

Translate this page into:
Genetic association study of NOD2 and IL23R amino acid substitution polymorphisms in Saudi Inflammatory Bowel Disease patients
⁎Corresponding authors at: Pediatric Gastroenterology Unit, Department of Pediatrics, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia (O.I. Saadah). Department of Genetic Medicine, Faculty of Medicine, King Abdulaziz University, Jeddah-21589, Saudi Arabia (B. Banaganapalli). osaadah@kau.edu.sa (Omar Ibrahim Saadah), bbabajan@kau.edu.sa (Babajan Banaganapalli)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background
Inflammatory Bowel Disease (IBD) is a complex autoimmune disease whose genetic basis is not well explored in the highly consanguineous Saudi population. Therefore, this study aims to evaluate the relevance of NOD2 and IL23R genes polymorphisms, selected from Caucasian Genome-wide association study (GWAS) findings, as risk markers for IBD pathology in Saudi patients.
Materials and Methods
The genetic status of NOD2 and IL23R polymorphisms of 97 IBD patients and 100 healthy individuals was determined through real-time PCR based TaqMan Allelic Discrimination Assay. Genotype calls of TaqMan assay were further validated by the direct DNA sequencing method. The pathogenicity of the variants was further explored by computational predictions and by 3D structural modelling methods.
Results
Our study found no evidence for a significant association of Caucasian IBD risk alleles, i.e., NOD2 (P268S and R702W) and IL23R (G149R and R381Q) with IBD pathology in Saudi Arabian patients under dominant, recessive or co-dominant genetic models. Moreover, SNP-SNP interaction analysis with multidimensionality reduction assays have also confirmed the above findings. Our computational predictions of the variants suggest the variable deleterious nature and minor structural drifts on the tertiary protein structures.
Conclusion
This study expands the genetic heterogeneity of IBD and concludes that Caucasian genetic risk markers have limited or of no use in estimating the risk of IBD development in Saudi patients. Future genetic association studies in Saudi Arabia need to employ genetic markers whose minor allele frequencies exceed 10% in Arab population to have robust outcomes.
Keywords
NOD2
IL23R
Inflammatory Bowel Disease
Crohn’s disease
Polymorphism

- IBD
-
Inflammatory Bowel Disease
- CD
-
Crohn’s disease
- UC
-
Ulcerative colitis
- GWAS
-
Genome-wide association study
- IL23R
-
Interleukin 23 Receptor
- NOD2
-
Nucleotide Binding Oligomerization Domain Containing 2
Abbreviation
1 Introduction
Inflammatory Bowel Disease (IBD) [OMIM: 266600] is a chronic autoimmune disease caused by complex interactions between genetic susceptibility of individuals, dysfunction of the immune response to normal microbial flora and exposure to external environmental factors. The two major subtypes of IBD, i.e., Crohn’s disease (CD) and Ulcerative colitis (UC) share similar pathological features and clinical manifestations (Dias‐Santagata et al., 2010). Overall, in the last two decades, prevalence of IBD has been increasing, especially in North America and Europe compared to Asian countries (Yamada et al., 2017). IBD has become an economic burden on human society due to the high cost of biological therapy and loss of productivity due to patient disability (Buckley et al., 2013; van der Valk et al., 2014). Nonetheless, the exact etiology of IBD is yet to be fully understood (Ek et al., 2014).
The increased prevalence of IBD etiology among twins, family members, and various ethnic groups highlights the potential contribution of genetic factors to disease etiology. (de Lange et al., 2015). While IBD is mostly considered a multifactorial disease, few rare monogenic types with an underlying defect in the IL-10 signaling pathway are also found in a subset of IBD patients during childhood (Bianco et al., 2015). On the other hand, a polygenic etiology, where hundreds of genetic susceptibility loci contribute to disease development, is seen in the late-onset IBD patients (Mesbah-Uddin et al., 2015). The advent of high-throughput microarray-based genotyping methods has allowed to conduct genome-wide association studies (GWAS) for many complex traits, including IBD. GWAS has identified different disease susceptibility loci markers, and vast majority of them are high frequency allelic variants. (Manolio, 2010). Few of those risk alleles are missense variants mapped to coding regions of genes like Nucleotide-binding Oligomerization Domain containing 2 (NOD2), Autophagy-related 16 Like 1 (ATG16L1), and Interleukin 23 Receptor (IL23R) (Wijmenga and Zhernakova, 2018). A large-scale GWAS study among Caucasians extended the number of IBD susceptibility loci to more than 200 (Uhlig and Muise, 2017). Approximately 87% of these risk markers are intronic variants, while the remaining 13% are mapped to the coding regions (Bianco et al., 2015; Mirkov, et al., 2017; Naser et al., 2012; ).
In general, majority of the GWA studies on IBD have been conducted in Caucasian or Asian populations. Thus, population-specific genetic variations in culturally and ethnically different disease groups like Arab populations living in Middle Eastern Asian countries are not well explored. The high prevalence of consanguinity in the Arab population makes this group unique, which provides an opportunity to reveal the novel genetic basis of complex disorders due to autozygosity (Al-Aama et al., 2017). Recent study has confirmed that consanguinity is extremely common in Saudi IBD patients (Mosli et al., 2017). Moreover, its potential impact on the emergence of pediatric onset UC in patients with known high familial aggregation of IBD was also reported (Al-Hussaini et al., 2016). Therefore, this study has aimed to evaluate the relevance of NOD2 (P268S and R702W) and IL23R (G149R and R381Q) polymorphisms, selected from Caucasian GWA study findings, as genetic markers for IBD pathology in Saudi patients.
2 Material and methods
2.1 SNPs selection
The DisGeNET (https://www.disgenet.org/) and GWAS Catalog (https://www.ebi.ac.uk/gwas/home) databases were used to identify the genetic biomarkers associated with IBD. The GWAS catalog has been used to find all SNPs associated with IBD. Non-synonymous missense variants showing association (p-value ≤5 × 10−8) with IBD risk in Caucasian populations were further filtered (Wijmenga and Zhernakova, 2018). The selected SNPs population frequencies were validated in Ensembl Human GRCh38.p13 genome (https://www.asia.ensembl.org/index.html), the Saudi Genome Project (SGP) database (https://www.sso.saudigenomeproject.com/), and the Greater Middle East (GME) Variome Project (GME Variome) (http://www.igm.ucsd.edu/gme/data-browser.php).
2.2 Subjects and Ethical approval
This research work was executed at the Department of Genetic Medicine, Faculty of Medicine, and PACER-HD labs of King Abdulaziz University. Ethical approval for this proposal was obtained through the Institutional Review Board (IRB) at King Abdulaziz University Hospital. The inclusion criteria for recruitment of IBD patients include confirmed clinical and pathological diagnosis of IBD, free from the family history of IBD, and Saudi nationality. All non-Saudi patients or Saudis with a positive family history of IBD were excluded. Healthy Saudis with no reported family history of IBD or any other autoimmune, gastrointestinal, or metabolic diseases were recruited as controls.
2.3 Sampling
IBD patients were recruited from pediatric and adult gastroenterology clinics based on their confirmed diagnosis, clinical presentations, endoscopic and histopathological characteristics, and gastrointestinal imaging. All study participants (97 IBD patients and 100 healthy controls) or their parents/guardians (for children less than 18 years of age) were thoroughly informed about the study’s aims and significance before recruitment and they signed the informed consent forms voluntarily. The clinical data, such as symptoms and age at the onset, gastrointestinal manifestations, and extra-intestinal manifestations, were also obtained from the hospital records of each participant.
2.4 Genomic DNA isolation
Two milliliters of peripheral blood were obtained from each participant in EDTA vacutainers and stored at −20°C until further processing was done. The DNA quality and quantity analysis was done by NanoDrop™ 2000c Spectrophotometer.
2.5 Genetic analysis
After extracting and quantifying DNA samples, Realtime PCR based TaqMan Allelic Discrimination Assay was carried out for genotyping 4 SNPs [rs76418789, rs11209026, rs2066842, rs2066844] using ABI7500HT Sequence Detection System (Applied Biosystems).
2.6 Validation
Sanger sequencing was done to verify the genotypes obtained by real-time PCR. Three samples from different genotype categories (homozygous major allele, homozygous minor allele and heterozygous alleles) of all four SNPs were sequenced using a 3500 Genetic Analyzer machine (Applied Biosystems). The PCR product sizes of NOD2 gene SNPs, i.e., rs2066842 and rs2066844, were 574 bp and 558 bp respectively, and IL23R gene SNPs, i.e., rs76418789 and rs11209026, were 390 bp, and 480 bp, respectively . The primers for the Polymerase Chain Reaction (PCR) for selected SNPs were designed using Primer3 (http://primer3.ut.ee/), and their specificity was validated using UCSC (https://genome.ucsc.edu/) and Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi) (.
2.7 Statistical analysis
GraphPad QuickCalcs, version 6.0 (GraphPad Software Inc., USA) online software was used to perform statistical analysis of the categorical (in percentage form) and continuous variable (in mean and standard deviation form) data. The statistical significance of allele and genotype frequency distributions was tested with the help of Pearson's standard chi-square test, odds ratio (OR), and 95% confidence interval (CI). Additionally, Hardy-Weinberg equilibrium was checked by the contingency table of observed versus predicted genotype distributions by using a chi-squared test with one degree of freedom. Epistasis between four examined polymorphisms and IBD occurrence was tested using the open-source multifactor dimensional reduction (MDR) program (MDR version 2.0 beta 5 http://www.epistasis.org/). The MDR (0.4.9 alpha) permutation test module performs more than 1000 permutation experiments. In this study, we compared the observed test accuracy with the predicted results to analyze the statistical significance. A p-value of <0.05 was considered as statistically significant.
2.8 Pathogenicity prediction
We used the variant effect predictor (VEP) webserver (http://asia.ensembl.org/Tools/VEP) hosted in Ensembldatabase to determine the variant pathogenicity with many computational prediction methods (McLaren et al., 2016). VEP accepts different input options including chromosome and nucleotide numbers, allelic variant, strand of the variant in human genome variation society (HGVS) identification format. From the VEP’s output, pathogenicity prediction scores of Sorting Intolerant from Tolerant (SIFT), Polyphen2, CADD, and Condel, were selected. The deleterious prediction (x) scoring criteria adopted by these different prediction tools is as follows; x < 0.05 for SIFT, x > 0.5 scores for PolyPhen2, x > 25 score for CADD and x > 0.5 score for Condel.
2.9 The 3-dimensional protein modelling and stability analysis
We have also explored the variant induced effects on structural features of proteins at molecular level. In this regard, to build the molecular 3D model of proteins we used I-Tasser webserver (Yang and Zhang 2015), which generates five models based on multiple-threading alignments and iterative template fragment assembly simulations. Out of these five models, the best model (selected based on the confidence (C) score, TM-score and RMSD values) was considered for downstream structural analysis. The selected models were further subjected to energy minimization to remove bad contacts in the protein structures by the SPDBV software. The energy minimized models of wild NOD2 and IL23R proteins have been used as a template to build the mutated NOD2 and IL23R structural models, followed by mutant protein stability analysis by the DUET web server (http://biosig.unimelb.edu.au/duet/) (Pires et al., 2014). The Pymol tool was used to evaluate the structural divergence between wildtype and mutant models of NOD2 and IL23R (Janson et al., 2017).
3 Results
3.1 Clinical details
In this study, 97 patients (53 CD patients, 25 UC patients, and 19 indeterminate IBD) and 100 healthy control participants were recruited. The gender distribution of our IBD patients showed that there were more males (57.7%) than females (43.2%). The mean age of disease onset was 10.98 ± 7.39 years, and the average age was 16.84 ± 8.13 years. Most were diagnosed during their childhood (88.6%), and the rest (11.4%) in adulthood. Most of the CD patients had ileocolonic location (57%), and 16% had inflammation limited to the terminal ileum; but none of the participants had upper GI involvement. More than half (58%) of CD patients had a non-structuring and non-penetrating disease behavior, 28.6% structuring disease, and 13.4% penetrating disease behavior. In UC patients, (according to Paris classification) 33.8% had pancolitis, 33.8% Left-sided colitis, and about 32.4% with extensive UC. According to the Montreal classification, approximately 44.4% of UC patients had extensive UC, 38.3% ulcerative proctitis, and 17.3 % left-sided UC.
3.2 SNPs selection
Searching for the keywords “Inflammatory Bowel Disease” on the GWAS catalog database yielded 1150 SNPs. Using the DisGeNET database, we identified five genes or SNP with P-value (p-value ≤5 × 10−8). Concordance results between the DisGeNET and GWAS catalog resulted in 37 significantly associated missense SNPs with IBD. Out of 37 missense SNPs, four including two in NOD2 gene (rs2066842, rs2066844) and two in IL23R gene (rs76418789, rs11209026), which were previously reported in the Caucasian IBD patients were selected (Supplementary Table S1). The variant details like its chromosomal location, variant nature and minor allele frequency (MAF) were also obtained from Ensemble and SGP databases (Table 1). *Highest Population MAF is the highest minor allele frequency observed in any population depended on Ensemble database.
rs ID
Chromosomal position
Gene
Chr location
Region
Nucleotide change
Amino acid change
Highest Population MAF*
Saudi MAF
rs2066842
16:50710713
NOD2
16q21.1
Exon 4
802C > T
Pro268Ser
0.32
0.2350
rs2066844
16:50712015
NOD2
16q21.1
Exon 4
2023C > T
Arg702Trp
0.16
0.0064
rs76418789
1:67182913
IL23R
1p31.3
Exon 4
445G > A
Gly149Arg
0.10
0.00168
rs11209026
1:67240275
IL23R
1p31.3
Exon 9
1142G > A
Arg381Gln
0.10
0.0899
3.3 Genetic analysis
The genotyping results of all four tested SNPs [rs2066842, rs2066844, rs11209026, rs76418789] were consistent with Hardy-Weinberg equilibrium in Saudi cohort. For rs2066842 (c.802C > T: p.P268S, NOD2) polymorphism, the distribution of the T allele in both monoallelic and biallelic forms is almost similar in IBD patients and control individuals (p values are > 0.05 for all tests). Additional testing of T-allele under recessive and codominant models have also confirmed the above findings (p values are >0.05 for all tests). Hence, it is concluded that T allele is not associated with IBD development in Saudi population (p = 0.71, X2 = 0.13, OR (95% CI) = 1.09 [0.67–1.73]) (Table 2). (*)represents that the value is subjected to Yates correction.
Gene
Amino acid Variant
SNP ID
Genotype/Allele
IBD cases [n = 93] (%)
Controls [n = 100] (%)
OR (95% CI)
X2
p-Value
NOD2
Pro268Ser
rs2066842
CC
61 (65.5)
65 (65)
Reference
CT
24 (25.8)
30 (30)
0.85 [0.44–1.61]
0.23
0.62
TT
8 (8.6)
5 (5)
1.70 [0.5–5.48]
0.81
0.36
CC vs TT + CT
61 vs 32
65 vs 35
0.97 [0.53–1.76]
0.07
1
CT vs TT + CC
24 vs 69
30 vs 70
1.23[ 0.65–2.31]
0.42
0.51
TT vs CC + CT
8 vs 85
5 vs 95
0.55 [0.17–1.77]
0.09
0.31
C
146 (0.78)
160 (0.80)
Reference
T
40 (0.21)
40 (0.20)
1.09 [0.67–1.73]
0.13
0.71
Arg702Trp
rs2066844
CC
92 (98.9)
99 (99)
Reference
CT
1 (1.07)
1 (1)
1.07 [0.06–17.45]
0.03
1
TT*
0
0
1 [0.979–1.02]
0.001
1
CC vs TT + CT
92 vs 1
99 vs 1
1.07 [0.06–17.45]
0.03
1
CT vs TT + CC
1 vs 92
1 vs 99
0.92 [0.05–15.07]
0.03
1
TT* vs CC + CT
0 vs 93
0 vs 100
1.07 [0.02–53.6]
0.01
1
C
185 (0.99)
199 (0.99)
Reference
T
1 (0.054)
1 (0.054)
1.07 [0.06–17.32]
0.03
1
IL23R
Gly149Arg
rs76418789
GG
89 (98.8)
98 (100)
Reference
AG*
0
0
0.99 [0.97–1.02]
0.002
1
AA
1 (1.11)
0
0.98 [0.96–1.01]
1.61
0.20
GG vs AA + AG
89 vs 1
98 vs 0
0.98 [0.96–1.01]
1.61
0.20
AG vs AA + GG*
0 vs 90
0 vs 98
1 [0.97–1.02]
0.002
1
AA vs GG + AG
1 vs 89
0 vs 98
0.012 [−0.01–0.033]
1.61
0.20
G
178 (0.98)
196 (1)
Reference
A
2 (0.01)
0
0.98 [0.97–1.00]
3.23
0.07
Arg381Gln
rs11209026
GG
80 (94.11)
93 (93)
Reference
AG
5 (5.88)
6 (6)
1.01 [0.99–1.03]
1.31
0.25
AA
0
1 (1)
5.81 [0.66–50.7]
3.19
0.07
GG vs AA + AG
80 vs 5
93 vs 6
0.96 [0.28–3.29]
0.03
1
AG vs AA + GG
5 vs 80
6 vs 94
1.02 [0.3–3.47]
0.01
1
AA vs GG + AG
0 vs 85
1 vs 99
−0.01 [−0.03–0.01]
1.31
0.25
G
165 (0.97)
192 (0.96)
Reference
A
5 (0.03)
8 (0.04)
0.72 [0.23–2.26]
0.30
0.58
For rs2066844 (c.2023C > T: p.R702W, NOD2) polymorphism, the homozygous C allele is the predominant genotype (99%) in the Saudi Arabia population. Though the homozygous T allele was completely absent, one individual in both case and control groups has possessed this allele in heterozygous form. The extreme rare prevalence of the minor allele (T) in the Saudi population indicates that this polymorphism is not associated with IBD development (p = 1, X2 = 0.03, OR (95% CI) = 1.07 [0.06–17.32]) (Table 2).
The major allele is predominant in both case and control group subjects for rs76418789 (c.445G > A: p.G149R, IL23R) polymorphism. The homozygosity of minor allele was found in one IBD patient, but absent in controls. Moreover, the heterozygous AG allele is also missing in both cohorts (p values are >0.05 for all tests). Both recessive and co-dominant models have further confirmed that there is no statistical association of this ‘A’ allele with disease risk in Saudi IBD patients (p = 0.07, X2 = 3.23, OR (95% CI) = 0.98 [0.97–1.00]) (Table 2).
For rs11209026 (c.1142G > A: p.R381Q, IL23R) polymorphism, the minor allele is completely absent in the IBD patient group but found in one control subject. The distribution of heterozygous and homozygous (major allele) genotypes was comparable in study cohorts. Both dominant and recessive models have also not shown any significant difference in their distribution (p values are >0.05 for all tests). Hence, it is concluded that the A allele is not associated with the IBD risk in Saudi population (p = 0.58, X2 = 0.30, OR (95% CI) 0.72 [0.23–2.26]) (Table 2).
3.4 Epistatic interaction between NOD2 and IL23R SNPs and IBD risk
The MDR test was used to identify SNP-SNP interactions in IBD cases and controls. MDR results suggested that none of the SNP-SNP combinations of NOD2 and IL23R genes is associated with a statistically significant risk of IBD (Table 3).
Genotype Model
Cross-Validation consistency
Testing accuracy
X2
OR
95% CI
p-Value
rs2066842
9
0.5224
0.2879
1.3557
0.4781–3.8441
0.5915
rs11209026/rs2066842
8
0.5308
0.5565
1.6357
0.5202–5.1546
0.4557
rs76418789/rs11209026/rs2066842
10
0.5347
0.5163
1.3964
0.5438–3.5860
0.4724
rs76418789/rs11209026/rs2066842/rs2066844
10
0.5353
0.5311
1.4030
0.5465–3.6019
0.4661
3.5 Sanger sequencing validation of TaqMan allelic discrimination assay results
The success rate of TaqMan assay genotyping for NOD2 SNPs i.e., rs2066842 (P268S) and rs2066844 (R702W) in IBD cases is 95.87% (93/97) and 100% (100/100) in controls. For IL23R, the success ratio of genotyping the rs76418789 (G149R) SNP in the case group is 91.75% (89/97), and in the control group, it is 98% (98/100). The rs2066844 (R702W) SNP was successfully genotyped in 87.62% (85/97) of cases and 100% (100/100) of control group subjects. The TaqMan genotype calls were further validated by directly sequencing target DNA regions harboring NOD2 and IL23R SNPs. The Sanger sequencing chromatograms of these 4 SNPs are shown in Figs. 1, 2. Our real-time PCR and Sanger sequencing results have shown a 100% match in genotype calling of IBD SNPs.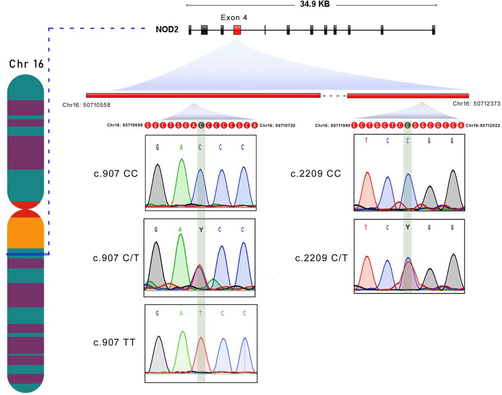
NOD2 gene and its exon localization, the Sanger sequencing chromatograms of NOD2 SNPs (rs2066842 and rs2066844).
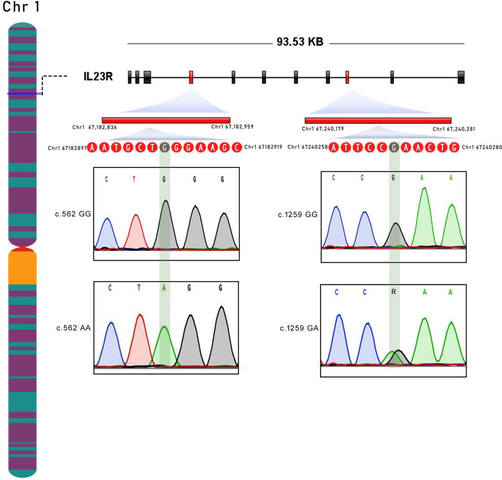
Sanger sequencing chromatograms of IL23R SNPs (rs76418789 and rs11209026) GG (wild-type homozygote); GA (heterozygote), and AA (variant homozygote).
3.6 Computational pathogenicity predictions of SNPs
Table 4 shows the pathogenicity prediction scores of IL23R and NOD2 SNPs. The variant rs2066842 in the NOD2 gene, was predicted to be benign by SIFT, PolyPhen2, CADD and Condel prediction tools. Whereas the other variant (rs2066844), was tested by all the four tools and predict a deleterious functional effect on the NOD2 protein. All four prediction tools have attributed the deleteriousness to rs76418789 and rs11209026 to the structure and functional features of IL23R protein (Table 4). 1* SIFT: Value < 0.05 (Deleterious), Value > 0.05 (Benign). 2* Polyphen2: Value > 0.5 (Deleterious), Value < 0.5 (Benign). 3* CADD: Value > 25 (Deleterious), Value < 25 (Benign). 4* Condel: Value > 0.5 (Deleterious), Value < 0.5 (Benign).
Variation
SYMBOL
Exon
cDNA position
Amino Acid
Codon
SIFT*
PolyPhen2*
CADD*
Condel*
rs2066842
NOD2
4/12
802
P268S
CCC/ TCC
0.56
0.003
0.049
_
rs2066844
NOD2
4/12
2104
R702W
CGG/TGG
0.01
0.72
23.6
0.667
rs76418789
IL23R
4/11
562
G149R
GGG/AGG
0
1
24.9
0.945
rs11209026
IL23R
9/11
1259
R381Q
CGA/CAA
0.02
0.995
26.0
0.841
3.7 3-D modeling of NOD2 and IL23R
NOD2 and IL23R protein crystal structures are not fully available in the Protein Data Bank (PDB). Therefore, the full-length NOD2 and IL23R protein was constructed depending on the hierarchical protein structure modeling approach by the I-TASSER webserver. The best NOD2 protein model predicted by this server was chosen based on the maximum C-score (0.08), template modeling (TM) score (0.72 ± 0.11), and RMSD (8.8 ± 4.6 Å), which indicates good structural similarities between template and the query proteins. The human NOD2 gene encodes a 1040 residue long protein of a 110 kDa that contains two domains in its N- terminal: caspase recruitment domain (CARD) and NACHT domain. A helical linker known as NOD2 winged-helix domain links the N-terminal domain with a central NOD domain that known as an NLRC4 helical domain HD2. The C-terminal consists of three repeated LRR domains. Fig. 3 shows the 3D molecular structure of NOD2 protein.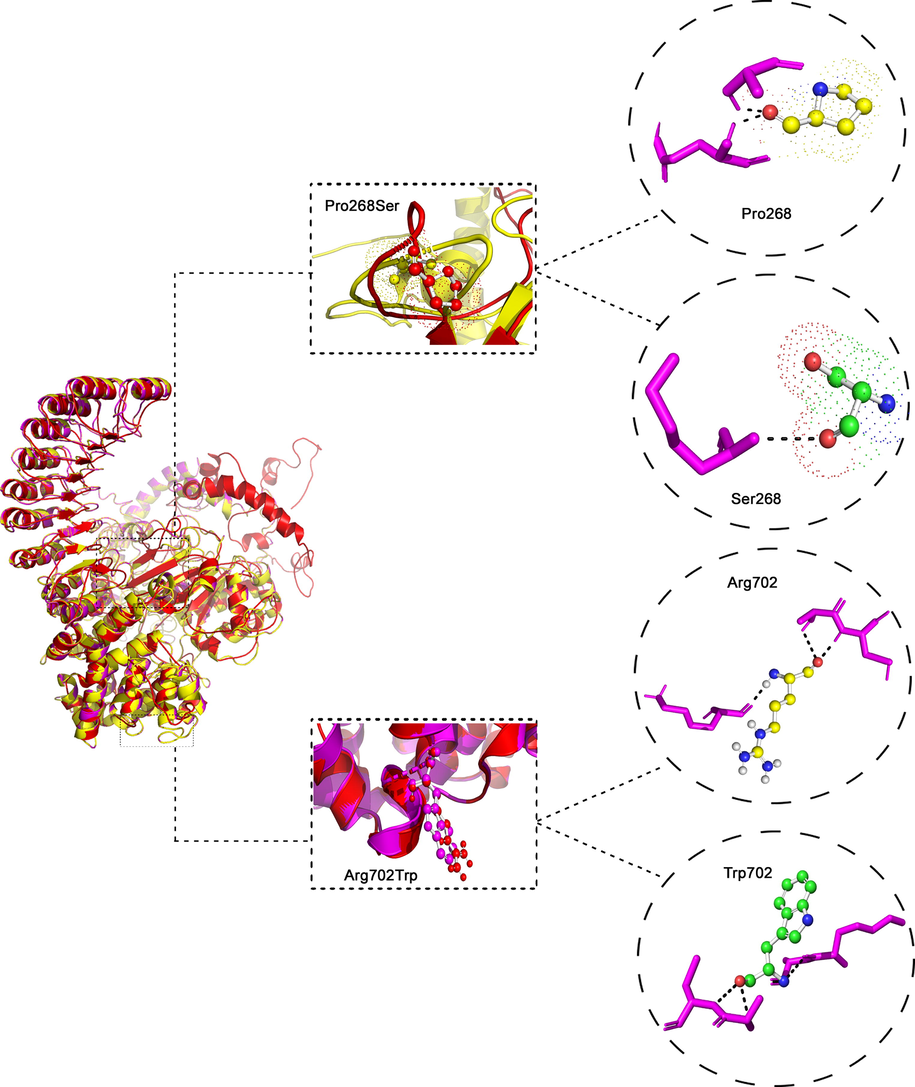
The 3-dimensional Protein Structure of NOD2 protein. In the middle Superpose of Pro268Ser and Arg702Trp in NOD2 protein. The wildtype Proline 268 and mutant Ser 268 amino acid interactions with surrounding amino acids in the NOD2 protein. The wildtype Arg 702 and mutant Trp 702 amino acid H- Bond interaction with surrounding amino acids in the NOD2 protein.
The best model of IL23R protein has been chosen based on the maximum C-score (-1.39) that demonstrates the predicted model's reliability, Template Modeling (TM) score (0.54 ± 0.15), and RMSD (11.1 ± 4.6 Å), indicate the structural similarities between template and the query proteins. The C-score value ranged between −5 to 2, which reflect the good quality of predicted protein models. The human IL23R gene encodes a 629-residue protein that weighs 71.7 kDa and consists of Interleukin-6 receptor alpha chain binding domain, fibronectin type-III domain, cytokine interleukin-12p40 C-terminus, and Ig-like C2-type domain. Fig. 15-b shows the 3D molecular structure of IL23R protein. NOD2 and IL23R generated protein models were further energy minimized using steepest descent algorithm and models were minimized around 1500 cycles.
3.8 Stability analysis of NOD2 and IL23R missense mutations
Table 5 shows free energy (ΔΔG) values for NOD2 (P268S and R702W) and IL23R (G149R and R381Q) variants by DUET webserver, which calculates the combined and consensus predictions of both mCSM and SDM methods (Pires et al., 2014). As per DUET results, all the 4 variants were predicted to be destabilizing to their corresponding proteins owing to their free energy values. The rs2066842 variant results in the substitution of Proline to Serine amino acid at the 268th residue of NOD2 protein. Pro268 Relevant Solvent Accessibility (RSA) property is 66%, and the Absolute Surface Accessibility (ASA) degree is 93 Å. The Pro268 binds to Ala266 by a total of 2.78 hydrogen bonds. The RSA property percentage of Serine is 56%, the ASA degree is 65 Å, and it binds to Asp262 by 2.62 hydrogen bonds. The total RMSD values of the c-alpha atoms in each amino acid of wild and mutant NOD2 protein models have been calculated to estimate the structural drifts at both amino acid residues and polypeptide chain levels. The RMSD value of the normal NOD2 protein model is 0.0 Å, and the RMSD value of the mutant model is 0.02 Å. The alteration in hydrogen bonds and RMSD values may alter the structure and function of the protein (Fig. 3).
SNPs
Amino acid
changemCSM Predicted Stability Change (ΔΔG)
SDM Predicted Stability Change (ΔΔG)
DUET Predicted Stability Change (ΔΔG)
Secondary structure
rs2066842
Pro268Ser
−1.732 Kcal/mol (Destabilizing)
0.31 Kcal/mol (Stabilizing)
−1.348 Kcal/mol (Destabilizing)
Loop or irregular
rs2066844
Arg702Trp
−0.062 Kcal/mol (Destabilizing)
0.1 Kcal/mol (Stabilizing)
−0.218 Kcal/mol (Destabilizing)
Loop or irregular
rs76418789
Gly149Arg
−0.468 Kcal/mol (Destabilizing)
0.18 Kcal/mol (Stabilizing)
−0.193 Kcal/mol (Destabilizing)
Loop or irregular
rs11209026
Arg381Gln
−0.442 Kcal/mol (Destabilizing)
−0.53 Kcal/mol (Destabilizing)
−0.421 Kcal/mol (Destabilizing)
Loop or irregular
In case of rs2066844 variant, the native Arg is replaced by Trp at 702th residue of NOD2 protein. The RSA property percentage of the Arg702 is 35%, the ASA degree is 81Å, and this amino acid forms a strong H-bond (3.02Å and 3.01Å) with Lys698 and Cys706 , respectively. The RSA property percentage of the Trp702 is 18%. The ASA degree of this amino acid is 42 Å, and it binds to Lys698 and Cys706 by 3.05Å hydrogen bonds. The RMSD value of the NOD2 normal model is 0.0 Å, and the mutant model is 0.00 Å. No significant difference between the RMSD values in both models at the protein level (Fig. 3). The rs76418789 variant substitutes Gly to Arg at position 149 of the IL23R protein. The Gly149 RSA property percentage is 28%, the ASA degree is 22 Å, and this amino acid binds with Asp126 by 2.83Å hydrogen bonds. The RSA property percentage of the Arg149 is 43%, and the ASA degree is 98 Å. The Arg149 binds with Lys126 by 2.88Å hydrogen bonds. The normal model’s RMSD value is 0.0 Å, and the value of the mutant model is 0.01 Å. The alteration in hydrogen bonds and RMSD values may change the structure and function of the protein (Fig. 4).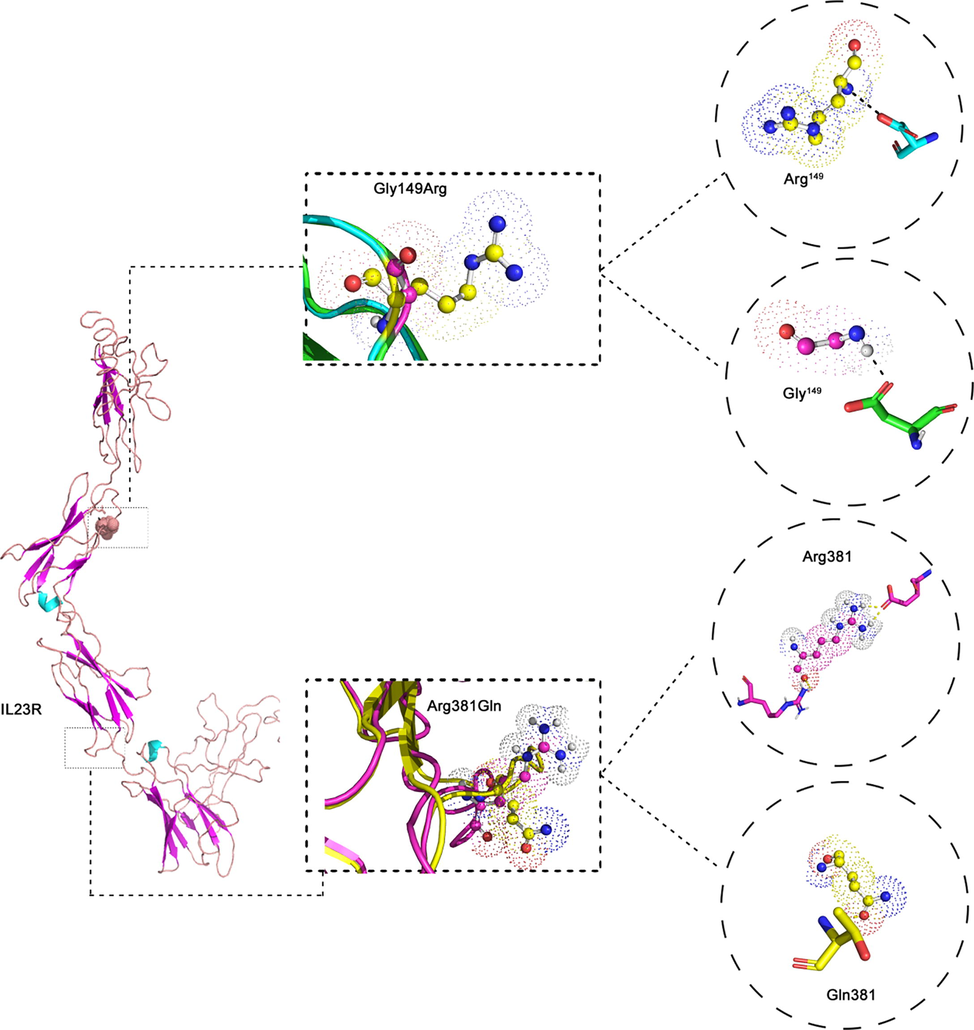
The 3D view of IL23R protein structure. In the middle Superpose of Gly149Arg and Arg381Gln in IL23R protein. The Wildtype Gly 149 and mutant Arg 149 forming H-bonds with surrounding amino acids. c) The wildtype Arg 381 and mutant Gln 381 forming H-bonds with surrounding amino acids of IL23R.
The normal amino acid in the rs11209026 variant is an Arginine at position 381of the IL23R protein, and the mutant amino acid is Glutamine. The RSA property percentage of Arg381 is 60%, and the ASA degree is 138 Å. The Arg381 links with Gln459 in two regions by 2.99 and 2.98 hydrogen bonds. On the other hand, the RSA property percentage of the Gln381 is 63%, the ASA degree is 112 Å, and this amino acid does not link with any other amino acids in the same protein. The RSMD value of the normal IL23R protein model is 0.0 Å, and the value of the mutant model is 0.01 Å. The changes in hydrogen bonds and RMSD values may alter the functional and structural properties of the protein (Fig. 4).
4 Discussion
In Saudi Arabia, the steadily increasing number of new pediatric IBD cases being diagnosed over the past decade implicates genetic background as the primary factor in disease etiology (Saadah et al., 2016). The latest study from Saudi Arabia has concluded that IBD is associated with environmental exposures like gut microbiome, childhood vaccinations, and maternal use of oral contraceptives (Algethmi et al., 2020). But, the influence of genetic polymorphisms and lifestyle factors are also known to vary across different ethnic groups (Uhlig and Muise, 2017). Therefore, the IBD genetic risk markers identified in one ethnic population can’t be directly recommended for ethnic populations with different cultural backgrounds, as they could potentially lead to false findings (Wijmenga and Zhernakova, 2018). Our study identified 3 out of 4 SNPs [rs2066844, rs76418789, rs11209026] as rare in the Arabian population, both in controls and IBD patients. Genetic association analysis showed insignificant differences between controls and cases. This negative association may be due to the limitations of small sample size as three of the variants are rare. These variants require thousands of samples to test for association. Since all four polymorphisms have shown conformity with the Hardy-Weinberg equilibrium in both groups, we are confident that our findings were not skewed by genotyping errors or population stratification.
The rs2066842 polymorphism in NOD2 has historically been associated with CD, Blau’s syndrome, and other autoimmune diseases (Caso et al., 2015). Globally, the minor allele frequency (MAF) is 0.1, the highest population MAF found among northern and western European ancestry peoples, 0.32, and the MAF of Saudis in Saudi Human Genome Project (SHGP) is 0.23. In the present study, we report numerically higher MAF scores than the world population, but with no statistical difference between controls and IBD patients. Several GWAS studies have reported a positive association of rs2066842 in CD patients in different populations including Italians (p = 0.018; OR (95%CI) = 2.022 [1.119–3.654]) and in Polish patients (p = 0.005, OR (95% CI) = 2.52 [1.34–4.75]) (Gaj et al., 2008a). Also, an Indian study found association of this variant with UC and no association with CD (Kedia and Ahuja, 2017). The second genetic polymorphism in NOD2 (rs2066844) has been associated with CD and Blau’s syndrome (Choteau et al., 2016). The Global MAF of this variant is 0.01, in northern and western European populations it is 0.16, but its frequency is extremely low in Saudi population (MAF is 0.0064). This variant is significantly associated with CD in Polish population (p = 0.0013, OR (95% CI) = 6.65 [1.99–22.17]), and with IBD in other Caucasians (p < 0.001) (Gaj et al., 2008b).
Further, we screened two risk markers in the IL23R gene among Saudi IBD patients. The rs76418789 polymorphism has been associated with CD and UC in Korean population (p = 0.0096, OR(95%CI) = 1.22 [1.12–1.32]) and Japanese population (p = 6.29E−7, OR(95%CI) = 0.89 [0.85–0.93]) (Okamoto et al., 2020; Ye et al., 2016). The global MAF is 0.01, whereas in Japanese it is 0.1, but in Saudis it is extremely rare i.e., 0.0016. The global MAF score of the other IL23R variant, rs11209026 is 0.02, but its prevalence in Europeans is higher (MAF is 0.1) comparable to Saudis (MAF is 0.09). This polymorphic change has a strong protective effect against CD in both the Jewish (p = 7.95 × 10−4, OR (95% CI) = 0.45[0.27–0.73]) and non-Jewish populations (p = 5.05 × 10−9, OR (95%CI) = 0.26[0.15–0.43]) (Duerr et al., 2006).(). A German study has also reported the significant differences in the distribution of minor allele frequency of the SNP rs11209026 (p.Arg381Gln) which suggested the strong protective role against the development of CD (P = 8.04 × 10−8, OR (95%CI) = 0.43[0.31–0.59]) (Glas et al., 2007). Another study of Czech IBD patients found a weak protection of this polymorphism against IBD (p = 0.02, OR (95%CI) = 0.56 [0.33–0.93]) (Dusatkova et al., 2009).
We propose a few explanations for the heterogeneous outcomes related to the association of the four SNPs in Saudi Arabian IBD patients. Firstly, the low MAF scores of Caucasian IBD GWAS alleles in a Saudi population could be due to sampling size differences. GWAS studies are conducted on thousands of patient and control populations. In our case, due to the small population size in Saudi Arabia, we were able to screen only a small patient cohort, and this could affect their robust statistical significance with disease etiology. Lastly, the variants selected in the study had a minor frequency of less than 5%, so they are considered rare variants. The consanguinity factor in the population increases the stretches of homozygosity in the genome. A SNP array analysis of Saudi Arabians showed stretches of homozygosity ranging from 2 to 65 MB in children born to consanguineous unions (Mohamoud et al., 2018). This factor might explain the loss of heterozygosity for the variants we tested.
In this study, we determined the pathogenicity of NOD2 (P268S and R702W) and IL23R (G149R and R381Q) variants using SIFT, PloyPhen2, CADD, and Condel algorithms. Our predictions results showed that NOD2 (R702W) and IL23R (G149R and R381Q) variants were deleterious in nature, which further support their role as IBD risk markers. Interestingly, none of those three SNPs were associated with IBD risk in Saudi patients, which underscores the need to carefully interpret the genetic association markers. Only one variant [rs2066842] was predicted as benign based on all four pathogenicity prediction tools. However, the prediction ability of these computational tools is often variable with both consensus and contradictions, mostly due to the differences in operating principles and datasets used in training those algorithms (Shaik and Banaganapalli, 2019; Masoodi et al., 2019). The 3D protein modelling of these 4 variants revealed the minor drifts on the tertiary structural features in the proteins, which could be due to change in hydrogens or peptide bonds. The structural drifts measured by RMSD value differences could in turn affect the distribution of hydrogen bonds and hydrophilic or hydrophobic properties of the affected amino residue or its surrounding amino acids, which maintains the molecular structure, including active site conformation of the protein molecules (Shaik et al., 2020). Protein stability is an important character that determines its functional properties (Pandurangan, 2020). In this study, we identified that NOD2 (P268S and R702W) and IL23R (G149R and R381Q) variants were destabilizing to the proteins. Hence, its most likely that these variants contribute to the dysfunction of NOD2 and IL23R proteins in IBD patients.
We conclude the insignificant association of the NOD2 and IL23R genetic risk markers in Saudi IBD patients . This study suggests the careful assessment of genetic risk markers identified in other populations before applying them in highly consanguineous Arab populations. However, this study provides background data to understand better GWAS loci's genetic heterogeneity in IBD patients with different ethnic backgrounds. Future genetic association studies in Saudi Arabia need to be searching for risk loci based on common genetic markers of Arabs, preferably with minor allele frequencies that exceed 10%. The findings of this study emphasize the need to identify and develop robust population-specific genetic markers which could aid in disease risk estimation, diagnosis, and prognosis of complex diseases.
CRediT authorship contribution statement
Raneem Saadi Alharbi: Data curation, Formal analysis, Funding acquisition, Methodology, Project administration, Writing – original draft. Noor Ahmad Shaik: Conceptualization, Software. Hadiah Almahdi: Methodology, Writing – original draft. Hanan Abdelhalim ElSokary: Writing – original draft. Bassam Adnan Jamalalail: Writing – original draft. Mahmoud H. Mosli: Writing – original draft. Hadeel A. Alsufyani: . Jumana Yousuf Al-Aama: . Ramu Elango: Software, Writing – original draft. Omar Ibrahim Saadah: Conceptualization, Software, Writing – original draft. Babajan Banganapalli: Conceptualization, Data curation, Funding acquisition, Software, Software, Writing – original draft.
Acknowledgements
This project was supported under STU-Graduate Research Program by King Abdulaziz City for Science and Technology – the Kingdom of Saudi Arabia – award number (1-17-03-009-0009). All the authors sincerely acknowledge the KACST for its financial support.
Data Availability Statement
All datasets analyzed for this study are included in the article/Supplementary Material.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Whole exome sequencing of a consanguineous family identifies the possible modifying effect of a globally rare AK5 allelic variant in celiac disease development among Saudi patients. PLoS ONE. 2017;12(5):e0176664.
- [CrossRef] [Google Scholar]
- Clinical pattern of early-onset inflammatory bowel disease in Saudi Arabia: A multicenter national study. Inflamm. Bowel Dis.. 2016;22(8):1961-1970.
- [CrossRef] [Google Scholar]
- Algethmi, W., Baumann, C., Alnajjar, W., Sroji, A., Alsahafi, M., Jawa, H., . . . Mosli, M. 2020. Environmental exposures and the risk of inflammatory bowel disease: a case-control study from Saudi Arabia. Eur. J. Gastroenterol. Hepatol., 32(3), 358-364. doi:10.1097/MEG.0000000000001619.
- Genetics of inflammatory bowel disease from multifactorial to monogenic forms. World J. Gastroenterol.. 2015;21(43):12296.
- [Google Scholar]
- The burden of comedication among patients with inflammatory bowel disease. Inflamm. Bowel Dis.. 2013;19(13):2725-2736.
- [Google Scholar]
- Autoinflammatory granulomatous diseases: from Blau syndrome and early-onset sarcoidosis to NOD2-mediated disease and Crohn’s disease. RMD Open. 2015;1(1):e000097.
- [Google Scholar]
- Polymorphisms in the mannose-binding lectin gene are associated with defective mannose-binding lectin functional activity in Crohn’s disease patients. Sci. Rep.. 2016;6(1):1-11.
- [Google Scholar]
- Understanding inflammatory bowel disease via immunogenetics. J. Autoimmun.. 2015;64:91-100.
- [Google Scholar]
- Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol. Med.. 2010;2(5):146-158.
- [Google Scholar]
- A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314(5804):1461-1463.
- [Google Scholar]
- Dusatkova, P., Hradsky, O., Lenicek, M., Bronsky, J., Nevoral, J., Kotalova, R., . . . Cinek, O., 2009. Association of IL23R p. 381Gln and ATG16L1 p. 197Ala with Crohn disease in the Czech population. J. Pediatric Gastroenterol. Nutr., 49(4), 405-410.
- The history of genetics in inflammatory bowel disease. Ann. Gastroenterol.. 2014;27(4):294.
- [Google Scholar]
- Lack of evidence for association of primary sclerosing cholangitis and primary biliary cirrhosis with risk alleles for Crohn's disease in Polish patients. BMC Med. Genet.. 2008;9(1):81.
- [Google Scholar]
- Lack of evidence for association of primary sclerosing cholangitis and primary biliary cirrhosis with risk alleles for Crohn's disease in Polish patients. BMC Med. Genet.. 2008;9(1):1-8.
- [Google Scholar]
- rs1004819 is the main disease-associated IL23R variant in German Crohn's disease patients: combined analysis of IL23R, CARD15, and OCTN1/2 variants. PLoS ONE. 2007;2(9):e819.
- [CrossRef] [Google Scholar]
- Epidemiology of inflammatory bowel disease in India: the great shift east. Inflammat. Intest. Dis.. 2017;2(2):102-115.
- [Google Scholar]
- Structural prediction, whole exome sequencing and molecular dynamics simulation confirms p. G118D somatic mutation of PIK3CA as functionally important in breast cancer patients. Comput. Biol. Chem.. 2019;80:472-479.
- [CrossRef] [Google Scholar]
- In-silico analysis of inflammatory bowel disease (IBD) GWAS loci to novel connections. PLoS ONE. 2015;10(3):e0119420.
- [CrossRef] [Google Scholar]
- Genetics of inflammatory bowel disease: beyond NOD2. Lancet Gastroenterol. Hepatol.. 2017;2(3):224-234.
- [Google Scholar]
- A missense mutation in TRAPPC6A leads to build-up of the protein, in patients with a neurodevelopmental syndrome and dysmorphic features. Sci. Rep.. 2018;8(1)
- [CrossRef] [Google Scholar]
- A cross-sectional survey of multi-generation inflammatory bowel disease consanguinity and its relationship with disease onset. Saudi J. Gastroenterol.. 2017;23(6):337.
- [CrossRef] [Google Scholar]
- Role of ATG16L, NOD2 and IL23R in Crohn’s disease pathogenesis. World J. Gastroenterol.. 2012;18(5):412.
- [Google Scholar]
- Okamoto, D., Kakuta, Y., Takeo, N., Moroi, R., Kuroha, M., Kanazawa, Y., . . . Hirano, A. 2020. P822 Genetic analysis of ulcerative colitis in Japanese individuals using population-specific SNP array. J. Crohn's Colitis, 14(Supplement_1), S638-S639.
- Saadah, O. I., El Mouzan, M., Al Mofarreh, M., Al Mehaidib, A., Al Edreesi, M., Hasosah, M., . . . AlSaleem, K. 2016. Characteristics of pediatric Crohn’s disease in Saudi children: a multicenter national study. Gastroenterol. Res. Pract., 2016.
- Molecular insights into the coding region mutations of low-density lipoprotein receptor adaptor protein 1 (LDLRAP1) linked to familial hypercholesterolemia. J Gene Med. 2020;22(6)
- [CrossRef] [Google Scholar]
- Computational molecular phenotypic analysis of PTPN22 (W620R), IL6R (D358A), and TYK2 (P1104A) gene mutations of rheumatoid arthritis. Front. Genet.. 2019;10:168.
- [CrossRef] [Google Scholar]
- Clinical genomics in inflammatory bowel disease. Trends Genet.. 2017;33(9):629-641.
- [Google Scholar]
- The importance of cohort studies in the post-GWAS era. Nat. Genet.. 2018;50(3):322-328.
- [Google Scholar]
- Yamada, A., Komaki, Y., Patel, N., Komaki, F., Aelvoet, A. S., Tran, A. L., . . . Cannon, L. 2017. Risk of postoperative complications among inflammatory bowel disease patients treated preoperatively with vedolizumab. Am. J. Gastroenterol., 112(9), 1423-1429.
- Identification of ten additional susceptibility loci for ulcerative colitis through immunochip analysis in Koreans. Inflamm. Bowel Dis.. 2016;22(1):13-19.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2021.101726.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




