λ2) ρ, that is, non-covalent interactions appear in areas of low density and low gradient. The sign of the second sign of the Hessein eigenvalue (λ2) is introduced to distinguish between different types of non-covalent interactions, and the density ρ represents the strength of interactions (Becke and Edgecombe, 1990). The force interactions in the molecular system, which indicate a stronger attractiveness of blue and a push of red, is analyzed using Multiwfn and VMD software.
The RDG scatter graphs of Biuret-(H2O)(1−10) clusters were indicated in Fig. 8. Based on the color scale of RDG and VMD visual representation, we can identify each type of interaction. The red, the green and the blue colors were respectively matched to steric effect, van der Waals interaction and hydrogen bonding contact. As clearly seen from RDG graphs, the hydrogen interactions appear in the range (-0.05)-(-0.02) a.u. The van der Waals interaction ranging from −0.02 to 0.01 a.u. While to range between 0.01 and 0.05 a.u was concerned to ring steric effect.
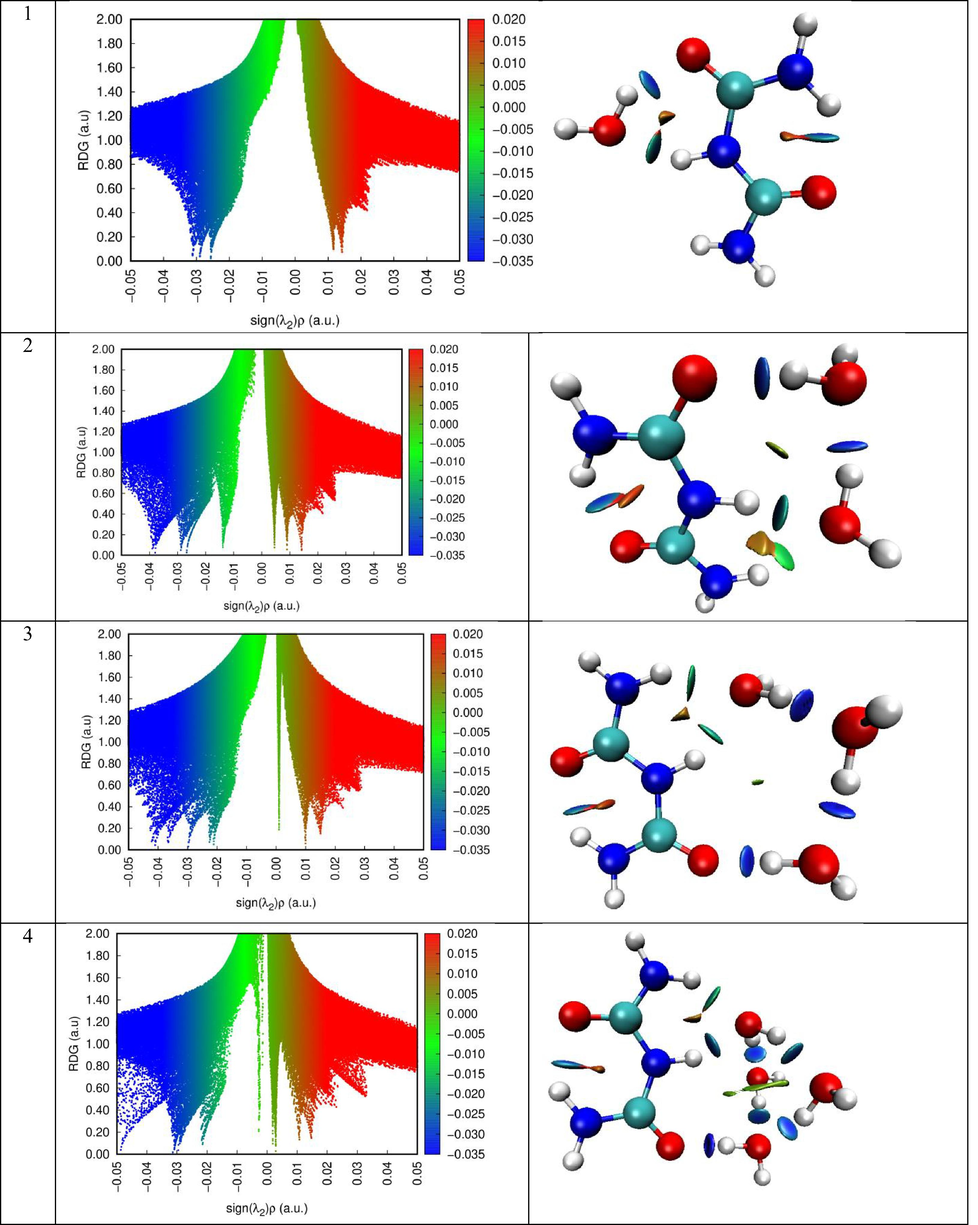
- RDG map along with VMD representation of the different clusters.
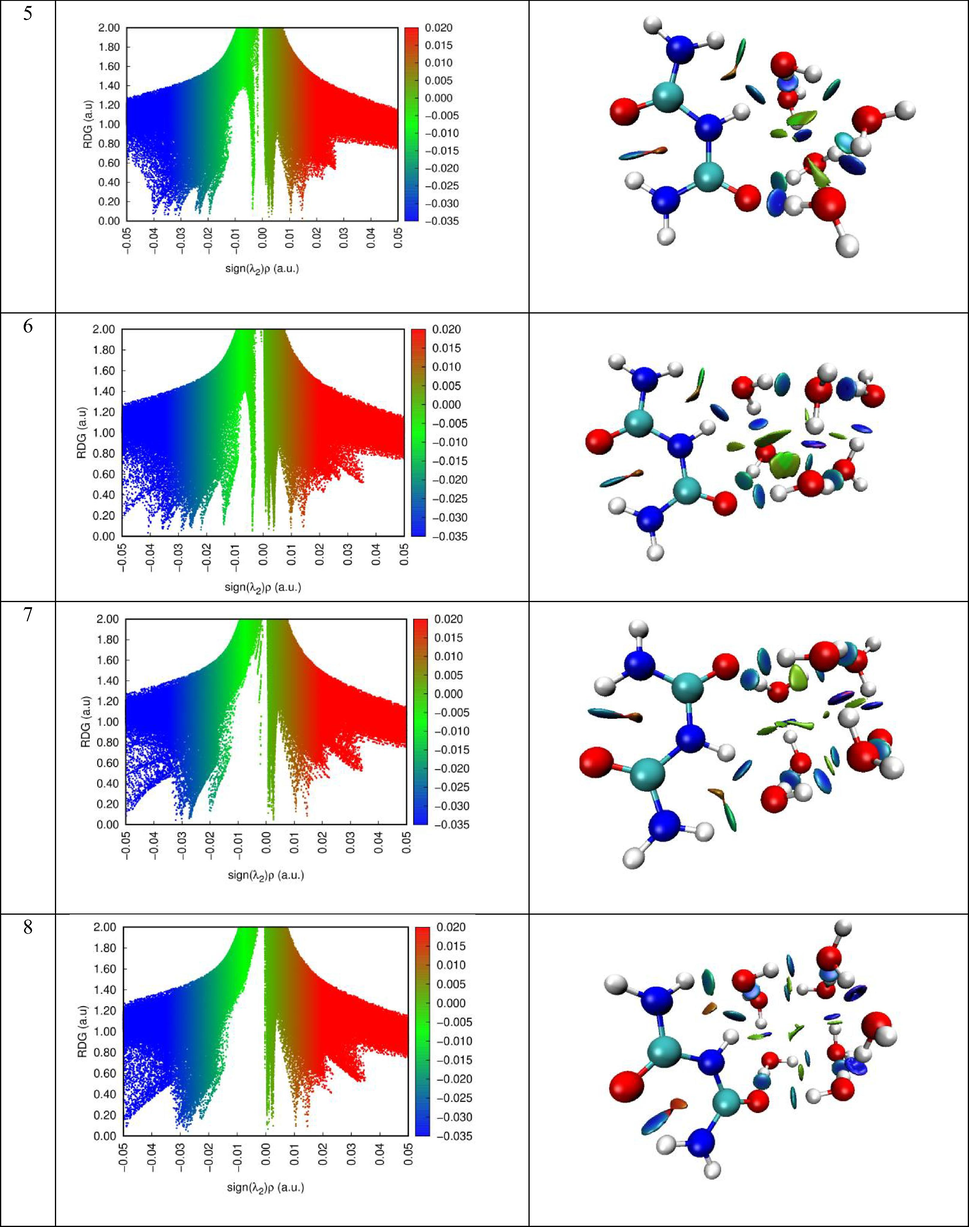
- RDG map along with VMD representation of the different clusters.
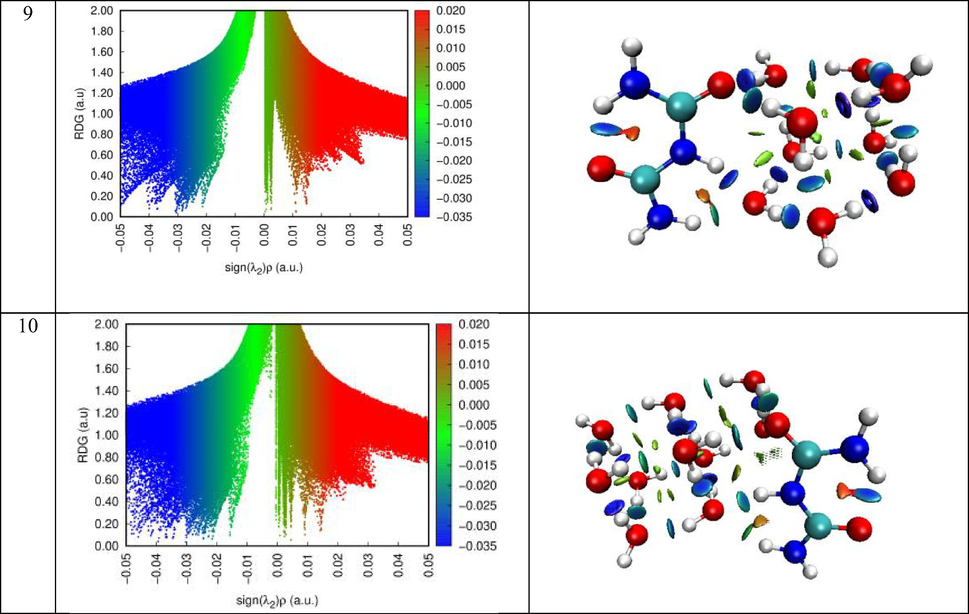
- RDG map along with VMD representation of the different clusters.
Important methods for studying the electronic structure of molecules free from an arbitrary choice of molecular orbitals are topological analysis of the electron density by Bader (AIM) (Johnson et al., 2010) and topological analysis of ELF (Michalski et al., 2019). The electronic structure of a molecule described by ELF is represented by maxima (attractors) and its field localization region η (r), which characterize covalent bonds, lone pairs, nuclear regions, and valence shells in atoms (Fuster et al., 2000).
The spatial position of these attractors makes it possible to differentiate the core basins and the valence basins (Kazachenko et al., 2021). Heart basins are located around nuclei (except for the hydrogen atom). The valence basins are classified according to their connectivity with the core basins. Topological analysis of the localization function of electrons constitutes the suitable mathematical model for the characterization of chemical bonds. The electron density, ELF diagrams of the different biuret-water clusters are shown in Fig. 9. The ELF color code range between blue and red, as shown in Fig. 9. The minimum Pauli repulsion correspond to blue regions. While the areas with maximum Pauli repulsion were colored by the red. In addition, the charge delocalization regions with ELF < 0.5 were mapped as blue spots. Whereas, the red electron localization areas with ELF > 0.5 were associated to covalent bonds.
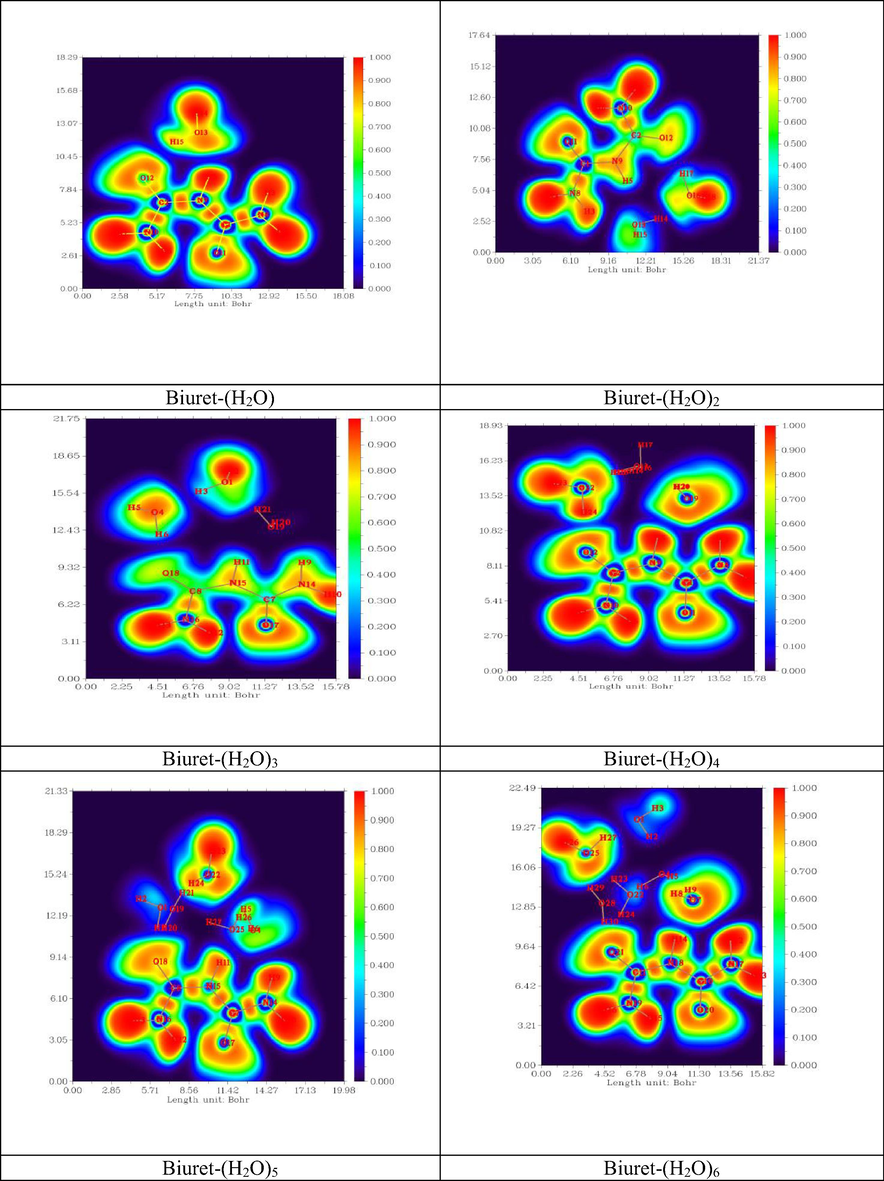
- 2D ELF representation of Biuret-water clusters.
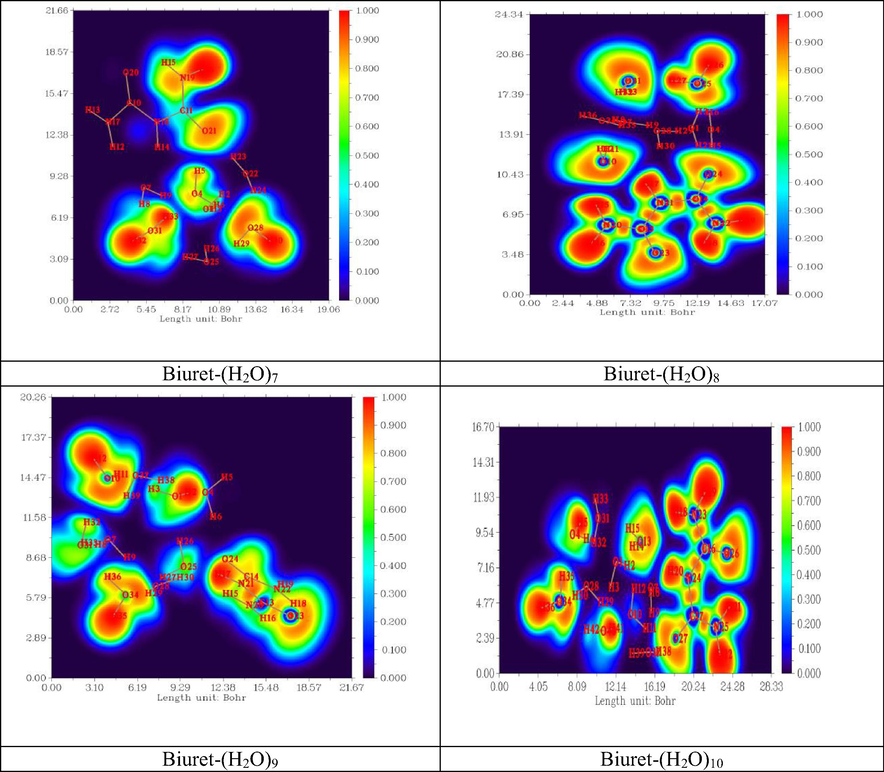
- 2D ELF representation of Biuret-water clusters.
3.6 Electrostatic potential (ESP) analysis of Biuret-water clusters
Calculation of the electronic characteristics of various materials is important for understanding their functionality and reactivity (Timmer and Mooibroek, 2021).
Electrostatic potential (ESP) or molecular electrostatic potential (MEP) has been actively used in scientific research for several decades (Sharma and Tiwari, 2016). Surface analysis by electrostatic potential (ESP) is one of the factors that can play an important role in the design of various substances (Drissi et al., 2015). Molecular electrostatic potential is the potential that a single positive charge will experience at any point surrounding a molecule, due to the distribution of electron density in the molecule. Electrostatic potential is considered to be a predictor of chemical reactivity, since areas of negative potential are expected to be sites of protonation and nucleophilic attack, while areas of positive potential may indicate electrophilic sites [50].
Different electrostatic potential values are indicated by different colors in the ESP (Fig. 10). A decreasing order potential is expressed as follows: blue > green > yellow > orange > red. Negative values are shown in red and are associated with the area of electrophilic attack and mainly with oxygen atoms in biuret, water, and the biuret-water cluster. The nucleophilic attack area (positive area) is shown in blue and is mainly associated with the hydrogen and nitrogen atoms in the biuret and the biuret-water cluster. As clearly seen from Fig. 10, the oxygen atoms of biuret-water cluster were colored with red color (nucleophilic sites). Whereas, the electrophilic sites were localized on hydrogen atoms (blue color).
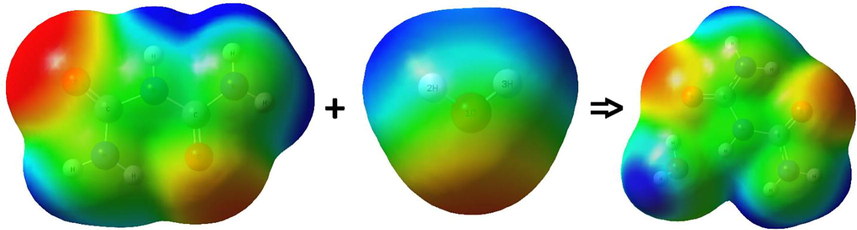
- The electrostatic potential (ESP) analysis of biuret, water and biuret-water cluster (with 1 water molecule).
4 Conclusions
In this work, water clusters of biuret (n = 1–10) were investigated by FTIR, XRD, AIM, DFT, RDG, ELF, NLO methods. It is shown that the introduction of water molecules into the biuret cluster leads to an increase in the intensity of the FTIR spectra. The inclusion of water molecules in a biuret crystal leads to a significant increase in the intensity of most peaks in the range from 14 to 65 2⊖ (deg). All calculations of biuret clusters - (H2O) (1–10) were carried out in the gas phase at the level of the B3LYP / 6–31 + G (d, p) theory. The strength of hydrogen bond interactions is discussed using the AIM topological analysis. We also studied the NLO properties for each cluster using the RB3LYP functionality. It was shown that the values of E (Thermal), Heat Capacity (Cv), Entropy (S) increase linearly with an increase in water molecules in the biuret cluster.
Declarations
Compliance with ethical standards.
5 Ethics approval
N/A. In the course of work on this article, the authors did not conduct research on animals and humans in any form.
Acknowledgments
Experimental work was conducted within the framework of the budget plan # 0287-2021-0017 for Institute of Chemistry and Chemical Technology SB RAS using the equipment of Krasnoyarsk Regional Research Equipment Center of SB RAS. Theoretical work was supported Researchers Supporting Project number (RSP -2021/61), King Saud University, Riyadh, Saudi Arabia. The authors are grateful to G.N. Bondarenko for the X-ray study and E.V. Elsuf’ev for recording the FTIR spectra.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Intermolecular hydrogen bond interactions in the thiourea/water complexes (Thio-(H2O)n) (n = 1, 5): X-ray, DFT, NBO, AIM, and RDG analyses. J. Mol. Model.. 2020;26:161.
- [CrossRef] [Google Scholar]
- Atoms in Molecules – A Quantum Theory. Oxford: Oxford University Press; 1990.
- A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys.. 1990;92:5397-5403.
- [Google Scholar]
- Hydrolysis and thermolysis of urea and its decomposition byproducts biuret, cyanuric acid and melamine over anatase TiO2. Appl. Catal. B.. 2012;115–116:129-137.
- [CrossRef] [Google Scholar]
- Structure and dynamics of urea/water mixtures investigated by vibrational spectroscopy and molecular dynamics simulation. J. Phys. Chem. B.. 2013;117(42):13291-13300.
- [Google Scholar]
- Acyl thiourea derivatives: A study of crystallographic, bonding, biological and spectral properties. Chem. Phys. Lett.. 2019;715:64-71.
- [Google Scholar]
- GaussView, Version 5. Shawnee Mission KS: Semichem Inc.; 2010.
- Efficient synthesis of polymeric g-C3N4 layered materials as novel efficient visible light driven photocatalysts. J. Mater. Chem.. 2011;21:15171-15174.
- [CrossRef] [Google Scholar]
- Theoretical and Experimental Electrostatic Potential around the m-Nitrophenol Molecule. Molecules.. 2015;20(3):4042-4054.
- [CrossRef] [Google Scholar]
- Computational evaluation on molecular structure (Monomer, Dimer), RDG, ELF, electronic (HOMO-LUMO, MEP) properties, and spectroscopic profiling of 8-Quinolinesulfonamide with molecular docking studies. Computat. Theoret. Chem.. 2021;1198:113169.
- [Google Scholar]
- Frontier Orbitals and Organic Chemical Reactions. London: Wiley; 1976.
- Feed Grade Biuret as a Protein Replacement for Ruminants. A Review. J. Animal Science.. 1975;40:1150-1184.
- [CrossRef] [Google Scholar]
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, Gaussian 09, Revision D.01,(2013) Gaussian Inc., Wallingford.
- Topological Analysis of the Electron Localization Function (ELF) Applied to the Electrophilic Aromatic Substitution. J. Phys. Chem. A. 2000;104(4):852-858.
- [CrossRef] [Google Scholar]
- A proton transfer compound template phenylethylamine: Synthesis, a collective experimental and theoretical investigations. J. Mol. Struct.. 2019;1191:183-196.
- [CrossRef] [Google Scholar]
- Melek Hajji, Jamelah S. Al-Otaibi, Marwa Belkhiria, Selma Dhifaoui, Mohamed A. Habib, Salima Moftah H Elmgirhi, Hasan Mtiraoui, Radhouane Bel-Hadj-Tahar, Moncef Msaddek, Taha Guerfel, Structural and computational analyses of a 2-propanolammonium-chlorocadmate(II) assembly: Pivotal role of hydrogen bonding and H—H interactions, J. Molecul. Struct., 1223, 2021, 128998.
- Taha Guerfel, Computational chemistry methods for modelling non-covalent interactions and chemical reactivity— An overview. J. Indian Chem. Soc.. 2021;98(11):100208
- [Google Scholar]
- Intermolecular hydrogen bonds in urea-water complexes: DFT, NBO, and AIM analysis. Comput. Theor. Chem.. 2015;1070:40-47.
- [Google Scholar]
- The Crystal Structure of Biuret Hydrate. Acta Crystallogr.. 1961;14:345-352.
- [CrossRef] [Google Scholar]
- J. Am. Chem. Soc.. 2010;132:6498-6506.
- Intermolecular hydrogen bonds interactions in water clusters of ammonium sulfamate: FTIR, X-ray diffraction, AIM, DFT, RDG, ELF, NBO analysis. J. Molecul. Liquids.. 2021;342:117475.
- [Google Scholar]
- Optimization of guar gum galactomannan sulfation process with sulfamic acid. Biomass Convers. Biorefin. 2021
- [CrossRef] [Google Scholar]
- Theoretical and experimental study of guar gum sulfation. J. Mol. Model. 2021;27:5.
- [CrossRef] [Google Scholar]
- Sulfamic acid/water complexes (SAA-H2O(1–8)) intermolecular hydrogen bond interactions: FTIR, X-ray, DFT and AIM analysis. J. Molecul. Struct.. 2022;1265:133394.
- [Google Scholar]
- Urea-SCR: a promising technique to reduce NOx emissions from automotive diesel engines. Catal. Today.. 2000;59:335-345.
- [CrossRef] [Google Scholar]
- B. Kunkle, J. Fletcher, D. Mayo. Florida Cow-Calf Management, 2nd Edition - Feeding the Cow Herd. IFAS Extension, University of Florida. Publication #AN117. (2013).
- Simple pyrolysis of urea into graphitic carbon nitride with recyclable adsorption and photocatalytic activity. J. Mater. Chem.. 2011;21:14398-14401.
- [CrossRef] [Google Scholar]
- Dynamics of urea-water mixtures studied by molecular dynamics simulation. J. Molecul. Liquid.. 2020;300
- [Google Scholar]
- Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem.. 2012;33(5):580-592.
- [CrossRef] [Google Scholar]
- Water: From Clusters to the Bulk. Angew. Chem. Int. Ed.. 2001;40(10):1808-1827.
- [CrossRef] [Google Scholar]
- Topological analysis of the electron localisation function (ELF) applied to the electronic structure of oxaziridine: the nature of N-O bond. Struct. Chem.. 2019;30:2181-2189.
- [CrossRef] [Google Scholar]
- Palladium-Catalyzed Cross-Coupling Reactions in Total Synthesis. Angew. Chem. Int. Ed.. 2005;44:4442-4489.
- [CrossRef] [Google Scholar]
- Quantum chemical studies on molecular structure, AIM, ELF, RDG and antiviral activities of hybrid hydroxychloroquine in the treatment of COVID-19: Molecular docking and DFT calculations. J. King Saud Univers. – Sci.. 2021;33(2):101334.
- [Google Scholar]
- Urea versus biuret in a roughage diet for steers. J. Animal Science.. 1969;29:816-822.
- [CrossRef] [Google Scholar]
- A complete basis set model chemistry. II. Open‐shell systems and the total energies of the first‐row atoms. J. Chem. Phys.. 1991;94(9):6081-6090.
- [Google Scholar]
- A complete basis set model chemistry. I. The total energies of closed‐shell atoms and hydrides of the first‐row elements. J. Chem. Phys.. 1988;89(4):2193-2218.
- [Google Scholar]
- Comparative computational analysis of electronic structure, MEP surface and vibrational assignments of a nematic liquid crystal: 4-n-methyl-4́-cyanobiphenyl. J. Mol. Liq.. 2016;214:128-135.
- [CrossRef] [Google Scholar]
- Small Water Clusters in Crystalline Hydrates. J. Clust. Sci.. 2003;14:337-366.
- [CrossRef] [Google Scholar]
- Intermolecular π–π Stacking Interactions Made Visible. J. Chem. Educ.. 2021;98(2):540-545.
- [CrossRef] [Google Scholar]
- Communication: Single crystal x-ray diffraction observation of hydrogen bonding between 1-propanol and water in a structure II clathrate hydrate. J. Chem. Phys.. 2011;134(12):121104
- [CrossRef] [Google Scholar]
- Biuret complexes of copper(II) and nickel(II) J. Indian Chem. Soc.. 1975;52:585-588.
- [Google Scholar]
- Infrared spectra of succinimide and maleimide in the crystalline state. Bull. Chem. Soc. Japan. 1962;35:276-283.
- [Google Scholar]
- Synthesis, Structures and Properties of Two New Coordination Polymers with Unprecedented Water Cluster. J. Clust. Sci.. 2015;26(3):959-972.
- [Google Scholar]
- Synthesis, Optical Characterization, and Thermal Decomposition of Complexes Based on Biuret Ligand. Int. J. Optics 2016:1-8.
- [CrossRef] [Google Scholar]
- Ozone-Water Interaction Revisited Through [(O3)m···(H2O)n] Clusters. J. Clust. Sci.. 2017;28:1693-1708.
- [CrossRef] [Google Scholar]
- Urea Derivatives as Functional Molecules: Supramolecular Capsules, Supramolecular Polymers, Supramolecular Gels, Artificial Hosts, and Catalysts. Chem. Eur. J.. 2021;27:5601-5614.
- [CrossRef] [Google Scholar]
- ABCluster: the artifcial bee colony algorithm for cluster global optimization. Phys. Chem. Chem. Phys.. 2015;17(37):24173-122418.
- [CrossRef] [Google Scholar]
- Chemistry for Chemical and Biological Sciences. Sausalito, CA: University Science Books; 2000.




