

Translate this page into:
Exploring the non-covalent interactions, vibrational and electronic properties of 2-methyl-4-hydro-1,3,4-triazol-thione-5 in different solutions
⁎Corresponding authors. utkirxolikulov12@gmail.com (Utkirjon Holikulov), masrur_hodiev@mail.ru (Masrur Khodiev), issaoui_noureddine@yahoo.fr (Noureddine ISSAOUI)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
In this study, the optimal geometry and vibrational assignments of 2-methyl-4-hydro-1,3,4-triazol-thione-5, one of the triazole derivatives, were analysed by the DFT approach and vibrational spectroscopy. PED values were calculated, and vibrational assignments were determined. Experimental results showed that interactions between 2-methyl-4-hydro-1,3,4-triazol-thione-5 and solvent (acetone, acetonitrile, dioxane, and DMF) molecules lead to a red shift of the N-H stretching vibrational band. The computations were performed at the B3LYP/6-311++G(d,p) functional set. The molecular electrostatic potential surface was used to distinguish between electrophilic and nucleophilic regions. The reactivity of the molecular complexes was determined by examining their frontier molecular orbitals. Topological investigations revealed the existence of N-H…N and N-H…O-type hydrogen bonds between 2-methyl-4-hydro-1,3,4-triazole-thiol-5 and solvent molecules. The red-shift of the N-H stretching band and H-bond strength between solute–solvent molecules are in the order of acetonitrile, acetone, dioxane, and DMF.
Keywords
H-bond
Vibrational spectra
DFT
Solvent
AIM

1 Introduction
Hydrogen bonding produces complexes of varied compositions and structures, which influence many chemical and physical characteristics of substances (Grabowski, 2006; Kazachenko et al., 2022). The presence of an intermolecular hydrogen bond causes a redistribution of electron density in the molecules that comprise the material, resulting in a change in its overall characteristics. While hydrogen bonds are not covalent, they possess the ability to support chemical reactions and various biochemical processes. This is primarily due to the variety of intermolecular interactions, the generality of which becomes a decisive factor in the chemistry of biological objects, because in specific intermolecular interaction processes, the ability of molecules to form hydrogen bonds is a measure of their reactivity and activity. As a result, hydrogen bonds play a crucial role in numerous biological and chemical processes (Pimentel and McClellan, 1971).
Among the substances with the ability to form H-bonds, heterocyclic compounds, which are used more and more recently in practise, occupy a special place. Particular consideration should be given to pharmacology, at the request of which many new effective drugs are synthesised. In this regard, understanding the H-donor and H-acceptor characteristics of substituted triazoles is both fundamental and practical (Dixit, 2021; Mulloev, 2016). The existence of a heteroatom in the ring of heterocyclic compounds influences many aspects of their intermolecular interaction. Heterocyclic substances are distinguished by their ability to operate as both H-donors and H-acceptors.
Currently, heterocyclic compounds have wide practical applications (Matin, 2022; Pagniez, 2020; Bozorov, 2019). These include primarily nitrogen-containing five-membered heterocycles. Among the triazole derivatives of this group of molecules, 2-methyl-4-hydro-1,3,4-triazole-thiol-5 (MHTT) is of particular interest. MHTT is an organic heterocyclic molecule with a 5-membered planar ring.
The drug discovery process includes study on the physicochemical properties of novel pharmaceuticals. Specifically, these qualities include the ability of molecules to form hydrogen complexes. The efficiency of forming H-complexes, which determines the degree of participation of these molecules in intermolecular hydrogen bonding, affects the reactivity of these molecules, which determines their biological activity (Foldes and Sandorfy, 1970; Jumabaev, et al., 2022; Ahmad, 2023; Bharathy, 2021). In this regard, the study of physico-chemical, especially donor–acceptor properties of heterocyclic compounds belonging to the azole group is an important and urgent task.
Vibrational spectroscopy is one of the common physical methods for studying the relationship between the molecular nature and molecular structure of heterocyclic compounds and their thermodynamic, structural, and physical properties (Sevvanthi, 2020; Thirunavukkarasu, 2021; Amul, 2019). This is due to the fact that, with the help of vibrational spectroscopy, it is possible to study the parameters of the spectrum of complex heterocyclic compounds in different states of aggregation and their changes with environmental changes.
Despite the long history of studying the manifestation of intermolecular interactions in vibrational spectra, a systematic study of the effectiveness of the interaction of molecules with a solvent belonging to the class of heterocyclic compounds, that is, with amphoteric properties, has not been carried out. The purpose of this work is to study the intermolecular interactions of MHTT in several solvents such as acetonitrile (ACN), acetone (AC), dioxane (DO), and dimethylformamide (DMF). Vibrational spectroscopy and Density Functional Theory (DFT) methods were used to achieve this goal. The solvents effects on the N-H vibrational spectrum of MHTT were evaluated. Topological analyses (AIM, NCI-RDG, and ELF-LOL) are used to investigate the nature of intermolecular hydrogen bonds.
2 Experimental and computational details
FTIR spectra of binary solutions of title compound in non-polar (CCl4) and polar (ACN, AC, DO, and DMF) solvents at concentrations of 0.0047 mol/l and 0.46 mol/l were recorded using an IRAffinity-1 infrared spectrophotometer in the range from 2000 to 3700 cm−1 at a temperature of T = (298 ± 2) K. To improve the accuracy of the experimental results, absorption bands were recorded in a wide frequency range and 2–3 times for each sample. The position and width of narrow and single bands were determined with an accuracy of 2 cm−1 for complex bands with a wide maximum of 5 cm−1.
Density functional theory (DFT) method with B3LYP/6–311++G(d,p) function set were used to optimize MHTT and its molecular complexes formed with solvent (acetonitrile, acetone, dioxane and dimethyformamide) molecules. The B3LYP/6–311++G(d,p) functional is well-known in the quantum-chemical literature for providing an accurate description of harmonic vibrational wavenumbers for small and medium-sized molecules (Kazachenko, 2023; Julie, 2021). All computations were performed using the Gaussian 09 W program package (Frisch et al., 2010). To determine the topological parameters at the bond critical points of H-bonded complexes and visualise non-covalent effects, the MultiWFN (Lu and Chen, 2012), GaussView (Dennington et al., 2016) and VMD (Humphrey, 1996) tools were employed.
3 Results and discussion
3.1 Molecular structure
Fig. 1a shows the optimal geometric structure of MHTT calculated at the B3LYP/6–311++G(d,p) level by the DFT method. Table S1 presents the geometric parameters (bond length, angles) corresponding to the optimal structure of MHTT. The 2D potential energy surface of MHTT (C4-N3-O12-H13) was calculated along the deviation of the dihedral angle in the range 0–360 Degrees (Fig. 2). Calculations showed that C4-N3-O12-H13 has the minimum potential energy at a value of 0 Degree dihedral angle.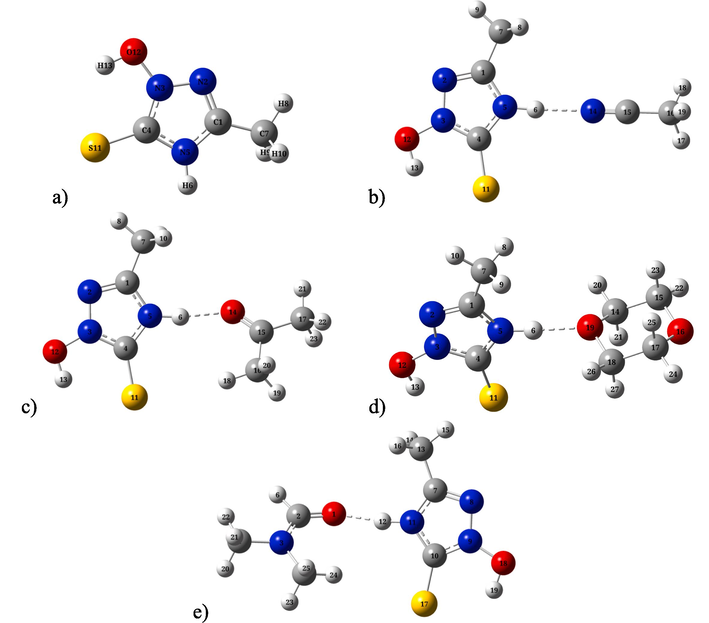
The optimized molecular structures of of 2-methyl-4-hydro-1,3,4-triazole-thiol-5 (a) and its complexes of with acetonitrile (b), acetone (c), dioxane (d), DMF (e) molecules calculated B3LYP/6–311++G(d,p) level.
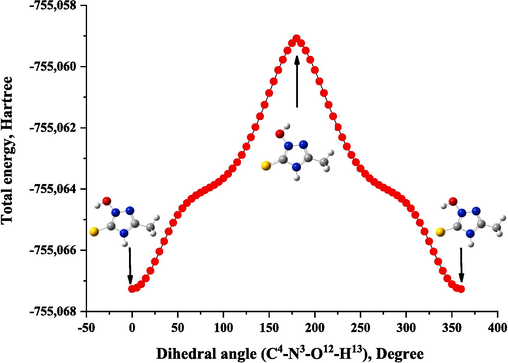
Potential energy surface scan for dihedral angle C4-N3-O12-H13 of MHTT.
A scan of potential energy surface (PES) was used to determine the preferred position of the MHTT molecule. A search to identify low-energy structures was performed for the (C4-N3-O12-H13) dihedral angle of MHTT, which is also a relevant coordinate for intramolecular conformational flexibility. In calculations, all geometric parameters were simultaneously relaxed and and rotation angles ranged from 0 to 360 degrees. The minimum energy curve for this rotation is −755.067265 Hartrees, with the minimum energy at 0 Degree. Therefore, in this study, we concentrated on the most stable form of the MHTT molecule to clarify the assignment of the molecular structure and vibrational spectra.
The optimal geometries of complexes of MHTT with solvent molecules such as acetonitrile, acetone, dioxane, and DMF are depicted in Fig. 1b–e. MHTT molecules form complexes with solvent molecules using N-H…N≡C (with acetonitrile), N-H…O = C (with acetone), N-H…O (with dioxane), and N-H…O = C (with DMF) hydrogen bonds. The hydrogen bond lengths are 1.919 Å (MHTT + ACN), 1.796 Å (MHTT + AC), 1.789 Å (MHTT + DO), and 1.742 Å (MHTT + DMF), respectively. Intermolecular interaction energies in molecular complexes are 7.15 kcal/mol (MHTT + ACN), 5.55 kcal/mol (MHTT + AC), 5.13 kcal/mol (MHTT + dioxane), and 5.93 kcal/mol (MHTT + DMF), respectively. The intermolecular interaction energies in molecular complexes were determined based on ΔE = Ecomplex – EMHTT – Esolvent where Ecomplex, EMHTT, Esolvent are the total energies of the complex, MHTT and solvent molecules, respectively (Allangawi, 2024; Sajid, 2021).
3.2 Vibrational analysis
The title molecule contains 13 atoms, and the vibrational spectrum shows 33 fundamental vibrational modes. The Raman and IR spectra of MHTT calculated at the DFT: B3LYP/6–311++G(d,p) in the gas phase are shown in Fig. 3a,b. VEDA software (Jamroz, 2004) was used to analyse the potential energy distribution (PED) of vibrations connected to each group of atoms in the molecule. The calculated vibrational frequencies, scaled values, Raman activity, IR intensities and PED assignments are all listed in Table 1. PED can predict the vibrational mode for every vibrational frequency (Thamarai, 2020). ν(C1-C7) 10 % + ν(N2-N3) 20 % + ν(S11-C4) 19 % + δ(N5-C1-N2) 27 %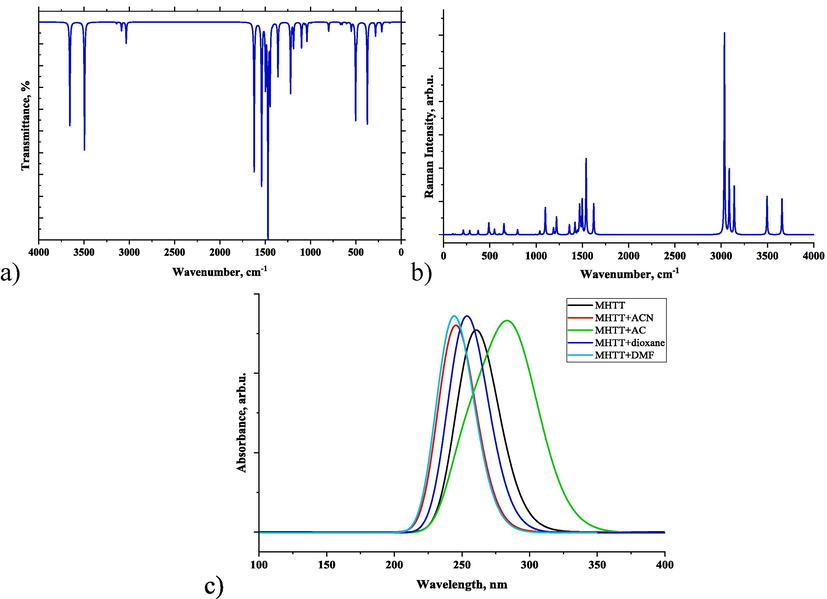
Simulated vibrational (Raman (a) and IR (b)) and UV–Vis (c) spectra of MHTT.
Mode
calculated DFT/B3LYP/6–311 + G(d,p)
Assignments (PED ≥ 10 %)a
Unscaled
Scaledb
IR intensity
Raman activity
1
3657
3536
49
63
ν(N5-H6) 100 %
2
3495
3379
61
64
ν(O12-H) 100 %
3
3141
3037
1
74
ν(C7-H8) 87 % - ν(CH2) 13 %
4
3086
2984
4
98
νasym(CH2) 100 %
5
3035
2934
10
302
νsym(CH3) 100 %
6
1622
1568
71
33
- ν(C1-C7) 52 % + δ(N5-C1-N2) 16 % - δ(H6-N5-C4) 10 %
7
1540
1489
77
79
ν(N3-C4) 12 % + δ(H13-O12-N3) 62 %
8
1498
1448
30
35
- ν(N5-C1) 10 % - δ(H6-N5-C4) 12 % - δ(H8-C7-H9) 18 % + δ(H8-C7-H10) 21 %
9
1478
1429
6
10
- δ(H8-C7-H9) 35 % - δ(H8-C7-H10) 37 % - τ(H8-C7-C1-N5) 13 % + τ(H9-C7-C1-N5) 13 %
10
1470
1421
100
29
ν(N3-C4) 26 % - ν(C1-C7) 13 %
11
1447
1399
37
3
- ν(N5-C1) 15 % + ν(C1-C7) 26 % - δ(H6-N5-C4) 10 % + δ(H8-C7-H9) 12 %
12
1420
1373
1
12
δ(CH3) 81 %
13
1360
1315
26
10
δ(N5-C1-N2) 10 % - δ(C4-N3-N2) 17 % - δ(N3-N2-C1) 13 %
14
1220
1179
34
17
ν(N3-C4) 22 % - ν(N2-N3) 10 % + δ(H6-N5-C4) 37 %
15
1188
1149
12
6
- ν(C1-C7) 26 % + δ(N5-C1-N2) 21 % + δ(H6-N5-C4) 12 % - δ(C4-N3-N2) 10 %
16
1100
1063
12
26
ν(N2-N3) 66 %
17
1064
1029
1
0
δ(H8-C7-H9) 12 % + δ(H8-C7-H10) 12 % - τ(H8-C7-C1-N5) 35 %
18
1040
1005
9
3
- ν(N5-C1) 21 % - ν(C1-C7) 10 % + ν(N2-N3) 10 % + δ(N3-N2-C1) 26 %
19
994
961
0
0
ν(N5-C1) 30 % - τ(H8-C7-C1-N5) 23 % - τ(H9-C7-C1-N5) 23 %
20
800
773
4
4
21
670
648
1
1
τ(H6-N5-C4-N3) 16 % + τ(N3-N2-C1-N5) 71 %
22
653
631
1
9
ν(C1-C7) 45 % + δ(N3-N2-C1) 22 % + δ(C4-N3-N2) 13 %
23
601
581
1
1
τ(C1-N2-N3-C4) 92 %
24
550
532
4
5
- ν(N2-N3) 24 % + δ(C-N-N) 51 %
25
503
486
46
1
τ(H6-N5-C4-N3) 85 %
26
489
473
7
10
ν(N2-N3) 21 % + δ(C4-N3-N2) 42 % + δ(C4-N3-N2) 22 %
27
374
362
48
3
τ(H13-O12-N3-C4) 85 %
28
283
274
6
3
δ(C7-C1-N2) 78 %
29
271
262
1
0
τ(H8-C7-C1-N5) 11 % - γ(C7-N2-N5-C1) 70 %
30
214
207
4
4
δ(C7-C1-N2) 82 %
31
200
193
0
0
τ(N3-N2-C1-N5) 11 % - γ(C7-N2-N5-C1) 15 % - τ(C1-N2-N3-C4) 64 %
32
126
122
0
0
γ(C7-N2-N5-C1) 67 % + τ(C1-N2-N3-C4) 17 %
33
100
97
0
1
τ(H8-C7-C1-N5) 80 % + γ(C7-N2-N5-C1) 12 %
It is known from the literature (Sagaama, 2022; Bellamy and Williams, 1957) that the N-H stretching vibration frequencies lie in the range of 3300–3500 cm−1. For the MHTT molecule, the calculated N-H stretching vibration frequency in the gas phase is 3657 cm−1 (Ghatfaoui et al., 2021). Fig. 5 shows the FTIR spectra of N-H stretching vibration of binary solutions of MHTT in different solvents. The N-H stretching vibration of MHTT in carbon tetrachloride (CCl4) solution is 3396 cm−1 (Fig. 4). The figure shows that the NH stretching band maximum of MHTT shifts to the low frequency region up to 3249 cm−1 (acetonitrile), 3201 cm−1 (acetone), 3168 cm−1 (dioxane) and 3063 cm−1 (DMF) in different solutions. Also, the half-width of the band and the integral intensity increase. The reason for this spectral change is due to the interaction between solvent–solute molecules in the solution.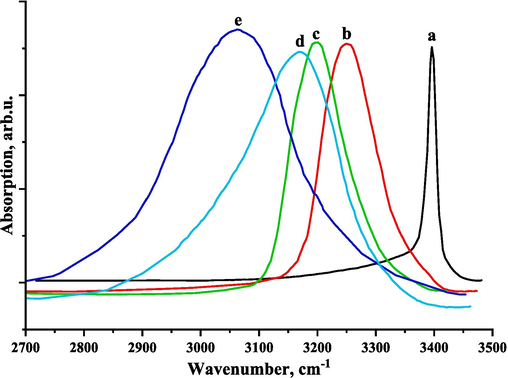
Experimental FTIR spectra of N-H stretching of the 2-methyl-4-hydro-1,3,4-triazole-thiol-5 in CCl4 (a), acetonitrile (b), acetone (c), dioxane (d), and DMF (e).
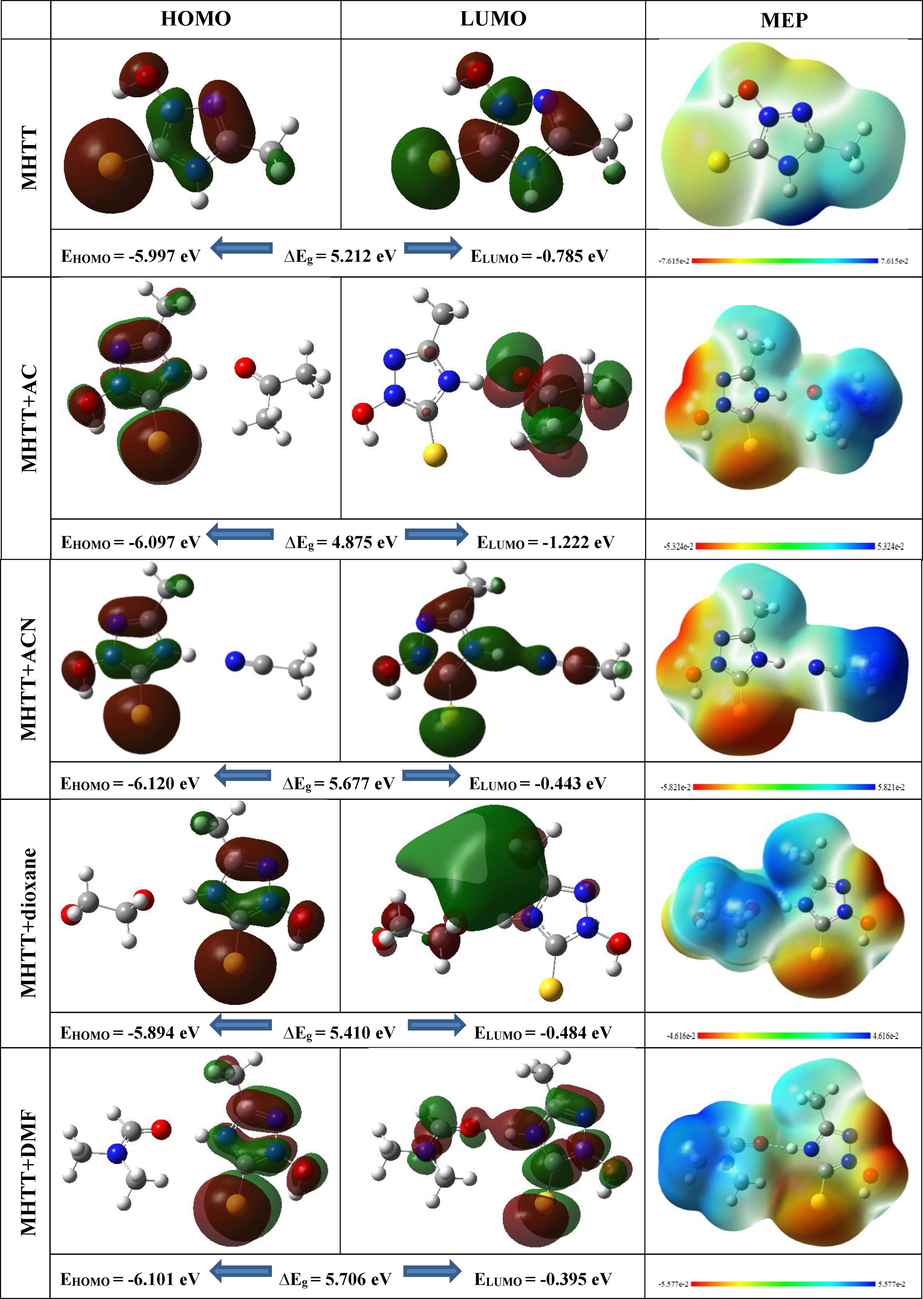
FMOs and MEP surfaces of MHTT and its complexes with different solvent molecules.
It is known from the literature (Mani, 2023) that the frequency of O-H stretching vibration is fined in the range of 3450–3600 cm−1. The calculated O-H stretching vibrational frequency for our title molecule is in the range of 3495 cm−1. Also, spectral lines at 1540 cm−1 belong to the combination of N-O-H bending and C-N stretching vibrations.
Analysis of the literature (Thamarai, 2020; Sagaama, 2022) showed that C-H stretching vibration frequencies are in the range of 2800–3300 cm−1. For our title molecule, the frequency of CH3 asymmetric stretching vibrations was found to be 3141 cm−1, the frequency of CH2 asymmetric stretching vibrations was 3086 cm−1, and the frequency of CH2 symmetric stretching vibrations was 3141 cm−1. CH3 bending vibration lines are located at 1420 cm−1.
3.3 Frontier molecular orbital (FMO) analysis
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are known as frontier molecular orbitals (FMO), and their values can be used to analyze intermolecular interactions and the electron donating and accepting abilities of molecules (Medimagh, 2023; Ullah, 2023). FMO also provides information on compound kinetic stability and chemical reactivity. The energy gap (Eg) of frontier orbital represents the energy difference between the HOMO and LUMO. The energy gap is a good predictor of a molecule's kinetic stability and chemical reactivity. A molecule with a low energy gap is polarizable, reactive, and soft. The high energy gap indicates the presence of a solid molecule. Table S2 presents the chemical reactivity parameters energy gap (Eg), hardness (η), chemical potential (μ), global electrophilic index (ω), ionisation potential (IP), and electron affinity (EA) calculated for the title molecule in the gas phase and its complexes with different solvent molecules (ACN, AC, dioxane, and DMF) using HOMO and LUMO energies. Fig. 5 indicates the HOMO and LUMO orbitals of the MHTT and its complexes with different solvent molecules. These values are determined using the following mathematical formulas based on Koopman's theorem (Koopmans, 1934):
3.4 Molecular electrostatic potential surface (MEPS) analysis
MEP surface of molecules and molecular systems is a helpful technique for studying hydrogen bonding interactions and the biological recognition process by identifying nucleophilic and electrophilic reaction sites (Koopmans, 1934; Fukui, 1982; Kazachenko, 2023). The MEPS has an essential role in characterizing the physicochemical properties of molecules, such as size, shape, electron density, delocalization, and chemical reactivity site (Ahmed, 2017).
Fig. 5 shows the MEP surfaces of the MHTT molecule and its complexes with various solvent molecules. Different colours on the MEPS show different levels of electrostatic potential. Blue > green > yellow > orange > red is in order of decreasing potential. The color code of the MEP map ranges from −7.615 a.u. (deepest red) to + 7.615 a.u. (deepest blue) for MHTT, respectively. It can be seen that in the MHTT molecule, a large electropositive potential is detected near the N-H group, and a small electropositive (blue) region is detected near the oxygen (O) atom. As a result of the formation of N-H…N and N-H…O hydrogen bonds during the formation of complexes, the electrostatic potential around the N-H group decreases and becomes neutral. In addition, the highly electronegative region is activated near the oxygen (O), sulphur (S), and nitrogen (N) atoms of the MHTT molecule. The surroundings of the CH3 group have an almost neutral electrostatic potential and are therefore inactive.
3.5 UV–vis absorption studies
The UV–Vis spectrum of MHTT was simulated using TD-DFT theory with several solvents. Table S3 displays the maximum absorption wavelength, oscillator strength, and transition probabilities for MHTT in solvent phases. Because each solvent provides various maximum absorption peaks, UV–Vis spectra for the MHTT molecule and its various complexes are shown in Fig. 3c in the 100–400 nm range. For the MHTT molecule, there are three absorption peaks in the gas phase: the primary absorption at 274 nm, the second absorption at 260.21 nm, and the third absorption at 259.73 nm. In this case, the oscillator strength of the second absorption is the largest (f = 0.1222), and the main contribution is 67 % from HOMO to LUMO + 1. The absorption is different for the complexes formed with different solvents. In MHTT + ACN and MHTT + DMF complexes, S0 → S3 absorption peaks are 245.15 and 243.62 nm, respectively, and have the largest oscillator strength. In these, the main transition contributions from HOMO to LUMO are 64 and 63 %, respectively. The first absorption peak for the MHTT + AC complex is 287.60 nm (f = 0.0039), and the main contribution is 71 % from HOMO to LUMO. The second absorption peak for the MHTT + DO complex is 253.23 nm, and the main contribution from HOMO to LUMO + 1 is 68 %. These indicate charge transfers within the complexes.
3.6 Atoms in molecules (AIM) analysis
The Atoms in Molecules (AIM) analysis, which is based on Bader's theory (Bader, 1985), is frequently used to understand intra- and intermolecular interactions in molecular complexes. Geometrical and topological characteristics are also important tools for determining the strength of hydrogen bonding. The geometric conditions for the presence of hydrogen bonds are as follows (Khan, 2015):
1. The distance between hydrogen (H) and acceptor (A) atoms is smaller than the sum of their van der Waals radii;
2. Hydrogen bond angle α(D-H∙∙∙∙A) > 900;
3. An increase in the D–H bond length is observed in the hydrogen bond.
The bond critical point (BCP) is a point on the bond path with the lowest electron density. Electron density (ρ), Laplacian of electron density (∇2ρ), energy density (H) and potential energy (V) in BCPs are important topological parameters describing the nature of hydrogen bonding (Asif, 2022). The following criteria are used to describe the presence of a hydrogen bond (Tang, 2006; Bader, 2010; Rozas, 2000):
-
BCP must be present between hydrogen (H) and acceptor (A) atoms;
-
Electron density and its Laplacian in BCPs should be in the interval of 0.002–0.040 a.u and 0.024–0.139 a.u, respectively.
The hydrogen bond energy is calculated using the relationship suggested by Espinosa et al. (Espinosa, 1998):
EHB = -V(r)/2
Fig. 6 shows the AIM plot for these compounds, and some calculated topological parameters in BCPs are presented in Table 2.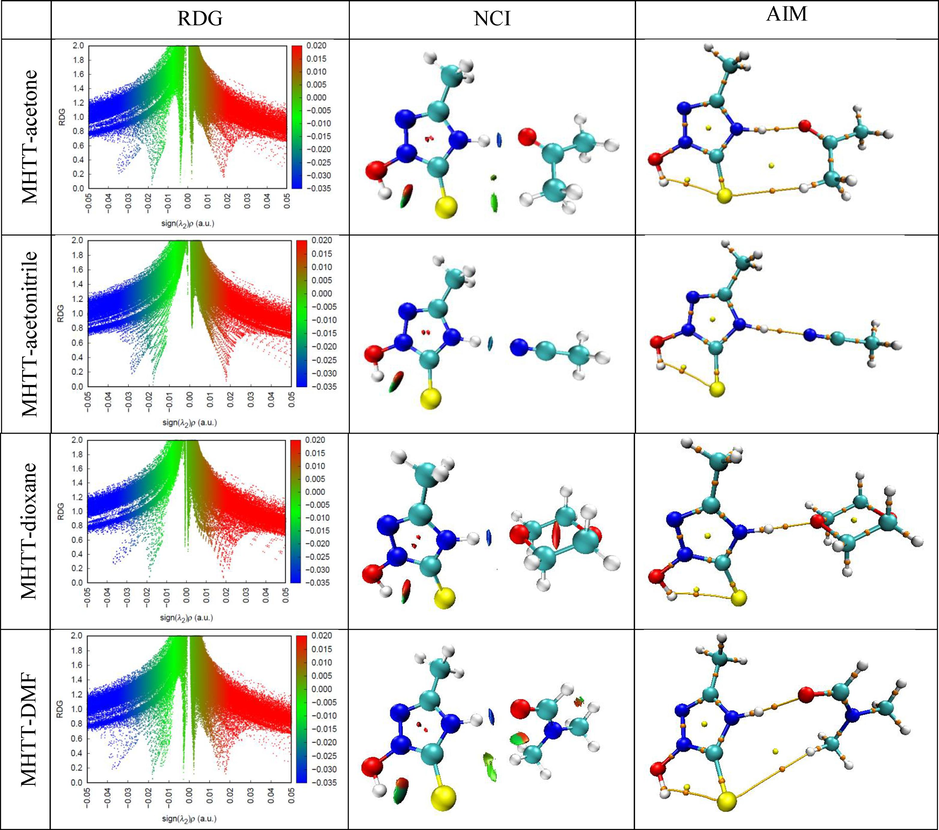
AIM plot, RDG scatter and NCI coloring maps of complexes.
H-bonds
Bond length r, Å
Density of all electrons
Lagrangian kinetic energy G(r)
Potential energy density V(r)
Energy density H(r)
Laplacian of electron density
Hydrogen bond energy EHB, kcal/mol
MHTT-AC
N5-H6…O14
1.796
0.0345
0.0289
−0.0288
0.0002
0.1164
9.036
MHTT-ACN
N5-H6…N14
1.919
0.0284
0.0214
−0.0203
0.0012
0.0906
6.369
MHTT-DO
N5-H6…O19
1.789
0.0362
0.0305
−0.0311
−0.0006
0.1194
9.758
MHTT-DMF
N11-H12…O1
1.742
0.0375
0.0332
−0.0337
−0.0004
0.1312
10.573
It can be seen from Table 2 that there is an N5-H6…O14 H-bond between MHTT and acetone molecules, and the values of electron density and Laplacian of electron density are 0.0345 a.u. and 0.1164 a.u., respectively. The hydrogen bond energy is 9,036 kcal/mol. Between MHTT and acetonitrile molecules, there is an N5-H6…N14 H-bond, and the electron density and its Laplacian values are 0.0284 a.u. and 0.0906 a.u., respectively. The energy of this H-bond is 6,369 kcal/mol. There is an N5-H6…O19 H-bond between MHTT and dioxane molecules, and the the electron density and its Laplacian values are 0.0362 a.u. and 0.1194 a.u., respectively. The energy of this H-bond is 9,758 kcal/mol. Also, there is a N11-H12…O1 H-bond between MHTT and DMF molecules, and the electron density and its Laplacian values are 0.0375 a.u. and 0.1312 a.u., respectively. This H-bond energy is 10,573 kcal/mol. The results of the analysis showed that the MHTT molecule forms a stronger hydrogen bond complex with the DMF molecule compared to other solvent molecules. If the energy density at BCPs is negative (H < 0), the hydrogen bond is covalent, while if it is positive (H > 0), it is electrostatic. It can be seen that the H-bonds in the MHTT-acetone and MHTT-acetonitrile complexes have an electrostatic nature, and the H-bonds in the MHTT-dioxane and MHTT-DMF complexes have a covalent nature.
3.7 RDG-NCI interaction analysis
It is very important to study the nature of intermolecular interactions between solute–solvent molecules. Reduced density gradient and non-covalent interaction (RDG-NCI) analysis was used to characterize weak interactions in MHTT-acetone, MHTT-acetonitrile, MHTT-dioxane, and MHTT-DMF complexes. These analyses are widely used to distinguish non-covalent interactions such as van der Waals, hydrogen bonding, and steric repulsion in molecular systems. RDG is a dimensionless quantity determined by electron density and its first derivative, and it is expressed using the formula follows (Rasul, 2023): The RDG scatter plot is obtained by estimating the electron density of the RDG-sign(λ2)ρ peaks. This plot describes the strength and kind of interactions in a molecule. If sign(λ2)ρ > 0, it represents a repulsive effect. If sign(λ2)ρ < 0, it represents an attractive effect. If sign(λ2)ρ≈0, Van der Waals represents the effect(Daghar et al., 2021). Furthermore, blue represents hydrogen bonding, green represents Van der Waals interactions, and red represents strong steric effect (Zahid, 2023).
Fig. 6 shows RDG scatter graphs and NCI coloring maps for MHTT-acetone, MHTT-acetonitrile, MHTT-dioxane and MHTT-DMF complexes. The blue spines in the range of −0.04 < sign(λ2)ρ < -0.03 in the RDG scatter graphs and the blue discs in the NCI coloring maps confirm the presence of intermolecular hydrogen bonds in the studied complexes.
3.8 Electron localisation function (ELF) and localized orbital locator (LOL) studies
For a better understanding of the electronic structure and chemical bonding in molecular systems, we conducted topological analyses of the electron localization function (ELF) and the localised orbital locator (LOL). Fig. 7 depicts ELF and LOL colour shade maps for the examined molecular complexes. ELF values range from 0.0 to 1.0, indicating a region with comparably bonding and nonbonding localised electrons. Fig. 7 shows a high ELF zone (in red) around the hydrogen atoms and a blue region with the carbon atoms, indicating a delocalized electron cloud. The LOL colour map reveals white in the centre of some hydrogen atoms, suggesting that the electron density exceeds the colour scale limit of 0.80. The complexes' hydrogen atom surrounds, as well as the centres of some carbon and nitrogen atoms, are coloured red. This location has high ELF and LOL values, indicating strong electron localization. The blue region surrounding the carbon atoms represents the delocalized electron cloud. The circular blue area surrounding the nitrogen and oxygen atoms represents the electron decrease area between the inner and valence shells.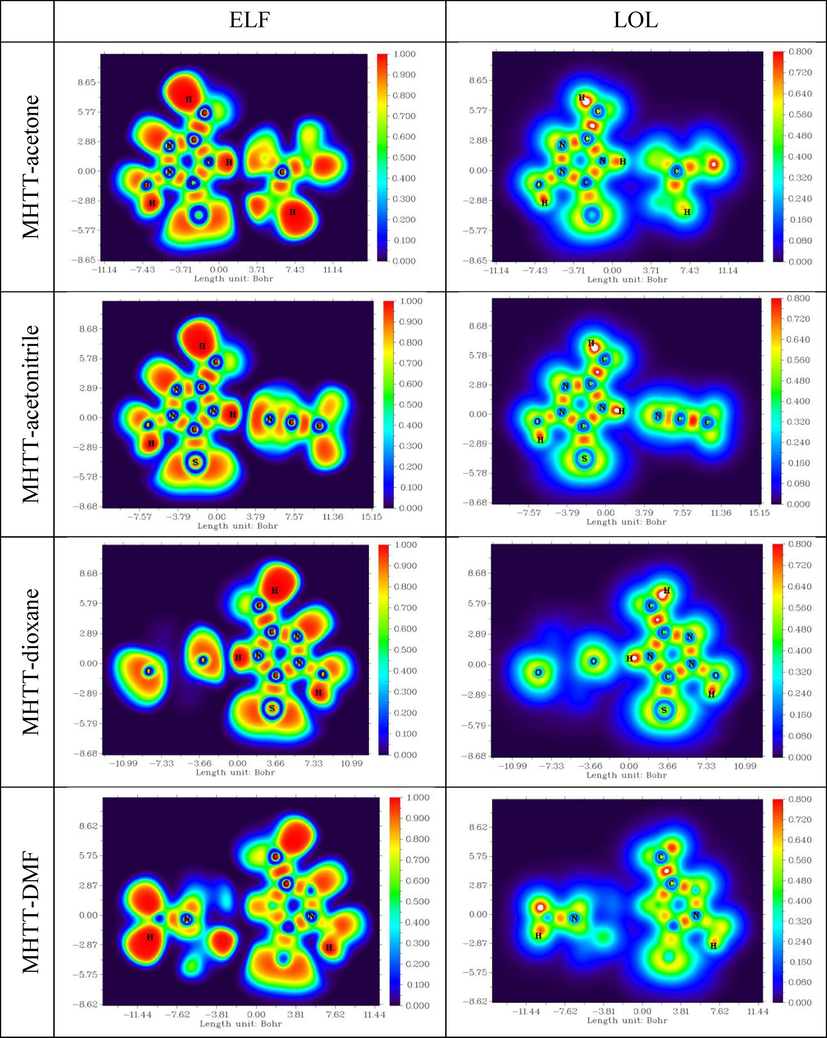
ELF and LOL diagrams of complexes of MHTT wit solvent molecules.
4 Conclusion
In this work, the optimal geometric structure and vibrational spectra of 2-methyl-4-hydro-1,3,4-triazol-thione-5 were investigated using the DFT method B3LYP/6–311++G(d,p) basis set.. The vibrational potential energy distribution (PED) of each group of atoms was calculated, and the vibrational assignments were evaluated. The N-H stretching vibrational band of 2-methyl-4-hydro-1,3,4-triazol-thione-5 shifts to a lower frequency and increases in half-width in acetonitrile, acetone, dioxane, and DMF solutions (ACN < AC < DO < DMF). Non-covalent interactions in solutions were analysed using AIM and RDG-NCI analyses. The energy of H-bonds between the title molecule and solvent molecules is 6.369 kcal/mol (with ACN), 9.036 kcal/mol (with AC), 9.758 kcal/mol (with DO), and 10.573 kcal/mol (with DMF). H-bonding forces explain the shift of the experimental N-H vibrational bands. In addition, the effect of the solvent on the electronic properties of 2-methyl-4-hydro-1,3,4-triazol-thione-5, such as MEP, FMO, ELF, LOL, and UV–Vis spectra, was studied.
CRediT authorship contribution statement
Utkirjon Holikulov: Conceptualization, Investigation, Writing – review & editing. Masrur Khodiev: Writing – original draft, Data curation, Investigation. Noureddine ISSAOUI: Supervision, Methodology, Writing – review & editing. Abduvakhid Jumabaev: Writing – review & editing, Formal analysis. Naveen Kumar: Methodology, Visualization, Software. Omar M. Al-Dossary: Validation, Funding acquisition.
Acknowledgements
The work was supported by the Projects No. FZ-20200929385 (Samarkand state University, Uzbekistan) and No. RSP2024R61 (King Saud University, Saudi Arabia).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Supramolecular assemblies of 3/4-chlorobenzoic acid and amino-chloropyridine derivatives: synthesis, X-ray Diffraction, DFT calculations, and biological screening. Crystals.. 2023;13:1663.
- [CrossRef] [Google Scholar]
- Synthesis, structural studies and biological activities of three new 2-(pentadecylthio)-5-aryl-1, 3, 4-oxadiazoles. J. Mol. Struct.. 2017;1129:50-59.
- [CrossRef] [Google Scholar]
- Transition metal loaded carbon penta-belt SACs for hydrogen and oxygen evolution reactions and identification of systematic DFT method to characterize the main interacting orbital for non-periodic system. Int. J. Hydrogen Energy.. 2024;53:989-998.
- [CrossRef] [Google Scholar]
- Spectral, DFT and molecular docking investigations on etodolac. J. Mol. Struct.. 2019;1195:747-761.
- [CrossRef] [Google Scholar]
- A first principles study on electrochemical sensing of highly toxic pesticides by using porous C4N nanoflake. J. Phys. Chem. Solids. 2022;160:110345
- [CrossRef] [Google Scholar]
- Definition of Molecular structure: by choice or by appeal to observation? J. Physics. Chem. A. 2010;114:7431-7444.
- [CrossRef] [Google Scholar]
- The NH stretching frequencies of primary amines. Spectrochim. Acta. 1957;9:341-345.
- [CrossRef] [Google Scholar]
- Evaluation of electronic and biological interactions between N-[4-(ethylsulfamoyl) phenyl] acetamide and some polar liquids (IEFPCM solvation model) with Fukui function and molecular docking analysis. J. Mol. Liq.. 2021;340:117271
- [CrossRef] [Google Scholar]
- 1,2,3-triazole-containing hybrids as leads in medicinal chemistry: a recent overview. Bioorg. Med. Chem.. 2019;27(16):3511-3531.
- [CrossRef] [Google Scholar]
- Intermolecular interaction study of L-threonine in polar aprotic solvent: Experimental and theoretical study. J. Mol. Liq.. 2021;338:116689
- [CrossRef] [Google Scholar]
- Empirical and computational studies on newly synthesis cyclohexylammonium perchlorate. J. Mol. Struc. 2021;1230:129820
- [CrossRef] [Google Scholar]
- GaussView, version 6. Shawnee Mission, KS: Semichem Inc.; 2016.
- A review on ‘triazoles’: their chemistry, synthesis and pharmacological potentials. J. Iran. Chem. Soc.. 2021;18:2535-2565.
- [CrossRef] [Google Scholar]
- Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett.. 1998;285:170-173.
- [CrossRef] [Google Scholar]
- Anharmonicity, solvent effects, and hydrogen bonding: NH stretching vibrations. Can. J. Chem.. 1970;48(14):2197-2203.
- [CrossRef] [Google Scholar]
- Gaussian 09. Wallingford CT: Gaussian Inc; 2010.
- Fukui, K., 1982. Role of Frontier Orbitals in Chemical Reactions, American Association for the, Advancement Sci. 218 747–754 (1982) 10.1126/science.218.4574.747.
- Synthesis, experimental and computational study of a non-centrosymmetric material 3-methylbenzylammonium trioxonitrate. J. Mol. Struc. 2021;1225:129132
- [CrossRef] [Google Scholar]
- Hydrogen bonding—New insight. The Netherlands: Springer; 2006.
- Jamroz, M.H., 2004. Vibrational Energy Distribution Analysis VEDA 4, Warsaw.
- Structural (monomer and dimer), wavefunctional, NCI analysis in aqueous phase, electronic and excited state properties in different solvent atmosphere of 3-{(E)-[(3, 4-dichlorophenyl) imino] methyl} benzene-1, 2-diol. J. Mol. Liq.. 2021;336:116335
- [CrossRef] [Google Scholar]
- Jumabaev A., et al., 2022. Interaction of valine with water molecules: Raman and DFT study, Ukr. J. Phys. 67, 8, 602-610. 10.15407/ ujpe67.8.602.
- Theoretical and experimental approach on investigation of ethylurea-water clusters. Z. Phys. Chem. 2023
- [CrossRef] [Google Scholar]
- Noncovalent interactions in N-methylurea crystalline hydrates. Z Phys Chem 2023
- [CrossRef] [Google Scholar]
- Sulfamic acid/water complexes (SAA-H2O(1–8)) intermolecular hydrogen bond interactions: FTIR, X-ray, DFT and AIM analysis. J. Mol. Struct.. 2022;1265:133394
- [CrossRef] [Google Scholar]
- Molecular structure, spectral analysis and hydrogen bonding analysis of ampicillin trihydrate: a combined DFT and AIM approach. New Journal of Chemistry. 2015;39(12):9800-9812.
- [Google Scholar]
- Über die zuordnung von wellenfunktionen und eigenwerten zu den einzelnen elektronen eines atoms. Physica. 1934;1:104-113.
- [CrossRef] [Google Scholar]
- Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem.. 2012;33:580-592.
- [CrossRef] [Google Scholar]
- Potential energy surface, effect of solvents in molecular level, experimental spectra (FTIR, raman, UV–visible & NMR), electronic, and dynamics simulation of isobavachalcone–Anti tuberculosis agent. J. Mol. Liq.. 2023;392:123465
- [CrossRef] [Google Scholar]
- Triazoles and their derivatives: chemistry, synthesis, and therapeutic applications. Front. Mol. Biosci.. 2022;9:864286
- [CrossRef] [Google Scholar]
- DFT and molecular docking study of the effect of a green solvent (water and DMSO) on the structure, MEP, and FMOs of the 1-ethylpiperazine-1, 4-diium bis (hydrogenoxalate) compound. J. Mol. Liq.. 2023;369:120851
- [CrossRef] [Google Scholar]
- Proton acceptor abilities of heterocyclic compounds determined by stretching vibration bands of the N-H groups of associated molecules. J. Struct. Chem.. 2016;57:1024-1026.
- [CrossRef] [Google Scholar]
- Biological exploration of a novel 1,2,4-triazole-indole hybrid molecule as antifungal agent. J. Enzyme. Inhib. Med. Chem.. 2020;35(1):398-403.
- [CrossRef] [Google Scholar]
- Alkali metals doped cycloparaphenylene nanohoops: promising nonlinear optical materials with enhanced performance. Heliyon. 2023;9(11)
- [CrossRef] [Google Scholar]
- Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. Am. Chem. Soc.. 2000;122(45):11154-11161.
- [CrossRef] [Google Scholar]
- Non covalent interactions analysis and spectroscopic characterization combined with molecular docking study of N′-(4-methoxybenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. Journal of King Saud University-Science. 2022;34(2):101778
- [CrossRef] [Google Scholar]
- Remarkable static and dynamic NLO response of alkali and superalkali doped macrocyclic [hexa-] thiophene complexes; a DFT approach. RSC advances. 2021;11(7):4118-4128.
- [CrossRef] [Google Scholar]
- PES, molecular structure, spectroscopic (FT-IR, FT-raman), electronic (UV-vis, HOMO-LUMO), quantum chemical and biological (docking) studies on a potent membrane permeable inhibitor: dibenzoxepine derivative. Heliyon. 2020;6(8)
- [CrossRef] [Google Scholar]
- Hydrogen bonds: relation between lengths and electron densities at bond critical points. Eur. Phys. J. D. 2006;37:217-222.
- [CrossRef] [Google Scholar]
- Molecular structure interpretation, spectroscopic (FT-IR, FT-raman), electronic solvation (UV–Vis, HOMO-LUMO and NLO) properties and biological evaluation of (2E)-3-(biphenyl-4-yl)-1-(4-bromophenyl) prop-2-en-1-one: Experimental and computational modeling approach. Spectrochim Acta A Mol Biomol Spectrosc.. 2020;226:117609
- [CrossRef] [Google Scholar]
- Computational spectroscopic investigations on structural validation with IR and raman experimental evidence, projection of ultraviolet-visible excitations, natural bond orbital interpretations, and molecular docking studies under the biological investigation on N-Benzyloxycarbonyl-L-Aspartic acid 1-benzyl ester. Chem. Dat. Coll.. 2021;31:100622
- [CrossRef] [Google Scholar]
- Potential of first row transition metal decorated graphtriyne quantum dots as single atom catalysts towards hydrogen evolution reaction (HER) Physica Scripta. 2023;98(11):115308
- [CrossRef] [Google Scholar]
- Unveiling the potential of B3O3 nanoflake as effective transporter for the antiviral drug favipiravir: density functional theory analysis. Molecules. 2023;28(24):8092.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2024.103164.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




