

Translate this page into:
Exploring particulate methane monooxygenase (pMMO) proteins using experimentation and computational molecular docking
⁎Corresponding authors. wafa.ali.11338@gmail.com (Wafa Ali Eltayb), mohnadabdalla200@gmail.com (Mohnad Abdalla), mahboobchem@gmail.com (Mahboob Alam)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Researchers had difficulty studying pure full-length pMMO due to the solubility problem and loss of enzymatic activity after its elimination from the native membrane. To study pMMO, we performed several bioinformatics tools to analyze the entire structure of it available in the PDB database. We also carried out molecular docking studies to prove that quinone and duroquinone can bind to several sites of eight pMMO proteins. However, some sites in the orientation are not required by the catalysis process. Furthermore, molecular docking was done for predicting the binding affinity of P450 with target enzymes. Interestingly, our analysis illustrated that pMMO can produce methanol in the presence of quinone and duroquinone and the absence of Cu. Moreover, pmoB1 can interact with P450. Consequently, our findings highlight, for the first time, the significance of studying the membrane of pMMO to provide valuable insights into its functions.
Keywords
Methanol
Molecular docking
Particulate methane monooxygenase
Quinone
Duroquinone

1 Introduction
Sustainable energy is arguably a very important challenge to society. (Hanson and Hanson 1996, Park et al., 2002). Methanol production is a standard outlet for natural gas. The production of methanol from methane has been receiving much attention for a long time and is still ongoing by various methods (Fig. 1). Effective production of methanol from methanotrophs requires inhibition of methanol dehydrogenase (MDH) (EC 1.1.1.244) to stop further methanol oxidation. The oxidation of methanol is required to generate reduction equivalents. Thus, inhibiting MDH needs the addition of an external reducing counterpart to drive methane oxidation. Generally, most studies focus on utilizing methane monooxygenase (MMO) (EC 1.14.13.25), but other choices include the use of ammonia-oxidizing bacteria (Ge et al., 2014). The toxicity of methanol to methanotrophic bacteria is a likely challenge that is often discussed for commercialization.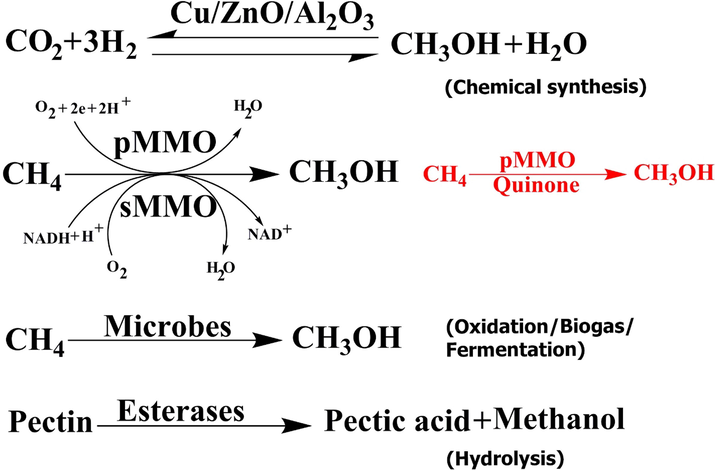
Production methods of methanol.
Methanotrophic bacteria use methane and convert it to methanol in the first step of their metabolic pathway; Methane is known as a considerable greenhouse gas (Hanson and Hanson 1996). Methanotrophs are gram-negative bacteria belonging to the phyla Proteobacteria and Verrucomicrobia, they are divided into three groups, Type I (also known as Gammaproteobacteria), Type II (Alphaproteobacteria) methanotrophs, and Verrucomicrobia. The methanotrophs convert methane to methanol, which is present in particulate or soluble form, and the MDHs catalyzed methanol to formaldehyde. After two more oxidation steps, the final intermediate products are used for carbon assimilation (Guerrero-Cruz et al., 2021). As the most important methane sinks in nature, these methanotroph bacteria are hopeful biological tools for biofuel production as well as methane remediation (Fei et al., 2014, Kalyuzhnaya et al., 2015, Lawton and Rosenzweig 2016). These bacteria activate a 105 kcal/mol C–H bond in methane utilizing metalloenzymes recognized as MMO (Zhu et al., 2022). They are categorized as membrane-bound (particulate, pMMO) (EC 1.14.18.3) or soluble (sMMO) (EC 1.14.13.25). MMO is the first enzyme in the metabolic pathway of methanotrophs, which are bacteria that utilize methane as their only source of carbon and energy (Hanson and Hanson 1996, Park et al., 2002). However, pMMO is involved in several biological processes (Supplementary Table 1), such as preponderant methane oxidation catalysts in living organisms. Unfortunately, pMMO is less characterized (Sirajuddin and Rosenzweig 2015, Ross and Rosenzweig 2017).
The pMMO methane oxidation should be understood to facilitate methanotroph engineering and to guide synthetic catalyst design (Balasubramanian et al., 2010, Sirajuddin et al., 2014, Sirajuddin and Rosenzweig 2015). Crystal structures of pMMO from eight different methanotrophs show approximately 300 kDa α3β3γ3 trimer, composed of three chains, namely, α (pmoA), β (pmoB), and γ (pmoC) (Lieberman and Rosenzweig 2005, Hakemian et al., 2008, Smith et al., 2011, Sirajuddin et al., 2014). α and γ chains are membrane subunits, and the β-chain is composed of two transmembrane helices and two periplasmic domains.
The crystal structures of eight pMMO proteins have been solved and are available in The Protein Data Bank (PDB). These structures have a copper site assigned as the active site located at the N-terminus of β chain, with two histidines from an HXH motif and the N-terminal histidine of PmoB1 or/and PmoB2 as ligands (Balasubramanian et al., 2010). The active site has been occupied with either one or two copper ions according to structures. However, the structure of pMMO from Methylococcus (Mcc.) capsulatus (Bath) has an additional pmoB monocopper site (Lieberman and Rosenzweig 2005). The γ chain subunit active site can be occupied by either copper or zinc, according to the crystallization conditions.
At present, our knowledge of the importance of microbial groups to undermine metabolic engineering strategies is limited. For effective metabolic engineering, the necessary knowledge about methanotrophy should be highlighted as well as how the knowledge gaps should be addressed. For instance, the difference in the structure between the pMMO and the binding of quinone and duroquinone is unknown.
2 Material and method
2.1 Cloning, expression, and purification of pMMO
The coding region of pMMO without the signal peptide (only pmoB 1&2) was PCR amplified from Methylococcus capsulatus genomic. Details about the cloning and expression method are shown by (Abdalla et al., 2016, Abdalla et al., 2018).
2.2 Bioinformatics analysis software
Multiple sequence alignment of pMMO was performed by employing the Clustal X version (1.83) with its default constraints (Larkin et al., 2007) and ESPript web server (Robert and Gouet 2014) to determine the sequence similarity. The phylogenetic tree was formed with MEGA version 7 by using the neighbor‐joining algorithm method (Kumar et al., 2016). Transmembrane protein was predicted utilizing the TMHMM software (Krogh et al., 2001) (https://www.cbs.dtu.dk/services/TMHMM-2.0/). Topology diagram of pMMO, obtained from PDBsum (https://www.ebi.ac.uk/).
2.3 Protein modeling and molecular docking
The Protein Data Bank (PDB) (https://www.rcsb.org/) (Berman et al., 2000, Westbrook et al., 2003) was used to obtain eight reference tertiary structures of pMMO wild-type (WT) pMMO (Methylococcus. capsulatus (PDB ID: 1YEW) (Lieberman and Rosenzweig 2005), Methylosinus trichosporium (PDB ID: 3CHX) (Hakemian et al., 2008), M. capsulatus (PDB ID: 3RGB) (Smith et al., 2011), Methylomicrobium alcaliphilum (6CXH) (Ro et al., 2018), Methylocystis sp. ATCC 49242(Rockwell) (PDB ID: 4PHZ, 4PI2, 4PI0) (Sirajuddin et al., 2014), and Methylocystis sp. strain M (PDB ID: 3RFR) (Smith et al., 2011), which represents a monomer form of pMMO β chain without Zn and Cu. To find possible binding sites of duroquinone (PubChem ID: 68238) and quinone (PubChem ID: 4650) (Kim et al., 2018) from the pMMO-encoded protein, a molecular docking and thermodynamic analysis were done using the web-based Swissdock program (https://www.swissdock.ch/docking) (Bitencourt-Ferreira and de Azevedo 2019, da Silveira et al., 2019). Swissdock predicts the possible molecular interactions between eight different methanotrophs and a small molecule by using the docking algorithm EADock DSS (Grosdidier et al., 2007, Alameen et al., 2022). A blind molecular docking was performed with the “Accurate” parameter or default parameters when no region of interest was defined. Binding energies were evaluated using Swissdock software. After all, the results were scored and ordered by full fitness (kcal mol−1), and the spontaneous binding was revealed by the estimated Gibbs free energy ΔG (kcal mol−1). The modeling and docking results were visualized using the PyMol software.
The structure of P450 (PDB code: 2J1M) was docked onto the pMMO β chain structure (PDB code: 4PI2) by using HADDOCK2.2 (Rodrigues et al., 2013). HADDOCK generated several structures in three clusters. The best cluster represents the most reliable model and had a HADDOCK score of − 95.2 and a Z score of − 1.2.
3 Result and discussion
3.1 pMMO crystal package
All pMMO crystal packages contain nine chains (three sets of αβγ); 6CXH contains three chains (one set of αβγ). Unexpectedly, these chains contain different sequences and have different 3D structures as well as binding atoms. For example, Zn ions are found in 1YEW, 3RFR, 3RGB, and 4PI2 but not in 3CHX, 4PHZ, 4PI0, and 6CXH. In addition, the pMMO crystal package contains different numbers of extra ligands (unknown peptide), such as 3CHX containing six extra ligand chains 4PHZ, 4PI0, 4PI2 containing three extra ligand chains, and 3RFR with only two extra ligand chains. Cu–Cu cluster is found in 1YEW, 3CHX, 3RFR, and 3RGB, and Zn–Zn cluster is found in 1YEW only (Supplementary Table 2).
3.2 Electrostatic surfaces of pmoB
The hydrophobicity of protein assists in reducing its surface area and the unfavorable interactions with water to maintain biologically active and stable protein. Here, we highlight the side views of the electrostatic surfaces of the eight pMMO β chains with completely modeled structures. Both positively and negatively charged surfaces are colored blue and red, respectively. The structures of pMMO (PDB ID: 1YEW, 3CHX, 3RFR, 3RGB, 4PHZ, 4PI0, 4PI2, and 6CXH). Mechanistically, the Cu–Cu cluster and Zn can be located in different positions for effective electron transfer by the electrostatically steered interactions and geometric complementation.
In all pMMO chains, strongly positively charged surfaces were detected in the Zn-chelating regions in 3RGR, 4PHZ, 4PI0, and 4PI2, which were found in strongly negatively charged surfaces in 1YEW and 3RGB. However, strongly negatively charged surfaces plus were detected in the Cu-chelating regions in 3RFR and neutrally charged surfaces in 3CHX. In addition, 6CXH was found in negatively and neutrally charged surfaces in 1YEW and 3RGB, respectively. Surprisingly, no Cu ion was found in the positively charged surfaces, and clear surfaces between the Cu and Zn binding sites were observed. The electrostatic potential on the β chain molecule shows the distribution of positive and negative charges on the surface. We hypothesize that Cu-Cu clusters surrounded by a negatively charged surface exist in the pMMO β chains, making them easily show high and broadband activities. Thus, this negative charge appears to be approximately harmonic with the pMMO flexibility of branch B (Fig. 2).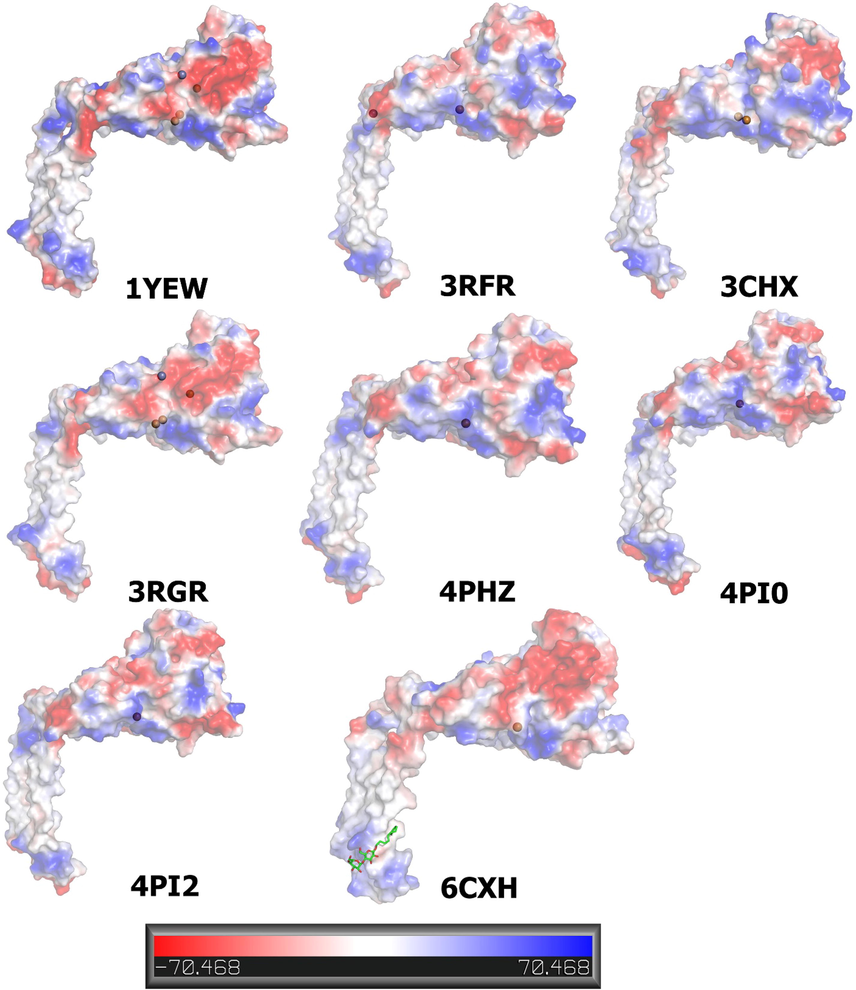
Comparison of the electrostatic potential on the molecular surface of pMMO β chains. Bound Cu and Zn are shown as a ball. CM5 is shown in 6CXH as a stick model. The electrostatic surface is presented as a color gradient in red (electronegative, ≤ −10 kT e-1) and blue (electropositive, ≥ −10 kT e-1); analysis of the 3D protein electrostatic surfaces derived from the Pymol software using vacuum electrostatic method.
3.3 pMMO topology
pMMO proteins consist of three chains α, β, and γ. According to the PDBsum database, the pMMO topology has a different number of α-helices and β-strands. In detail, the α chain of 1YEW and 3RGB consists of two α-helices and nine β-strands, 3RFR consists of three α-helices and nine β-strands, 3CHX consists of one α-helices and nine β-strands, and 6CXH consists of two α-helices and 10 β-strands (Fig. 3αc).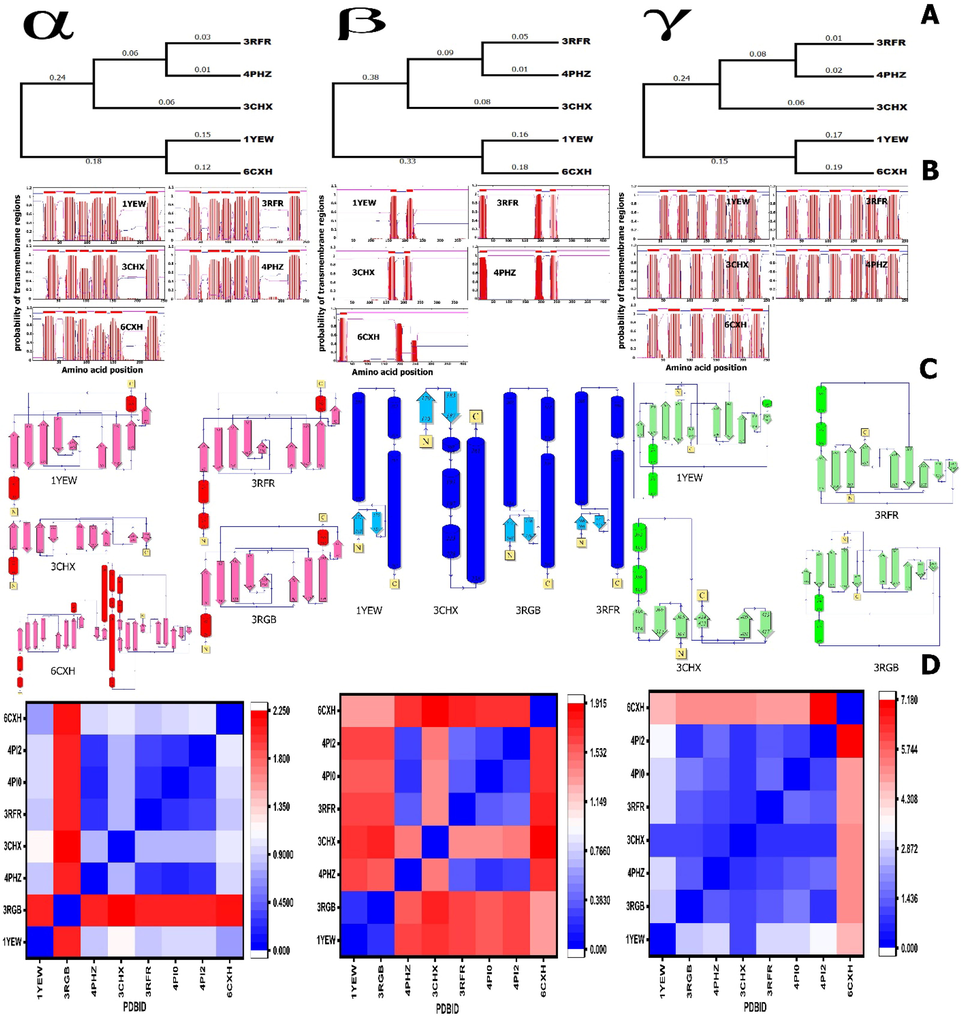
Phylogenetic tree. The tree was constructed from a complete alignment using the neighbor-joining method. B. Prediction of transmembrane regions of pMMOs. Transmembrane regions are in red, and other regions are predicted to be either outside (pink) or inside (blue) the membrane. Data were obtained from the TMHMM Server v. 2.0. X-axis, which refers to the amino acid position; Y-axis refers to the probability of the transmembrane regions. The pink line refers to intracellular proteins, and the blue line refers to extracellular proteins. C. Topology diagram of pMMO. Helices are represented by barrel, and strands of β-sheets are shown with arrow. D. RMSD diagram; the RMSD was calculated using the PyMol software.
In the case of γ chain, 1YEW consists of three alpha-helices and 10 beta-strands, 3RFR consists of two alpha-helices and nine beta-strands, 3CHX consists of two alpha-helices and six beta-strands, 3RGB consists of two alpha-helices and 10 beta-strands, and 6CXH consists of 10 beta-strands only (Fig. 3γc). In β chain 1YEW, 3RFR and 3RGB consist of three alpha-helices and two beta-strands, and 3CHX consists of four alpha-helices and two beta-strands (Fig. 3βc). Unfortunately, the data for all structure chains is not available in the PDB sum database. Our structural topology result can explain the wide ranges of RMSD between the pMMO structures. Long asymmetric loops in α and γ chains fold into alpha-helices, which may prevent [zn and cu] assembly and may lead to a high oligomerization tendency.
3.4 pMMO transmembrane
It is known that signaling protein regions have different types of functions. One of these functions results in the moving of the polypeptide into the endoplasmic reticulum (Abdalla et al., 2018), which is very important to identify. pMMO plays an essential role in the cell wall due to its attachment to the plasma membrane. The translocation of the protein is based on the interaction interface of different regions with the phospholipid membrane. Therefore, our analysis of pMMO transmembrane regions demonstrated that all pMMO protein contains αβγ chains.
Signal peptide prediction of β chains in 3RFR, 4PHZ, and 6CXH showed three signal peptides in each of them 1 localized at the N and 2 localized at the C-terminus, while in the case of 1YEW and 3CHX, there are only 2 signal peptides localized at the C-terminus (Fig. 3βb). In addition, methanotrophic bacteria pMMO transmembrane are predicted to α and γ chains, and results show six signal peptide with different intracellular and extracellular amino acid positions (Fig. 3 α and γ b).
Protein functions are generally confined to specific locations. Therefore, the prediction of protein localization allows knowing more about the mechanistic pathways. Localization analysis using the signal peptide server indicated that some parts of pMMO include cytoplasmic proteins. pMMO transmembrane localization has not been demonstrated previously, and this result can provide important details about the methanotrophic bacteria cell wall architecture.
3.5 Multiple sequence alignment and phylogenetic tree
In the present study, pMMO is analyzed by bioinformatics tools to identify sequence similarity. FASTA sequences belonging to the reference pMMO proteome are downloaded from the PDB database to highlight the conserved regions.
A high degree of similarity is shared between the pMMO sequences and the percentage sequence identity calculation in the process. Some conserved motifs and amino acids that are common to methanotroph species are highlighted. However, one or more differences may affect the entire protein function.
The sequences of and around the catalytic ligands His33, 48, 72, 137, 139, and/or Glu35 of pMMO β chain from different methanotrophs are shown in Fig. 4b. Although there is controversy in the community as to the actual active site of pMMO.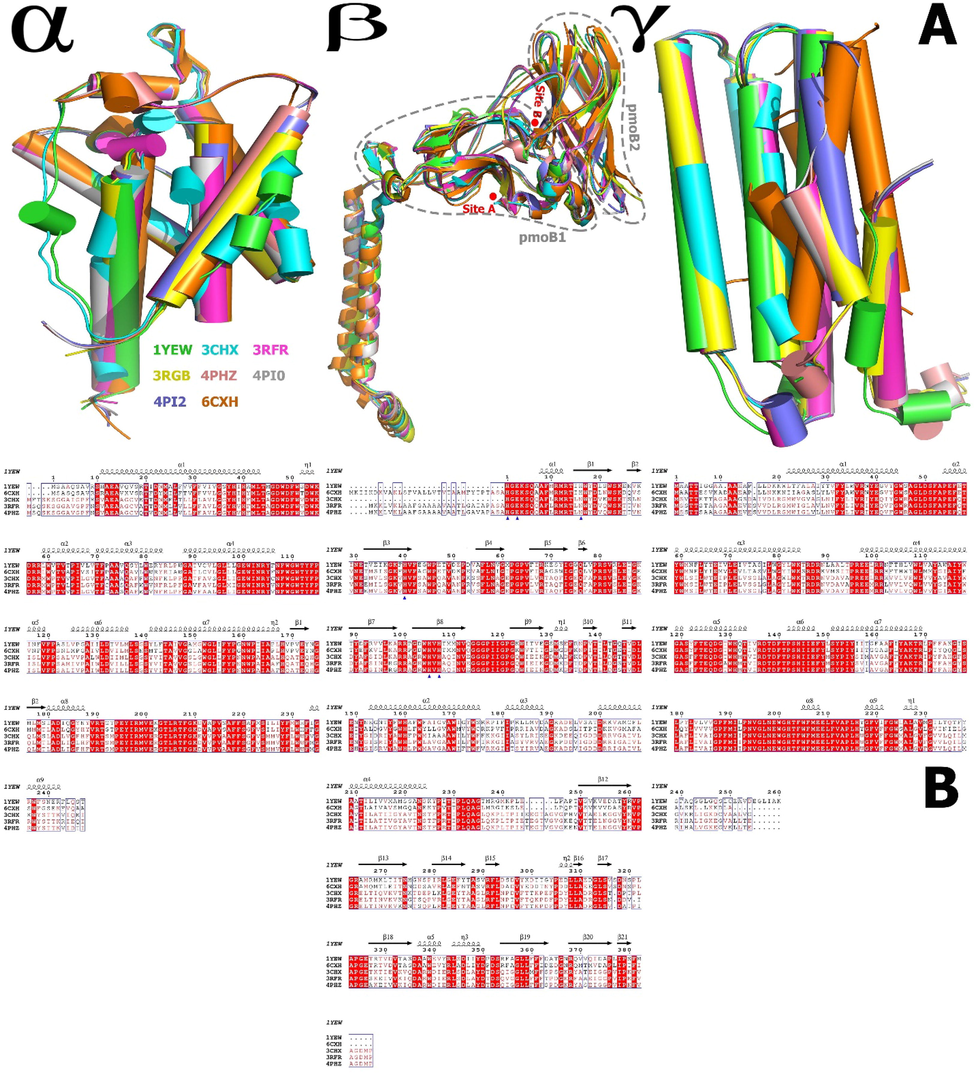
Cartoon representation of the structural alignment, and superposition of the structures of the pMMO α, β, and γ chains, where β chains pmoB1 and pmoB2 and the membrane-embedded region interact directly and make a catalytically active conformation, demonstrating the high conformational adaptability. The models were created by the Pymol. B. Sequence homology of and around the catalytic ligands of pmoB from different methanotroph species. Blue triangle. The sequence homology of and around the catalytic conformation (His33, His137, His 139, and/or Glu35 at site A and His48 and His72 at site B) of pmoB1 from different methanotroph species (M. capsulatus (PDB ID: 1YEW), M. trichosporium (PDB ID: 3CHX), M. alcaliphilulm (6CXH), Methylocystis sp. ATCC 49242(Rockwell) (PDB ID: 4PHZ), and Methylocystis sp. M (PDB ID: 3RFR), M. capsulatus (PDB ID: 3RGB), and Methylocystis sp. ATCC 49242(Rockwell) (PDB ID:4PI0 & 4PI2) are deleted because of the ambiguously aligned portions.
As a structural variation, we calculate the sequence identity in the phylogenetic tree (Fig. 3a). For example, in 1YEW and 6CXH, α = 78.41%, β = 67.39%, and γ = 63.79%. In 3RFR and 4PHZ, α = 96%, β = 4%, and γ = 96.8%. The average of sequence identity between the hole pMMO protein (in 1YEW, 6CXH, 3RFR, 4PHZ, and 3CHX) is α = 81.27%, β = 72%, and γ = 70.93%.
PDBFlex tool (Hrabe et al., 2016) has been used to characterize regions of structural flexibility present in the pMMO structure. These results indicate that two to three regions on the α and γ chains containing the loop reflect important structural flexibility. β chain has loops reflected at the C-terminal but are unimportant and do not interact with distinct ligands.
3.6 Root mean square deviation (RMSD) and superimposed structures
The structure of pMMO appears identical, as shown in the superposition of the structures (Fig. 4a). We calculated the superposition RMSD to assess the structural differences in the pMMO. These results revealed some structural dissimilarities.
RMSD measures the similarity between 3D structures of all pMMO structures in the PDB database. After optimal superposition, the values of all backbone atoms and the metal-binding site for only one monomer were calculated and shown in Fig. 3d). The average distance deviation between backbone equivalent atoms is between 0.2 and 1.91 Å in β chains and 0.17 to 2.2 in α chains and 0.8 to 7.1 Å in γ chains. A comparison of the RMSD values among the eight β structures shows that the 1YEW and 3RGB have the lowest RMSD 0.2 Å, and 3CHX and 6CXH have the highest RMSD 1.91 Å. The γ chains of the 6CXH structure exhibited a relatively larger RMSD value (∼7.18 Å) than that of 4PI2, probably due to the opposite residue sequence in the retro form of γ chains. Among the pMMO α chains, 4PHZ shows the smallest RMSD values having ∼ 0.171 Å compared with the 4PI0 tertiary structure.
Kim (Kim et al., 2019) suggested that all the designed pMMO show a significant difference in RMSD values by comparing pmoB with WT-pMMO for mono and di-copper models. Our results showed a difference in RMSDs between mono and di-copper for pmoB1 site A.
The influence of the configurational differences on the structural similarity between the structural motifs (pmoB1/pmoB2 domains) in the crystal structures of WT-pMMO can be found in the ability to bind the decoy molecule. This finding suggests that pMMO protein has differences in the binding pocket in the different structures. Approximately, the RMSD is not identical in the reduced and oxidized forms. This result suggests that the uneven structure may influence the enzymatic function of native pMMO.
3.7 High-yield insoluble recombinant pMMO
The culture of recombinant E. coli BL21(DE3) is transformed into PET28a and PET22b plasmid expression vectors. Notably, the expressed pmoB (pmoB1/pmoB2) was very high but unfortunately insoluble, as previously reported (Balasubramanian et al., 2010) (Supplementary Fig. 1). A recent report suggests that the interdomain interaction between pmoB1 and pmoB2 is necessary for the expression of soluble pmoB (Kim et al., 2019).
3.8 Molecular docking of quinone and duroquinone to pMMO β chains
Quinones are oxidized derivatives of aromatic compounds and are often readily prepared from reactive aromatic compounds bearing electron-donating substituents such as phenols and catechols. Duroquinol, which has low water solubility, acts as an electron donor either in dissolved form or as a dried powder. The protein–ligand interaction studies play an important role in enzyme engineering for industrial applications. pMMO is one of the most promising proteins for methanol production and commercial use.
A molecular docking was done to map the interactions between quinone and duroquinone to identify a putative binding site for these compounds with β chains of pMMO. The molecular interaction between β chains and two cofactor molecules (quinone and duroquinone) was examined through a Swissdock (Grosdidier et al., 2007, Bitencourt-Ferreira and de Azevedo 2019, da Silveira et al., 2019) (Fig. 5) analysis along with the corresponding 3D structure. Molecular docking identifies different sites of the protein.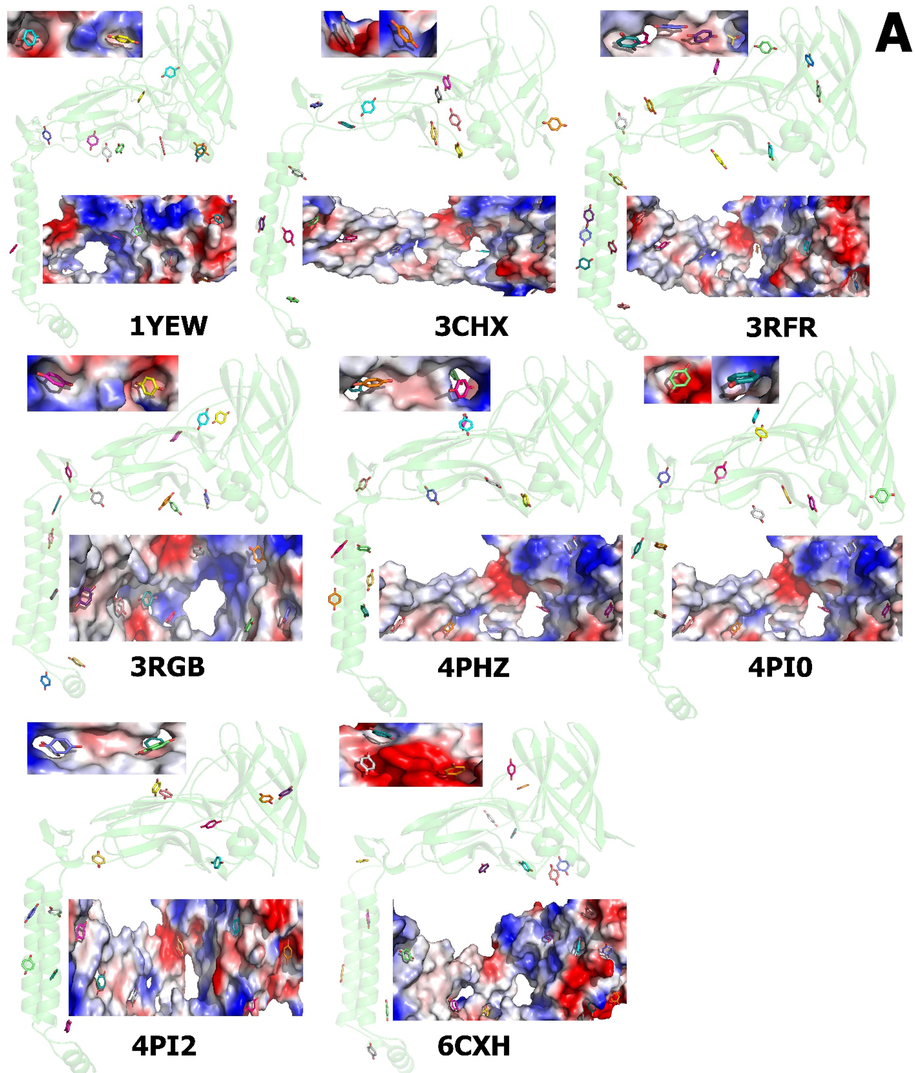
pMMO β chain proteins are shown in cartoon form by light green color; structure of pmmo B with quinone (PubChem ID: 4650) (5A) and duroquinone (PubChem ID: 68238) (5B). The predicted binding site for best energy conformations for quinone and duroquinone displayed is shown as a solid surface (at the top and bottom of the cartoon structure); both molecules are localized in the surface of pMMO β chains. Allocation of the electrostatic potential, where red regions represent the negative electrostatic potential and blue ones represent the positive electrostatic potential (from − 70 kcal mol−1e−1 for red, the most negative residue to white at 0.0, to + 70 kcal mol-1e-1 for blue, the most positive residue) and hydrophobicity on the surfaces of the pMMO β chains. The estimated locations of the quinone and duroquinone are illustrated. The models are created by the Pymol.

pMMO β chain proteins are shown in cartoon form by light green color; structure of pmmo B with quinone (PubChem ID: 4650) (5A) and duroquinone (PubChem ID: 68238) (5B). The predicted binding site for best energy conformations for quinone and duroquinone displayed is shown as a solid surface (at the top and bottom of the cartoon structure); both molecules are localized in the surface of pMMO β chains. Allocation of the electrostatic potential, where red regions represent the negative electrostatic potential and blue ones represent the positive electrostatic potential (from − 70 kcal mol−1e−1 for red, the most negative residue to white at 0.0, to + 70 kcal mol-1e-1 for blue, the most positive residue) and hydrophobicity on the surfaces of the pMMO β chains. The estimated locations of the quinone and duroquinone are illustrated. The models are created by the Pymol.
The highest-scoring cluster showed better full fitness; This parameter is determined by averaging the 30% most favorable effective energies of a cluster element, and the lower free energies for the potential binding sites and the Gibbs free energy (ΔG) of docking were evaluated. Thus, the thermodynamic parameters, full fitness, and free energy (ΔG) provided by Swissdock clearly showed that quinone and duroquinone can bind tightly to pMMO β chains.
From the docking of the ligands into pMMO protein, quinone shows the visualization of the most energetically favorable binding. Full fitness between − 1790.89 and − 2043.71 Kcal mol−1 and estimated ΔG between − 5.65 and − 6.51 Kcal mol−1, whereas duroquinone showed full fitness between − 1832.38 –and − 2106.54 kcal/mol and ΔG between − 5.89 to − 6.96 Kcal mol−1 for the most favorable interaction (Supplementary Table 3).
The interactions between quinone and duroquinone with pMMO normally occur through the voids and/or protrusions present on the pMMO surface. Swissdock's computational tools enable the identification of cavities and the characterization of their local geometric and chemical properties, the duroquinone has a higher free energy of a pMMO than quinone, which means duroquinone is more stable or rapidly intrepidity pMMO.
Several studies reported that duroquinol molecules were detected in the space between the pmoB1 and PmoB2 (Fig. 6). In addition, the numbers of hydroquinone and duroquinone in the region close to the copper sites were considerably reduced compared with that of duroquinol (Kim et al., 2019), particularly in some charged groove regions within roughly 1 nm. The hydrophobic residues were predominantly found at the same site and may provide polar and nonpolar interactions to bind amphiphilic quinone and duroquinone. Unfortunately, previous reports did not mention whether hydrogen bond occurs or how to bind to β chains. We believe that due to the same grove, they can be located at the surface of the protein. Molecular docking results indicate that these enzymes can bind duroquinol partially. However, the binding energy is smaller than that of the interactions with quinone and duroquinone.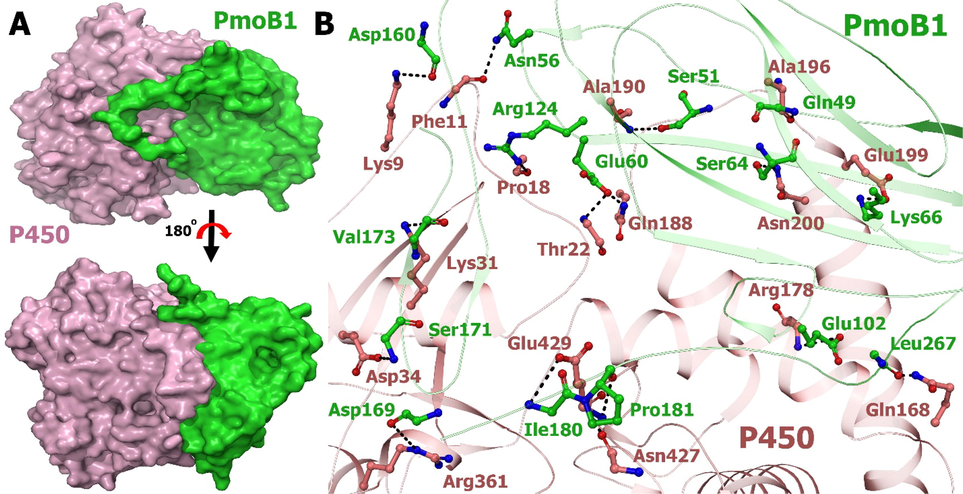
Interactions of pmoB1 (set as a ligand) and P450 (set as a receptor) are studied by HADDOCK2.2. A. The surface structures pmoB1 and P540 are colored tv_green and light pink, respectively. The top view of the same is visualized by rotating the molecule at 180° toward the reader. B. Stereo representation suggested binding mode. A hydrogen bond is displayed by black arrows. Residues involved in the interaction are shown as sticks.
We suggest that quinone and duroquinone binding to pMMO β chains can be detected unless unusually high or less concentrations were binding to the protein and depend on the 3D structure. Thus, quinone and duroquinone probably did not affect the structure, contrary to the enzymatic function of pmoB. This finding suggests that pmoB1 and membrane region are probably the best candidates for preserving the binding to quinone and duroquinone camper to pmoB2. For instance, the concentration of duroquinone in that region near site A significantly decreases when compared with the case of quinone. Furthermore, duroquinone can be released after oxidizing electron shuttling to copper sites, afterward refilling the pool with fresh quinone and duroquinone from the bulk solution. These results reveal that selecting the cofactor has supreme importance for appropriately reloading and releasing the cofactor in the pool essential for electron shuttling to copper sites.
Hydroquinone is another reducing agent and water-soluble with a similar reduction potential, but cannot stimulate methanol production, according to previous reports. The catalytic sites of pmoB probably bind directly to duroquinol (Lieberman and Rosenzweig 2005). Based on previous reports, the copper sites in the periplasmic domains of pmoB (1 and 2) but not pmoA or pmoC are key players in pMMO activity (Chan et al., 2007). Compared with the two copper sites in pmoB, site A is proposed to be more suitable for methane oxidation than site B (Yoshizawa and Shiota 2006). In a similar study, sites A and B are mutant. No methanol production is observed when site A is mutant, and methanol is found when site B is mutant. The turnover number and methanol production are decreased by the site-B mutant. Site B is not a catalytically activated site, but plays a remarkable role in completely activating site A (Kim et al., 2019). The analysis shows that quinone and duroquinone molecules are enriched more in the region near site A than site B.
The methanol production started after two to four hours from adding duroquinol as the dried powder to the reaction system. This finding is due to the dissolved duroquinol that binds quickly to pMMO active sites compared with the undissolved form. The methanol production was increased by more amount of dissolved duroquinol within the pMMO reaction system (Kim et al., 2019). Hence, this can prove that our result (pMMO β chains) can bind a high number of quinone and duroquinone through the surface when available. In addition, these data suggest that the alcohol group is not crucial for the interaction.
Quinone and duroquinone molecules can guide the pMMO to select the reaction type by orientation needed by the catalysis process the same as olefins, which can guide the p450 to select the reaction type. Therefore, pMMO (PDB 4PI2) is perhaps the top candidate for maintaining the structure and enzymatic function of native pMMO.
Therefore, RMSD is a measure of similarity between the experimental macromolecule and the computed macromolecule with docking ligand of the same crystal package, showing no different structure either with quinone or duroquinone. All the ligands in Fig. 5 and Supplementary Fig. 2 has almost the same binding energy.
3.9 Docking pmoB1 to P450 heme domain
Cytochromes P450 (CYPs) are enzymes containing heme as a cofactor that functions as monooxygenases (Gonzalez and Gelboin 1992) the same as pMMO. P450 has industrial applications and has been studied deeply. At present, a hundred mutation has been created but cannot be further modified. We estimate that the two oxygenase enzymes are combined with other oxygenase enzymes, such as pMMO. Substantially, super protein can be widely used and can catalyze more reactions and solve protein problems. Practically, pMMO can increase p450 stability even in high temperatures, and P450 can increase pMMO solubility. This combination can be more efficient as monooxygenases. We believe that it will be useful at the industrial level. For these reasons, we dock pMMO/pmoB1 to P450.
To gain structural insights into the increased pmoB1 and P450 binding affinity without the help of a corepressor, we perform docking experiments to assemble a complex model. Electrostatic potential analysis revealed that the P450 subunit of the interaction region has a strongly negative patch (Fig. 4C), in addition to the basic and neutral region in pmoB1. This finding strongly suggests that P450 and pmoB1 form a complex interaction between the complementary basic patch and acidic surface, as well as the hydrogen bond. Simulation with HADDOCK2.2 enabled us to build a stable complex of P450-pmoB1 with a buried interface area of ∼ 1219 Å. The distances between the heme and Cu in the most possible docking modes range from 44 to 47 Å. And in parallel, other complexes, especially those with the narrowest reactivity spectra, may have longer corresponding distances (>47 Å).
This putative binding site is established by residues located in the protein surface instead of the active site residues. In this regard, pmoB1 and P450 might have strong interaction because there is hydrogen, hydrophobic interactions, electrostatic energy, helix dipole energy, and cis bond energy, and no van der Waals, torsional, or backbone clash occurs. Therefore, the high free energy required hydrogen bonds suggested a strong nonspontaneous binding process for both proteins. The hydrogen bond in a distance of less than 3.5 Å occurs among the amino acids (Gln46, Ser51, Asn 56, Glu60, Ser64, Lys66, Glu102, Arg124, Asp160, Asp169, Ser171, Val173, Ile 180, Pro181, and Leu267) on PmoB1 and (Lys9, Phe11, Pro18, Thr22, Lys31, Asp34, Gln168, Arg178, Gln188, Ala190, Ala196, Glu199, Asn427, Gln429, Arg361) on P450. In addition, the distance between the heme and the Cu is 47 Å, the heme is inside the complex and the Cu is the surface. Moreover, residues in pmoB1 involved in binding Zn or Cu did not participate in binding because this residue lies in the protein. Therefore, it is difficult to the methanol production.
The structure of pMMO reveals that the enzymes have an open active site with multiple subsites. These data imply that the catalytic cavities of the studied pMMO have many properties. sMMO and p450 have narrow active site cavities, while pMMO has shallow and more open active site cavities. In addition, our data demonstrate that pMMO displays larger substrate specificity at multiple positions.
Reports have shown that the activity of a P450 enzyme could be enhanced by introducing a gene encoding a more efficient [2Fe-2S] into the bacterial genome (Zhang et al., 2018). Therefore, we assume that this state can also occur in Zn-Cu. Further experiments are required to test this industrially important hypothesis, which is currently being performed in our laboratory.
This knowledge can be used to design pmoB1-P450 complex for biological applications. Engineering a novel pmoB1-P450 complex, which has functions and regulatory mechanisms beneficial under specific conditions or can be integrated with other methane-oxidizing systems favorable for industrial applications, is an intriguing idea.
Further studies illustrating interactions with the microenvironment are proposed to confirm the pmoB1 and P450 interactions. Finally, HADDOCK and Swissdock return docking information based only on the thermodynamic spontaneity and favorable interactions by which molecular structures bind to the peptide. Therefore, no apparent binding affinity information is available, and further studies on binding affinities are proposed to improve the estimation interactions.
3.10 Future plan
Several future research directions emerge from this study. First, laboratory experiments are necessary to prove our results. Further analysis is also needed to uncover whether the quinone and duroquinone transporters are involved in the interaction with other compounds and whether they act indirectly to provide tolerance to the stress response to high environmental concentrations of methanol.
Second, designing linkers (rigid or flexible) between the pmoB1 and P450 is important to increase the complex folding, expression, and biological activity and to avoid any further aggregation during the absence of salt bridges or oligomerization.
Third, it is important to perform molecular dynamics simulations for other types of intermolecular interactions to assess the bulk effect of affinity and to thoroughly assess the influence of side-chain geometry and variability in target and ligand flexibility on the observed interactions.
4 Conclusions
This article is the first to present the comparison between the sequence and structural properties of pMMO belonging to methanotrophic bacteria. We found that the residues participating in the catalytic processes are highly conserved. In contrast, a few differences are found between the physicochemical properties of the site of catalytic domains in β chains. Furthermore, the distribution of charge is observed inside the catalytic cavities of site B, and they are positively charged in their surrounding in all pMMO β chains, except the 1YEW, 3RGR, and 6CXH, which are negatively charged. Moreover, we observed the distinct surfaces between the Cu and Zn-binding sites inside pMMO β chains. In addition, protein topology shows a diversity in the number of alpha-helices and beta-strands of three pMMO chains, namely, α, β, and γ. Thus, the signal and cytoplasmic peptide have different positions and numbers inside the pMMO protein sequence. Our result suggested that the RMSD is different in the reduced and oxidized forms. Thus, the uneven structure may influence the enzymatic function of native pMMO, and the configurational differences in the structure can affect the ability to bind the decoy molecule.
Our results also indicate that these enzymes can bind and process quinone and duroquinone, but with lower efficiency into duroquinone. This finding confirms the unimportance of the carbon group for the interaction with the pMMO β chains for the methanol process, and some quinones and duroquinones can bind to the active sites and other sites on the surface without the need for hydrogen bonds. Thus, the methanol process can bind more of the quinone and duroquinone in one reaction time. Quinone is likely more sufficient in methanol production than duroquinone and duroquinol.
At the same time, the docking structure reveals that pmoB1 can produce active complexes with P450. This complex is beneficial for industrial applications. No structural rearrangement is observed in the case of the complex with P450 or when binding to quinone and duroquinone.
Acknowledgments
The authors are grateful to the Researchers Supporting Project Number (RSP2023R147), King Saud University, Riyadh, Saudi Arabia. This work was supported by the CAS President’s International Fellowship Initiative Program to MA.
Ethical approval
This article does not contain any studies with human participants or animals.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Crystal structure of yeast monothiol glutaredoxin Grx6 in complex with a glutathione-coordinated [2Fe-2S] cluster. Acta crystallographica. Section F, Structural biology communications.. 2016;72(Pt 10):732-737.
- [CrossRef] [Google Scholar]
- Structure analysis of yeast glutaredoxin Grx6 protein produced in Escherichia coli. Genes and environment : the official journal of the Japanese Environmental Mutagen Society.. 2018;40:15.
- [CrossRef] [Google Scholar]
- Comparison of structures among Saccharomyces cerevisiae Grxs proteins. Genes and environment : the official journal of the Japanese Environmental Mutagen Society.. 2018;40:17.
- [CrossRef] [Google Scholar]
- In-silico studies of glutathione peroxidase4 activators as candidate for multiple sclerosis management. J. Saudi Chem. Soc.. 2022;26(6):101554.
- [CrossRef] [Google Scholar]
- Oxidation of methane by a biological dicopper centre. Nature. 2010;465(7294):115-119.
- [CrossRef] [Google Scholar]
- Redox Potentiometry Studies of Particulate Methane Monooxygenase: Support for a Trinuclear Copper Cluster Active Site. Angew. Chem. Int. Ed.. 2007;46(12):1992-1994.
- [CrossRef] [Google Scholar]
- Web Services for Molecular Docking Simulations. Methods Mol Biol.. 2019;2053:221-229.
- [CrossRef] [Google Scholar]
- Bioconversion of natural gas to liquid fuel: opportunities and challenges. Biotechnol. Adv.. 2014;32(3):596-614.
- [CrossRef] [Google Scholar]
- Biological conversion of methane to liquid fuels: status and opportunities. Biotechnol. Adv.. 2014;32(8):1460-1475.
- [CrossRef] [Google Scholar]
- Human cytochromes P450: evolution and cDNA-directed expression. Environ. Health Perspect.. 1992;98:81-85.
- [CrossRef] [Google Scholar]
- EADock: docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins. 2007;67(4):1010-1025.
- [CrossRef] [Google Scholar]
- Methanotrophs: Discoveries, Environmental Relevance, and a Perspective on Current and Future Applications. Front. Microbiol.. 2021;12:678057
- [CrossRef] [Google Scholar]
- The metal centers of particulate methane monooxygenase from Methylosinus trichosporium OB3b. Biochemistry. 2008;47(26):6793-6801.
- [CrossRef] [Google Scholar]
- PDBFlex: exploring flexibility in protein structures. Nucleic Acids Res.. 2016;44(D1):D423-D428.
- [CrossRef] [Google Scholar]
- Metabolic engineering in methanotrophic bacteria. Metab. Eng.. 2015;29:142-152.
- [CrossRef] [Google Scholar]
- PubChem 2019 update: improved access to chemical data. Nucleic Acids Res.. 2018;47(D1):D1102-D1109.
- [CrossRef] [Google Scholar]
- Biological conversion of methane to methanol through genetic reassembly of native catalytic domains. Nat. Catal.. 2019;2(4):342-353.
- [CrossRef] [Google Scholar]
- Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol.. 2001;305(3):567-580.
- [CrossRef] [Google Scholar]
- MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol.. 2016;33(7):1870-1874.
- [CrossRef] [Google Scholar]
- Clustal W and Clustal X version 2.0. Bioinformatics (Oxford, England). 2007;23:2947-2948.
- [CrossRef] [Google Scholar]
- Methane-Oxidizing Enzymes: An Upstream Problem in Biological Gas-to-Liquids Conversion. J. Am. Chem. Soc.. 2016;138(30):9327-9340.
- [CrossRef] [Google Scholar]
- Crystal structure of a membrane-bound metalloenzyme that catalyses the biological oxidation of methane. Nature. 2005;434(7030):177-182.
- [CrossRef] [Google Scholar]
- The effect of various environmental and design parameters on methane oxidation in a model biofilter. Waste management & research : the journal of the International Solid Wastes and Public Cleansing Association, ISWA.. 2002;20(5):434-444.
- [CrossRef] [Google Scholar]
- From micelles to bicelles: Effect of the membrane on particulate methane monooxygenase activity. J. Biol. Chem.. 2018;293(27):10457-10465.
- [CrossRef] [Google Scholar]
- Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res.. 2014;42(W1):W320-W324.
- [CrossRef] [Google Scholar]
- Defining the limits of homology modeling in information-driven protein docking. Proteins. 2013;81(12):2119-2128.
- [CrossRef] [Google Scholar]
- A tale of two methane monooxygenases. Journal of biological inorganic chemistry : JBIC : a publication of the Society of Biological Inorganic Chemistry.. 2017;22(2–3):307-319.
- [CrossRef] [Google Scholar]
- Effects of zinc on particulate methane monooxygenase activity and structure. J. Biol. Chem.. 2014;289(31):21782-21794.
- [CrossRef] [Google Scholar]
- Crystal structure and characterization of particulate methane monooxygenase from Methylocystis species strain M. Biochemistry. 2011;50(47):10231-10240.
- [CrossRef] [Google Scholar]
- The Protein Data Bank and structural genomics. Nucleic Acids Res.. 2003;31(1):489-491.
- [CrossRef] [Google Scholar]
- Conversion of methane to methanol at the mononuclear and dinuclear copper sites of particulate methane monooxygenase (pMMO): a DFT and QM/MM study. J. Am. Chem. Soc.. 2006;128(30):9873-9881.
- [CrossRef] [Google Scholar]
- Mechanistic Insights into Interactions between Bacterial Class I P450 Enzymes and Redox Partners. ACS Catal.. 2018;8(11):9992-10003.
- [CrossRef] [Google Scholar]
- Structure and activity of particulate methane monooxygenase arrays in methanotrophs. Nat. Commun.. 2022;13(1):5221.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2023.102634.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




