

Translate this page into:
Development of newer generation Vascular endothelial growth factor Receptor-2 Inhibitors: Pharmacophore based design, virtual Screening, molecular Docking, molecular dynamic Simulation, and DFT analyses
⁎Corresponding authors. subhasisbanerjee864@yahoo.com (Subhasis Banerjee), supriyo9@gmail.com (Supriyo Saha)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Vascular Endothelial Growth Factor (VEGF) has a greater impact on carcinogenesis, and it is the most significant receptor in mediating the mutagenesis and permeability of endothelial cells. Here, we report the identification of potential VEGFR-2 inhibitors as new putative anti-cancer agents. In this regard, a pharmacophore model was generated, considering established potent VEGFR2 inhibitors. This model was further applied for the virtual screening of the ZINC database and the feature-based design of another eight molecules (B1–B8). Examining these molecules using sequential computational approaches including molecular docking, molecular dynamic simulation, and DFT analysis leads to the identification of compounds B3, B5, and B7 as potential inhibitors that showed better binding affinity, stability, and interaction mechanisms concerning the reference control, Sorafenib. Further, the Lipinski rule filters and ADMET analysis support the selected compounds as drug candidates subjected to experimental validation.
Keywords
Pharmacophore
Virtual Screening
ZINC database
Molecular docking
MD Simulation
DFT
Drug Likelihood

- VEGFR
-
Vascular Endothelial Growth Factor Receptor
- RCC
-
Renal Cell Carcinoma
- HCC
-
Hepatocellularcarcinoma
- PDGF
-
Platelet-derived growth factor
- FGF-2
-
Fibroblast growth factor 2
- BFGS
-
Broyden-Fletcher-Goldfarb-Shanno
- MD
-
Molecular Dynamics
- FMO
-
Frontier Molecular Orbital
- MEP
-
Molecular Electrostatic Potential
- DFT
-
Density Functional Theory
- RMSD
-
Root Mean Square Deviation
- ADMET
-
Absorption, Distribution, Metabolism, Excretion, Toxicity
- RMSF
-
Root Mean Square Fluctuation
- SASA
-
Solvent Accessible Surface Area
- PDB
-
Protein Data Bank
- Rg
-
Radius of Gyration
- HOMO
-
Highest Occupied Molecular Orbital
- LUMO
-
Lowest Unoccupied Molecular Orbital
Abbreviations
1 Introduction
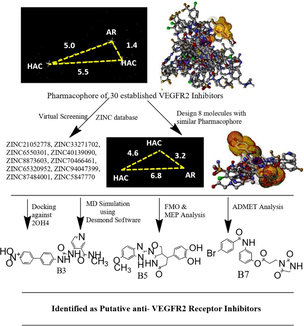
One of the key indicators of cancer and solid tumors is angiogenesis. Over the past ten years, the crucial significance of angiogenesis in cancer therapy has been proven by studies (Zhou et al., 2022). Tumors require enough nutrients, oxygen, and efficient methods of waste release to continue growing. Angiogenesis is the process through which existing endothelium-lined blood vessels within the tumor mass grow new blood vessels (Eguchi et al., 2022; Wang et al., 2020). This procedure could meet every requirement as the tumor grows. Tumor angiogenesis also contributes to the pathophysiology, development, and metastasis of cancer (Alanazi et al., 2021). The coordination of the angiogenesis process depends on the balance between angiogenic and anti-angiogenic elements (Liu et al., 2017; Sung et al., 2021). Angiogenesis happens when stimulator activity outpaces inhibitor activity. Vascular endothelial growth factor (VEGF) is the angiogenic factor with the greatest impact (Prasad et al., 2022). In the development of human cancer, VEGF is the main regulator of angiogenesis and metastasis. Angiogenesis is slowed and tumor growth is inhibited by treatments that disrupt VEGF signaling, as shown (Yakes et al., 2011; Ni et al., 2022). VEGFR1 and VEGFR2 significantly control how VEGF acts on endothelial cells (Ma et al., 2021). The two receptors mediate the mitogenesis and permeability of endothelial cells, but VEGFR2 has a more significant impact (Grüllich, 2014). Among the downstream signaling molecules that are phosphorylated as a result of VEGFR2 activation are mitogen-activated protein kinases (MAPK), serine/threonine kinases (AKT), and signal transducer and activator of transcription 3 (STAT3) (Adasme et al., 2020). Roxy-ZV-5 J demonstrated strong inhibition against CDK4 and VEGFR2 (Huang et al., 2019). Apatinib showed good anticancer activity by inducing cell cycle collapse at the G1 phase. Cabozantinib effectively inhibited the growth of hepatic cancer cells (Shang et al., 2021). Axitinib, sunitinib, sorafenib, and pazopanib effectively inhibited the VEGFR2 enzyme at nanomolar concentrations with IC50 values of 0.2 nM, 10 nM, 90 nM, and 30 nM, respectively (Sonpavde et al., 2008). Dovitinib inhibited both FGFR3 and VEGFR2, with IC50 values of 9 nM and 13 nM, respectively. Sorafenib, a VEGFR2 inhibitor, was used in the treatment of RCC and HCC (Sangande et al., 2020). The FDA recently approved regorafenib as a second-line treatment for HCC (Bruix et al., 2017; Personeni et al., 2018). Anlotinib prevented endothelial cells from migrating and forming capillary-like tubes in response to VEGF, PDGF, and FGF-2 (Yu et al., 2015). In this manuscript, we computationally developed newer generation VEGFR2 inhibitors using pharmacophore mapping, virtual screening, molecular docking, MD simulation, frontier molecular orbital analysis, molecular electrostatic potential analysis, and assessment of ADMET properties.
2 Materials and methods
2.1 Pharmacophore modeling, virtual screening and design of newer generation VEGFR2 inhibitors
Based on the structural diversity and potency, we selected 30 established VEGFR2 inhibitors from the PubChem database (Table S1), followed by pharmacophore model generation using the PharmaGist server (PharmaGist Webserver (tau.ac.il), accessed on November 25, 2023). The pharmacist server provides pharmacophores generated from a set of known bioactive molecules. The advantages of pharmacist pharmacophore are the flexibility of the aligned structures and their tolerance to any outlier to several binding modes. Ligand representation, pairwise alignment, multiple alignment, solution clustering, and output are the stages of pharmacophore generation. Assignment of rotatable bonds, hydrogen bond donor group, hydrogen bond acceptor group, and aromatic group are the representation factors of ligand. In the pairwise alignment section, one pivot and one flexible ligand are selected. Then target rigid was superimposed on the pivot. The outcomes were observed with a new pose on the pivot, followed by reassembling all the poses into a new alignment. The scoring function is the culmination of matched structures. If the type and distance between the pivot and target match, then pairwise alignment is observed. Multiple alignments were observed, with multiple pairwise alignments between pivot and target ligand molecules. In the final step, alignment with the maximum scoring value is selected as a pharmacophore. In the next step, pharmacophore output was added to the Zinc Pharmer online portal, ZINCPharmer (pitt.edu). Molecular weight less than 500, rotatable bonds less than 5, and RMSD less than 2.0 \AA were considered filters (Saha et al., 2023).
2.2 Molecular docking study
The virtually screened, designed molecules and standard sorafenib were considered for molecular docking studies. The X-ray crystallographic structure of the VEGFR-2 receptor (PDB ID: 2OH4) was obtained from the Protein Data Bank (https://www.rcsb.org) and belongs to Homo sapiens. As per the Ramachandran plot, 91.2 % of residues resided in the most favorable region (Figure S1). Methyl (5-{4-[({[2-Fluoro-5-(Trifluoromethyl)Phenyl]Amino}Carbonyl)Amino]Phenoxy}-1H- Benzimidazol-2-yl)Carbamate (GIG) was present as a complexed ligand. The resolution of the receptor was 2.05 Å. We considered Glide and AutodockVina for molecular docking studies (Dror et al., 2009).
2.2.1 Molecular docking study using Glide
2.2.1.1 Preparation of protein structure via protein preparation wizard
Schrödinger Suite 2018–4 was employed to address specific deficiencies observed in the protein structure obtained through X-ray crystallography, such as the absence of hydrogen atoms. The assignment of bond ordering was conducted, and any potential hydrogen atoms that were absent in the PBD structure were incorporated (Akash et al., 2023a). Furthermore, disulfide bonds were formed by interacting with two sulfur atoms nearby, while the remaining choices were maintained at their normal settings. In the last refinement stage, a comprehensive energy optimization was conducted utilizing the OPLS 2005 force field, with the criterion for the RMSD of heavy atoms set at 0.3 Å. The investigational compounds were imported into the Maestro molecular interface (version 11v5) of the Schrödinger suite. The authors built low-energy 3D conformers for each two-dimensional structure, ensuring that the bond lengths and angles were sufficient. The potential ionization states for each ligand structure were computed under a physiological pH of 7.2 ± 0.2 (Saha et al., 2022).
2.2.1.2 Molecular docking study parameters
The ligand located at the receptor active site was used to identify the grid box size at 32, 32, and 32, respectively, in x, y, and z coordinates (Akash et al., 2023b). Extra-precision (XP) docking and scoring methods were chosen above the conventional precision strategy. XP software aims to analyze ligand poses shown to have high scores using SP docking. To provide accurate predictions concerning the binding affinity between the newly created inhibitors and the protein crystal, the induced-fit docking (IFD) technology was utilized. In the docking phase, a subset of the docked poses is passed to the prime during refinement. The best receptor designs for every ligand are then sent back to Glide for ligand re-docking after prime side-chain minimization and prediction (Friesner et al., 2004).
2.2.2 Molecular docking study using AutoDock-Vina
2.2.2.1 Preparation of protein
The MGL Tools 1.5.6 package was used to simulate the protein. Monte-Carlo sampling with the BFGS local optimization method was applied in the AutoDock-Vina molecular docking process (Pagadala et al., 2017). The co-crystal ligand, water molecules, and heteroatoms were eliminated, followed by energy minimization using the Swiss PDB viewer (Trott & Olson, 2010). Later, partial gasteiger atomic charges were included along with the assignment of bond ordering, polar, and missing hydrogens. The H++ server assigns protonation states and missing hydrogens (Gordon et al., 2005).
2.2.2.2 Ligand preparation
Avogadro software was used to produce the 2D structures of virtually screened, designed, and standard sorafenib. The molecules were optimized using the molecular mechanics force field. All the structures were optimized with polar hydrogen and charges (Fatima et al., 2023a).
2.2.2.3 Molecular docking parameters
The optimal ligand binding site inside each protein was found using the Lamarckian genetic algorithm (Fatima et al., 2023b). Discovery Studio Visualizer 4.5 was used to visualize and analyze the docking results (Akash et al., 2023c). The grid box dimension of 2OH4 was center_x = 3.554, center_y = 35.43, and center_z = 19.878.
2.2.3 Validation of molecular docking
The active site was emptied by removing the co-crystallized-ligand GIG of the protein (PDB: 2OH4) and redocked within the active site of the protein (Al Mashud et al., 2023). The RMSD between the redocked conformation and the unprocessed crystallographic conformation was found to be < 1.5 Å. This ensures the reliability of the docking method in regenerating the experimentally observed binding mode for VEGFR2.
2.3 MD simulation
Schrodinger's Desmond program (Schrodinger Release 2019–4: Desmond) was used for MD simulation of atomic-level dynamics (Fatima et al., 2023c). Missing loops and side chains were added to the protein–ligand complex, followed by deleting water molecules (Jabir et al., 2021). The OPLS 2005 force field was applied for energy minimization (Akash et al., 2023d). The TIP3 water model was used to solvate the receptor-ligand complex in a cubic box of 10 Å. The total number of water molecules and the counterions used to neutralize the complex (Table S2). 100 ns MD simulations for each complex were performed at constant NPT (N = number of atoms, P = pressure, and T = temperature). The Martyna-Tobias-Klein barostat and Nose Hoover chain thermostat methods were used to apply system pressure and temperature, respectively (Akash et al., 2022). The bond length of 1 nm was preserved by short-range electrostatic and Van Der Waals interactions. The particle mesh Ewald (PME) summation process was used to calibrate the long-range Coulomb electrostatic interactions. The leap-frog algorithm was used to calculate the motion with a time step of 2 fs (Lokhande et al., 2019). The conformational changes in the receptor Cα backbone atoms after binding of ligand molecules were compared with the initial conformations of the crystal structure (PDB ID: 2OH4) in terms of RMSD (Akash et al., 2023e). Also, the amino acid fluctuations of the receptor after interacting with the ligand molecules were calculated using RMSF (Lokhande et al., 2022). Protein-ligand contacts, Rg, and SASA values were also calculated (Bharadwaj et al., 2022).
2.4 DFT analysis
2.4.1 Frontier molecular orbital (FMO) analysis
FMO analysis is the electrical and optical property analysis of a chemical structure using Beck’s (B) three-parameter hybrid model with Lee, Yang, and Parr’s (LYP) correlation with the B3LYP/6–31 + G(d,p) basis set. Sometimes atomic orbitals are distorted from their original shape and polarized with the effect of surrounding functional groups; in such a situation, a polarization factor is added, such as (d, p), where polarization is added in both d and p orbitals. In the case of large molecules, the nucleus loses control of certain functional groups, and those functional groups form a new orbital. To counter this factor, a diffuse formation factor was added. Structures with a lone pair of electrons, anions, or electronegative groups must require this diffusion factor (+) to calculate the orbital energy. As the chemical structures consisted of nitro, halogen, and other electronegative groups, we considered B3LYP/6–31 + G(d,p) as a basis set in the DFT calculation. The molecules have one multiplicity and zero charges. The highest molecular orbital (HOMO) can donate electrons. The leading empty innermost orbital lowest molecular orbital (LUMO), which is not fully occupied, acts as an electron acceptor. GAMESS WxMacMolPlt (version 7.7.3) was used to perform and visualize FMO analysis (Perri & Weber, 2014).
η(chemical hardness) = ; ζ (chemical softness) = ; μ (electronegativity index) = ; Ψ (electrophilicity index) = , where A and I are electron affinity and ionization potential. A = − ELUMO and I = − EHOMO.
2.4.2 Molecular electrostatic potential (MEP) investigation
The electrical charge distributions from protons, nuclei, and electrons were taken into consideration by MEP, which took interaction energy at a certain structural zone into account (Sumithra et al., 2023). High negative and positive potential attract positively and negatively charged species, respectively. The potentiality of the attacking zone decreases with the sequence of blue, green, yellow, orange, and red. The maximum negative area is displayed in red, where electrophiles can quickly attack, and the total positive area is indicated in blue, which is suitable for nucleophilic attack. The green color shows zero potential zones. MEP of B3, B5, and B7 was computed with the B3LYP-6–31 + G(d, p) level (Alexeev et al., 2012) using GAMESS software (version R2, released on September 30, 2023).
2.5 Calculation of drug likelihood
Drug likelihood and toxicity behaviors of the molecules were calculated using SwissADME (https://www.swissadme.ch/index.php, accessed on December 1, 2023) and Osiris version 2.9.1, respectively (Daina et al., 2017; Ertl and Schuffenhauer, 2009).
3 Results
3.1 Pharmacophore mapping, virtual screening and designing of newer generation VEGFR2 inhibitors
The pharmacophore mapping of the VEGFR2 inhibitors showed three main features, including aromatic and two hydrogen bond acceptors (Garcia-Gomez et al., 2013;Schneidman-Duhovny et al., 2008).
The pharmacophore model was then utilized to screen out around 21 million compounds from the ZINC database, and the top 10 compounds, including ZINC21052778, ZINC33271702, ZINC6550301, ZINC40139090, ZINC8873603, ZINC70466461, ZINC65320952, ZINC94047399, ZINC87484001, and ZINC5847770, based on the RMSD values, were chosen (Figure S2) (Koes and Camacho, 2012). Based on the pharmacophoric features of the established molecules, eight molecules (B1–B8) were designed. The pharmacophoric features of the designed molecules were kept nearly equal to those of the established molecules, such as one aromatic point and two hydrogen bond acceptors (Fig. 1) (Kaserer et al., 2015; Irwin et al., 2020).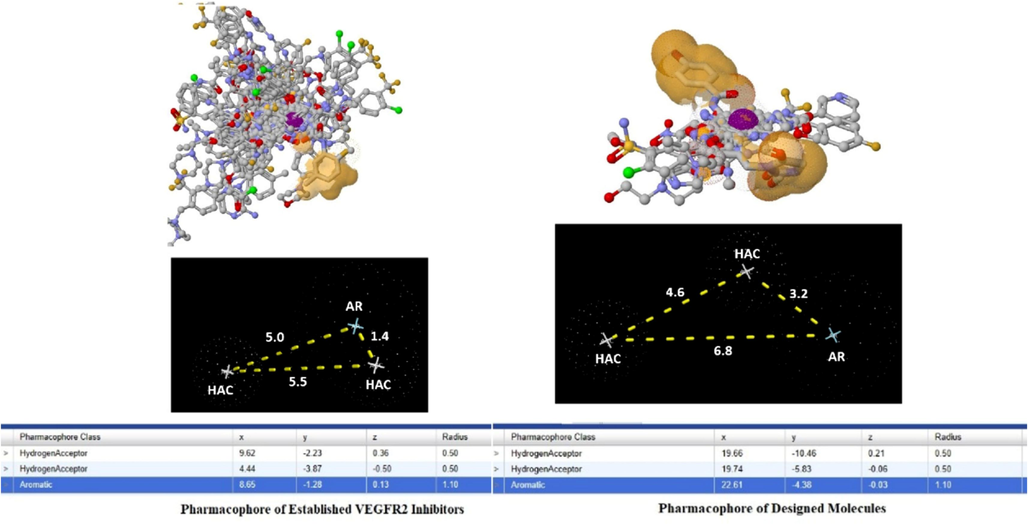
Pharmacophore of the established and designed VEGFR2 inhibitors.
3.2 Molecular docking studies data
As per the molecular docking study data using Glide software, among the virtually screened and designed molecules, B3, B5, and B7 showed good docking scores of −10.274 kcal/mol, −10.539 kcal/mol, and −9.515 kcal/mol, respectively. The docking score of standard sorafenib was −10.791 kcal/mol (Akash et al., 2023f). B3 showed interactions with GLU 883, CYS 917, and ASP 1044 using hydrogen bond interactions. B5 interacted with receptors via GLU 883 and ASP 1044 using hydrogen bonds, and PHE 1045 using pi-pi stacking interactions. B7 interacted with GLU 883, CYS 917, ASP 1044 by hydrogen bond interaction, and LYS 866 by pi-pi stacking interaction. The standard sorafenib interacted with GLU 883, CYS 917, and ASP 1044 using hydrogen bond interactions (Fig. 2).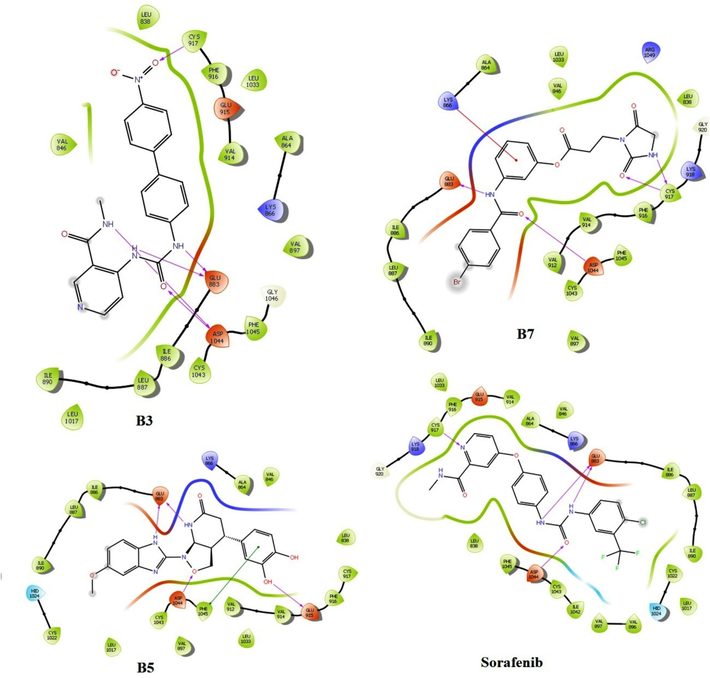
Molecular docking interaction image of B3, B5, B7 and Sorafenib with VEGFR2 (Glide software).
In the case of molecular docking interactions, AutoDockVina B3, B5, and B7 showed good docking scores of −10.6 kcal/mol, −10.2 kcal/mol, and −9.3 kcal/mol, respectively. Standard sorafenib showed a docking score of −9.4 kcal/mol (Table S3) (Giordano et al., 2022). Compound B3 showed proper interactions with LEU 887, VAL 897, VAL 846, LEU 838, ALA 864, and CYS 1043 via pi-pi interactions and a hydrogen bond with ASP 1044. B5 formed hydrogen bonds with CYS 917, ASN 921, and ARG 1049; pi-pi interactions with ALA 864; PHE 916; LEU 1033; and LEU 838. Compound B7 is involved in hydrogen bonding interactions with ASN 921, ARG 1030, ARG 1049, ASP 1062, and pi-pi interactions with LEU 838, VAL 846, ALA 864, Lys 866, CYS 1043, and GLU 883 (Alamri et al., 2023). The standard sorafenib interacted with receptors via hydrogen bonds with ASN 921, ARG 1030, and ARG 1049 and pi-pi interactions with ALA 864, PHE 916, LEU 1033, and LEU 838 (Fig. 3) (Akash et al., 2023g). The 3D interaction image of B3, B5, B7, and sorafenib with VEGFR2 was added to Figure S3.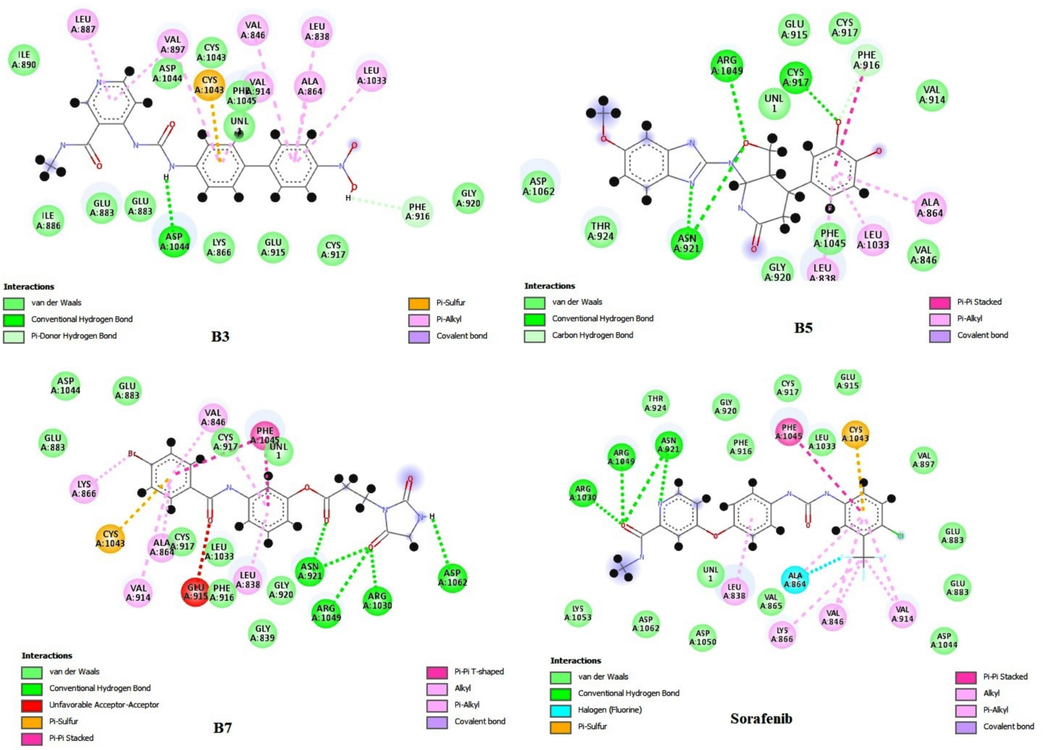
Molecular docking interaction image of B3, B5, B7 and Sorafenib with VEGFR2 (AutoDock Vina).
3.3 MD simulation data
In the case of B3, the ligand RMSD was within 2.0 °C. The protein structure of RMSD lies within (2.5–3.3) \AA (Fig. 4A). Both protein and ligand achieved a static value near 90 ns. Percent secondary structure values reflected the presence of an alpha helix and beta sheet in the structure and confirmed the structural integrity (Tripathi et al., 2023). The maximum beta-strand and alpha-helices were stable during the simulation because there was no sign of overlapping. Some residues exceeded 3.0 °C near the 100, 125, and 200 residue numbers in the protein RMSF map. The green bars near residue numbers 25, 30, 50, (70–80), 100, and (150–180) confirmed that at these points, protein interacted with ligand (Fig. 4B). The ligand RMSF value was within 1.5 °C, which confirmed the stability (Fig. 4C). The terminal nitro group fluctuated the most within the simulation (Kawsar et al., 2023). In protein–ligand interaction during simulation, four types of interactions, such as hydrogen bonding, hydrophobic interaction, ionic interaction, and water bridges, were observed. LEU 838, VAL 846, ALA 864, ILE 886, LEU 887, ILE890, VAL 897, VAL 914, PHE 916, LEU 1033, CYS 1043, and PHE 1045 interacted with protein structure by hydrophobic interactions; ALA 879 and SER 882 interacted with protein by water bridges; GLU 883, CYS 917, and ASP 1044 interacted with protein by hydrogen bonds and water bridges. GLU 883 and ASP 1044 showed maximum contacts (Fig. 4D). The urea residue and NH group of N-methyl pyridinamide interacted with GLU 883 and ASP 1044 by most stable interactions (93–99 %) using charged negative interaction during simulation (Fig. 4E). The terminal nitro group interacted with PHE 916 and CYS 917 through a less stable interaction using hydrophobic interaction during the simulation process (34–52 %), which confirmed that the nitro group did not positively contribute to the protein–ligand interaction. The radius of the gyration value attained a static value near 5.5 °C, which confirmed the protein stability during simulation (Fig. 4F).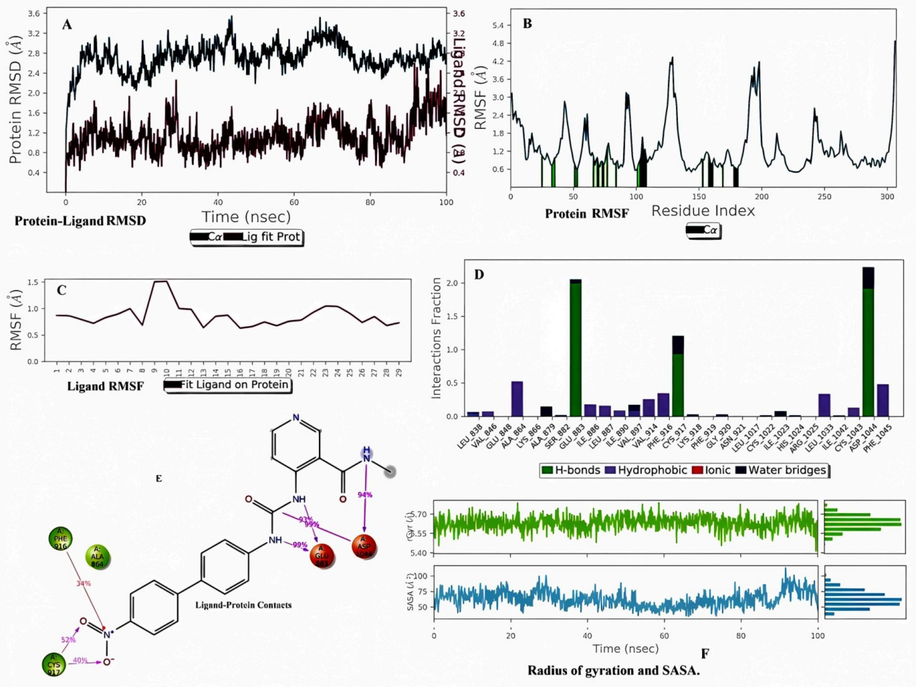
Molecular dynamics simulation analysis of B3 with VEGFR2. (A) Protein-Ligand RMSD. (B) Protein RMSF (C) Ligand RMSF. (D) Histogram represents the Protein-Ligand interactions. (E) Ligand Contacts. (F) Rg and SASA values.
In the case of B5, the ligand RMSD lies below 2.0 °C. The protein structure attained a stable form due to its RMSD lying within (2.0–3.8) °C. Both protein and ligand achieved a static value near 70–100 ns and intermingled near 10 ns and 30 ns. These RMSD values confirmed that the protein–ligand complex was within the active site (Fig. 5A). Percent secondary structure assessment of B5 showed that the maximum beta-strand and alpha-helices were stable during simulation because there was no sign of overlapping (Lokhande et al., 2021). In the case of protein RMSF, some residues exceeded 3.0 °C, near 125 and 180 residue numbers. The green bars near residue numbers 10, 20, 50, (70–80), 100, and (150–180) confirmed that at these connections, protein interacted with ligand (Fig. 5B). In ligand RMSF, the total molecule was stable during simulation because the RMSF value was within 1.5 °C (Fig. 5C). The terminal methoxy group fluctuated within the simulation. LEU 838, VAL 846, ALA 864, ILE 886, LEU 887, ILE 890, VAL 914, ILE 1033, CYS 1043, and PHE 1045 interacted with protein structure by hydrophobic interactions; LYS 866, GLU 883, GLU 915, CYS 917, and ASP 1044 interacted with protein by hydrogen bonds. GLU 883 and GLU 915 showed maximum stable contacts (Fig. 5D and 5E). The 3-hydroxy group of terminal catechol interacted with GLU 915 by stable hydrogen bond interaction (90 %), which confirmed that this interaction did not change during the protein–ligand interaction. The radius of gyration value attained a static value near 5.0 °C, which confirmed the protein stability during simulation (Fig. 5F) (Barcellos et al., 2019).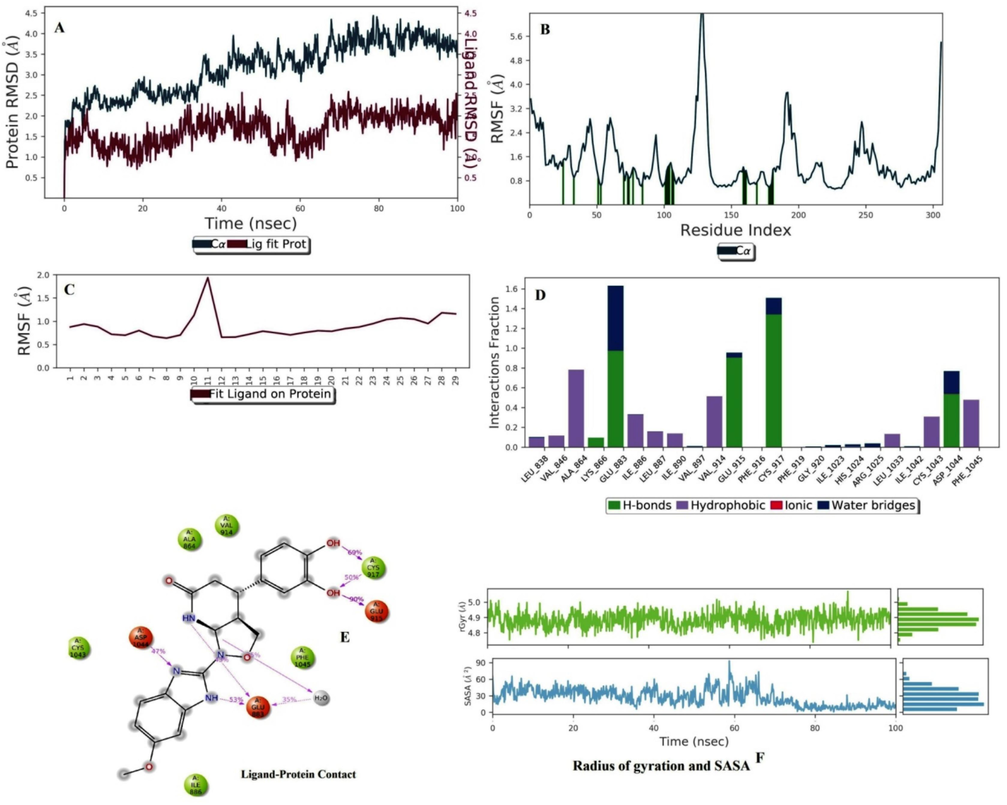
Molecular dynamics simulation analysis of B5 with VEGFR2. (A) Protein-Ligand RMSD. (B) Protein RMSF (C) Ligand RMSF. (D) Histogram represents the Protein-Ligand interactions. (E) Ligand Contacts. (F) Rg and SASA values.
In the case of B7, the ligand RMSD lies below 2.0 °C. The protein structure attained a stable form due to its RMSD lying within (2.0–3.5) \AA. Ligand attained a static value of 20 ns and intermingled near 10 ns, 20 ns, 30 ns, and 90 ns. The percent secondary structure assessment of B7 showed that the maximum beta strand and alpha helices were stable during simulation because there was no sign of overlapping (Fig. 6A). In the case of protein RMSF, some residues exceeded 3.0 °C, nearing 125 and 180 residue numbers (More-Adate et al., 2022). The green bars near residue numbers 10, 20, 50, (70–80), 100, and (150–180) confirmed that at these connections, protein interacted with ligand (Fig. 6B). In ligand RMSF, the total molecule was stable during simulation because the RMSF value was within 2.0 °C (Fig. 6C). The terminal ethylidene imidazole-2,4-dione group was most fluctuating within the simulation. LEU 838, VAL 846, ALA 864, LYS 866, ILE 886, LEU 887, ILE 890, VAL 896, VAL 897, VAL 914, HIS 1024, ILE 1033, and PHE 1045 interacted with protein structure by hydrophobic interactions; GLU 883, GLU 915, CYS 917, CYS 1043, and ASP 1044 interacted with protein by hydrogen bond interactions. GLU 883 and ASP 1044 showed a maximum period of stable contacts (Fig. 6D). The ketone of the –NHC = O group makes the maximum stable interaction with ASP 1044 (68 %), which confirms that this interaction has not changed frequently during the interaction (Fig. 6E). The radius of gyration values of B3, B5, and B7 were within the (5.5–6.6) range from (0–100) ns, below 5.0 °C from (0–100) ns, and [(6.0–6.1) °C from (0–50) ns and near 5.4 °C from (51–100) ns], respectively (Fig. 6F). The SASA values of B3, B5, and B7 fluctuated within (75–90) \AA2, below 30 \AA2, and (50–75) \AA2, respectively (Fig. 4F, 5F, 6F) (Méndez-Álvarez et al., 2023).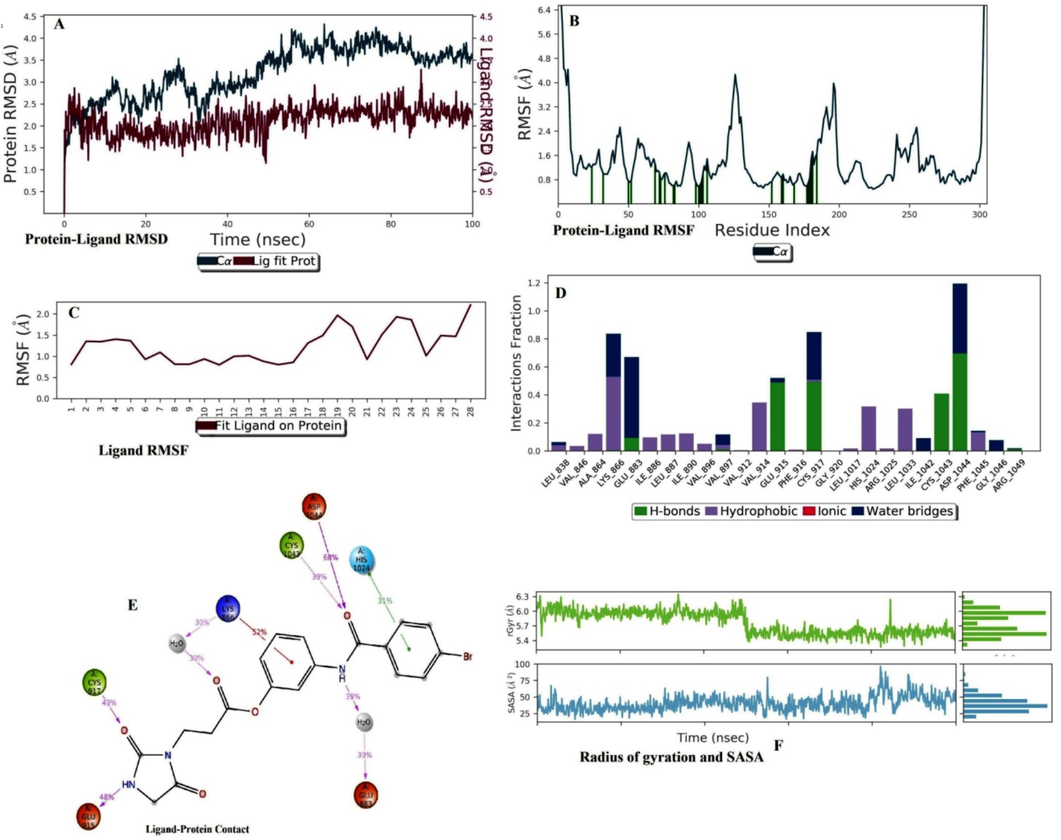
Molecular dynamics simulation analysis of B7 with VEGFR2. (A) Protein-Ligand RMSD. (B) Protein RMSF (C) Ligand RMSF. (D) Histogram represents the Protein-Ligand interactions. (E) Ligand Contacts. (F) Rg and SASA values.
3.4 DFT analysis
3.4.1 FMO analysis data
In the investigation of FMO analysis, B3, B5, and B7 were chosen. B3, B5, B7, and sorafenib have corresponding HOMO orbital energy (eV) values of −5.95, −5.46, −6.34, and −6.39. For B3, B5, B7, and sorafenib, the corresponding LUMO orbital energy (eV) values were −1.57, −0.73, −1.36, and −1.68. HOMO-LUMO orbitals energy gaps reveal the molecule's chemical strength and reactivity. B3, B5, B7, and sorafenib showed 4.38 eV, 4.73 eV, 4.98 eV, and 4.71 eV orbital energy distances, respectively (Table 1 and Figure S4) (Devereux et al., 2020). HOMO-LUMO orbitals of B3 were focused on the (4′-carbamamido[1,1′-biphenyl]-4-yl)(hydroxy)oxoammonium group. HOMO-LUMO orbitals of B5 were focused on the 6-methoxy-2-(1,2-oxazolidin-2-yl)-1H-benzimidazole and 4-methylbenzene-1,2-diol groups, respectively. HOMO-LUMO orbitals of B7 are focused on the 3-(4-bromobenzamido)phenylformate group.
Sr. No.
Molecules
EHOMO
(eV)
ELUMO
(eV)
ΔE gap (eV)
I
A
η
ζ
μ
Ψ
1.
B3
−5.95
−1.57
4.38
5.95
1.57
2.19
0.22
3.57
2.90
2.
B5
−5.46
−0.73
4.73
5.46
0.73
2.37
0.21
3.09
2.01
3.
B7
−6.34
−1.36
4.98
6.34
1.36
2.49
0.20
3.85
2.97
4.
Sorafenib
−6.39
−1.68
4.71
6.39
1.68
2.35
0.21
4.03
3.44
3.4.2 Molecular electrostatic potential (MEP) data
In the case of B3, the nitrogen of the terminal acetamide's O = C-NH group, the urea group, the biphenyl group, and the HO-N = O group are all vulnerable to nucleophilic and electrophilic assault, respectively. Regarding B5, the methoxy group and imidazole group are accountable for the nucleophilic assault and the electrophilic attack, respectively. For B7, the overall molecular composition strikes a balance between the nucleophilic and electrophilic attack sites. The 4-{4-[(4-chlorophenyl)carbamamido]phenoxy}-N-methylpyridine-2-carboxamide group is responsible for both nucleophilic and electrophilic assault in the instance of sorafenib (Fig. 7) (Al-Sanea et al., 2021; Xiao-Hong et al., 2015). A structure's chemical reactivity is increased when urea, methanimidamide, nitro, methoxy, and imidazolone groups are added to the parent structure.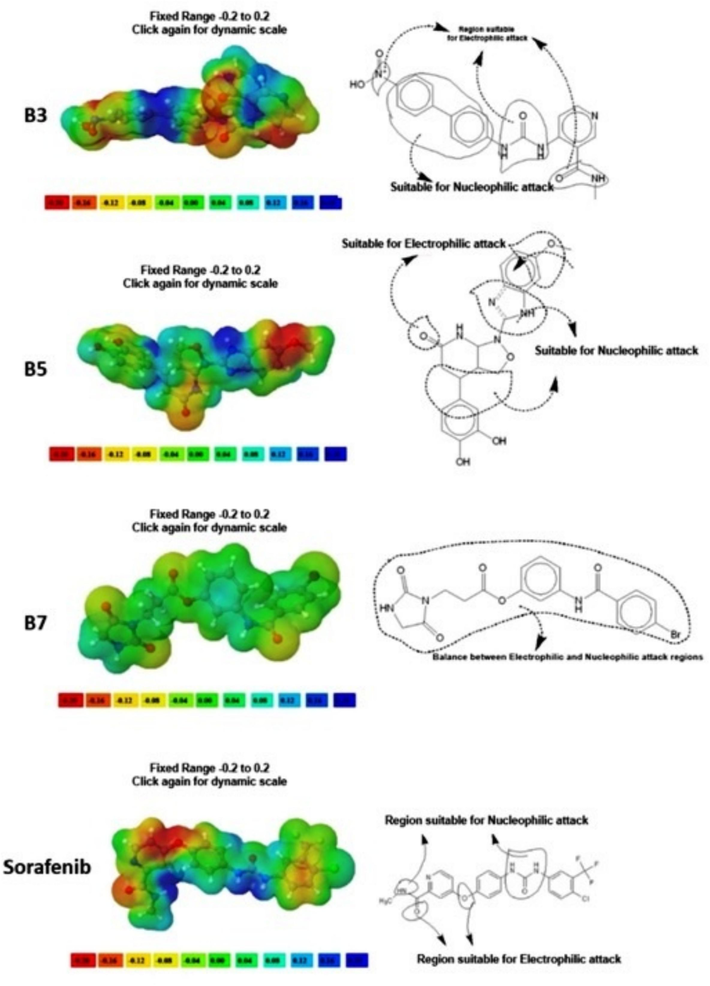
MEP analysis of B3, B5, B7, and Sorafenib.
3.5 Drug likelihood data
The drug likelihood calculations of the designed molecules showed that all the molecules, including sorafenib, passed the Lipinski rule with an oral bioavailability score of 0.55. The bioavailability score (Abbott Bioavailability Score) was designated based on Caco-2 cell permeability and polar surface area. A bioavailability score of 0.11 for anions means a polar surface area greater than 150 Å2, a bioavailability score of 0.56 observed with a polar surface area between 75 and 150 Å2, and a bioavailability score of 0.85 observed with a polar surface area less than 75 Å2. A bioavailability score of 0.55 was observed with molecules that passed the Lipinski rule of five, and if any molecules violated the Lipinski rule of five, the bioavailability score was 0.17 (Martin, 2005). Except for B2 and sorafenib, the rest of the molecules showed good absorption through the gastrointestinal tract. Swiss ADME data and the boiled egg diagram confirmed that only ZINC87484001 and B1 showed probable penetration of the blood–brain barrier. Most of the molecules were moderate to highly soluble in nature. The synthetic accessibility of ZINC21052778, ZINC33271702, ZINC6550301, ZINC40139090, ZINC8873603, ZINC70466461, ZINC65320952, ZINC94047399, ZINC87484001, ZINC5847770, B1, B2, B3, B4, B5, B6, B7, B8, and Sorafenib were 3.59, 4.22, 3.06, 2.86, 3.11, 2.88, 3.78, 2.97, 2.60, 2.47, 2.86, 3.94, 2.93, 3.47, 4.96, 4.41, 2.59, 3.30, and 2.87 (Table S4). Osiris toxicity prediction data of all the molecules showed that except B3, ZINC8873603, and ZINC70466461, the rest of the molecules were non-mutagenic in nature; except ZINC6550301, ZINC70466461, ZINC87484001, ZINC5847770, and B3 were non-tumorigenic in nature. Sorafenib showed non-mutagenic, non-tumorigenic, non-irritant, and non-reproductive toxicant properties (Table S5) (Jeyavijayan, 2015).
4 Discussion
Pharmacophore mapping and synthetic feasibility were among the top critical parameters considered during the design. The synthetic accessibility score of all the designed compounds was thoroughly monitored before fixing each one of them as an eligible candidate for an in silico study. The synthetic accessibility scores of B5 and B6 showed the highest score due to 1-(1H-benzo[d]imidazol-2-yl)hexahydroisoxazolo[3,4-b] pyridin-6(1H)-one and 7-benzoylimidazo[1,5-a]pyridin-4-ium-8a-ide nucleus (Fasiuddin et al., 2022). The higher synthetic accessibility score is based on structural complexity and structural fragments. Though computational chemistry helps to design the newer generation of VEGFR2 inhibitors with statistical evidence from a huge data library, it requires biological experiments to establish the design molecules (Lagorce et al., 2017; Elkaeed et al., 2022). Among the designed VEGFR2 inhibitors, B3, B5, and B7 showed good docking interactions. The distance between docking scores performed by Glide and AutoDock Vina for B3, B5, and B7 was 0.326, 0.339, and 0.215, respectively. The complexed ligand of the 2OH4 receptor connected GLU 883, CYS 917, and ASP 1044 by hydrogen bonding interactions; PHE 916 by pi-pi interaction; and LEU 838, VAL 846, ALA 864, LYS 866, LEU 887, and VAL 914 by pi-alkyl interactions (Figure S5). The similar interacting residues ASP 1044, GLU 883, and CYS 917 confirmed that all ligand molecules effectively docked within the receptor active site (Saha et al., 2022; Kumar et al., 2023). All protein–ligand interactions reached a static value in the RMSD, which confirmed that all ligands were well fitted into the receptor-active region. The structural integrity was not significantly hampered by RMS changes (Vishvakarma et al., 2022). There is a clear correlation between structural stability and the radius of gyration value. Less stability is indicated by high values, and vice versa. Greater stability is indicated by gyration radius values in the lower range. SASA values had a favorable impact on the binding energy, as demonstrated by the nearly consistent SASA values throughout the analysis. Here, the complexes with lower SASA values demonstrated greater stability. Overall outcomes stated that ligand molecules make a stable complex with receptors throughout the simulation process. Rg analysis confirmed that all atoms were well distributed from the center of mass, which confirmed the structural integrity of the protein–ligand complex. Finally, we confirmed that all the protein–ligand complexes were thermodynamically stable during simulation (Lokhande et al., 2023). The FMO study indicated that the molecules were stable and reactive in the order B3 > B5 > B7 (Srivastava, 2021). Because of its narrower energy gap, B3 was the softest molecule. B7 and B3 showed the highest levels of electronegativity and electrophilicity, respectively. Thus, among the designed molecules, B7 and B3 were observed as the most reactive molecules (Pradiba et al., 2018). MEP data displayed the electrophilic and nucleophilic attack sites of the molecules. One crucial factor in the systemic absorption of a drug molecule is its water solubility (Harder et al., 2016). Except B2, B5, ZINC6550301, ZINC40139090, ZINC8873603, ZINC65320952, ZINC94047399, ZINC87484001, and sorafenib, the rest of the molecules showed good aqueous solubility due to their Log S value ranges between −4 and 0.5 log mol/L. When a drug molecule has the right distribution coefficient, it can pass across biological membranes. The log P value of (0–3) log mol/L has the best value. Except for B1, B5, ZINC6550301, ZINC40139090, ZINC65320952, ZINC94047399, ZINC87484001, and sorafenib, the rest exhibited favorable distribution behavior. Only ZINC87484001 and B1 showed a potential propensity towards blood–brain barrier penetration. All the molecules showed good oral bioavailability with zero violation of the Lipinski rule and non-toxic properties (Kushwaha et al., 2021).
5 Conclusion
After performing all the major computational studies, B3, B5, and B7 showed the maximum probability of becoming next-generation VEGFR2 inhibitors. The next big challenge is to establish these molecules by in vitro-in vivo experiments as potential VEGFR2 inhibitors, which opens a new endeavor for targeting angiogenesis and cancer proliferation.
Funding
This study is supported via funding from Prince Sattam bin Abdulaziz University project number (PSAU/2023/R/1444).
CRediT authorship contribution statement
Mubarak A. Alamri: Funding acquisition, Validation. Mohammed Merae Alshahrani: Formal analysis, Visualization. Abdullah S. Alawam: Resources, Software, Validation. Souparno Paria: Data curation, Formal analysis, Methodology. Kalyan Kumar Sen: Conceptualization, Project administration. Subhasis Banerjee: Project administration, Validation, Writing – original draft. Supriyo Saha: .
Acknowledgments
The authors extend their appreciation to Prince Sattam bin Abdulaziz University for providing the necessary technical and computational tools to conduct this research. This study is supported via funding from Prince Sattam bin Abdulaziz University project number (PSAU/2023/R/1444).
References
- Structure-based drug repositioning explains ibrutinib as VEGFR2 inhibitor. PLoS One. 2020;15(5):e0233089.
- [Google Scholar]
- Development of new bioactive molecules to treat breast and lung cancer with natural myricetin and its derivatives: A computational and SAR approach. Front. Cell. Infect. Microbiol.. 2022;12:952297
- [CrossRef] [Google Scholar]
- Antimalarial drug discovery against malaria parasites through haplopine modification: An advanced computational approach. J. Cell. Mol. Med.. 2023;27(20):3168-3188.
- [CrossRef] [Google Scholar]
- Novel computational and drug design strategies for inhibition of human papillomavirus-associated cervical cancer and DNA polymerase theta receptor by Apigenin derivatives. Sci. Rep.. 2023;13(1):16565.
- [CrossRef] [Google Scholar]
- Mechanistic inhibition of gastric cancer-associated bacteria Helicobacter pylori by selected phytocompounds: A new cutting-edge computational approach. Heliyon.. 2023;9(10):e20670.
- [Google Scholar]
- Target specific inhibition of West Nile virus envelope glycoprotein and methyltransferase using phytocompounds: an in silico strategy leveraging molecular docking and dynamics simulation. Front. Microbiol.. 2023;14:1189786.
- [CrossRef] [Google Scholar]
- Akash, S., Mahmood, S., Ahamed, R., Bayıl, I., Dev Bairagi, R., Islam, M.R., Hosen, M.E., de Lima Menezes, G., S Almaary, K., Nafidi, H.A., Bourhia, M., Ouahmane, L., 2023. Novel computational and drug design strategies for the inhibition of human T-cell leukemia virus 1-associated lymphoma by Astilbin derivatives. J. Biomol. Struct. Dyn. 1-16. 10.1080/07391102.2023.2294376. Epub ahead of print.
- Anti-parasitic drug discovery against Babesia microti by natural compounds: an extensive computational drug design approach. Front. Cell. Infect. Microbiol.. 2023;13:1222913.
- [CrossRef] [Google Scholar]
- Design and development of new inhibitors against breast cancer, Monkeypox and Marburg virus by modification of natural Fisetin via in silico and SAR studies. Acta. Biochim. Pol.. 2023;70(3):599-600.
- [Google Scholar]
- Mechanistic inhibition of Monkeypox and Marburg virus infection by O-rhamnosides and Kaempferol-o-rhamnosides derivatives: a new-fangled computational approach. Front. Cell. Infect. Microbiol.. 2023;13:1188763.
- [CrossRef] [Google Scholar]
- Establishing the Role of Iridoids as Potential Kirsten Rat Sarcoma Viral Oncogene Homolog G12C Inhibitors Using Molecular Docking; Molecular Docking Simulation; Molecular Mechanics Poisson-Boltzmann Surface Area; Frontier Molecular Orbital Theory; Molecular Electrostatic Potential; and Absorption, Distribution, Metabolism, Excretion, and Toxicity Analysis. Molecules. 2023;28(13):5050.
- [CrossRef] [Google Scholar]
- RSC. Adv.. 2021;11(48):30315-30328.
- [CrossRef]
- GAMESS as a free quantum-mechanical platform for drug research. Curr. Top. Med. Chem.. 2012;12(18):2013-2033.
- [CrossRef] [Google Scholar]
- Identification of Novel Potential VEGFR-2 Inhibitors Using a Combination of Computational Methods for Drug Discovery. Life.. 2021;11:1070.
- [CrossRef] [Google Scholar]
- Pharmacophore and structure-based drug design, molecular dynamics and admet/tox studies to design novel potential pad4 inhibitors. J. Biomol. Struct. Dyn.. 2019;37(4):966-981.
- [CrossRef] [Google Scholar]
- Potent Bioactive Compounds From Seaweed Waste to Combat Cancer Through Bioinformatics Investigation. Front. Nutr.. 2022;9:889276
- [CrossRef] [Google Scholar]
- Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
- [CrossRef] [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [CrossRef] [Google Scholar]
- Extending the applicability of the ANI deep learning molecular potential to sulfur and halogens.J. Chem. Theory. Comput.. 2020;16:4192-4202.
- [CrossRef] [Google Scholar]
- Novel approach for efficient pharmacophore-based virtual screening: method and applications. J. Chem. Inf. Model.. 2009;49(10):2333-2343.
- [CrossRef] [Google Scholar]
- VEGF-Independent Angiogenic Factors: Beyond VEGF/VEGFR2 Signaling. J. Vasc. Res.. 2022;59(2):78-89.
- [CrossRef] [Google Scholar]
- Discovery of New VEGFR-2 Inhibitors: Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies. Molecules. 2022;27:6203.
- [CrossRef] [Google Scholar]
- Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform.. 2009;1(1):8.
- [CrossRef] [Google Scholar]
- Synthesis, spectral property, IEF-PCM solvation, anti-microbial evaluation and molecular docking studies of 6 amino-2-(4 nitrophenyl)-1H-benzimidazole. J. Mol. Liquid.. 2022;352:118756
- [CrossRef] [Google Scholar]
- Synthesis, Spectroscopic, Crystal Structure, DFT, Hirshfeld Surface and Molecular Docking Analysis of Hexahydroquinoline Derivative (HQ) Polycycl. Aromat. Compd.. 2023;43(5):4242-4270.
- [CrossRef] [Google Scholar]
- Synthesis, Characterization, Crystal Structure, Hirshfeld Surface, Electronic Excitation, Molecular Docking, and DFT Studies on 2-Amino Thiophene Derivative. Polycycl. Aromat. Compd.. 2023;43(2):1644-1675.
- [CrossRef] [Google Scholar]
- Experimental Spectroscopic, Structural (Monomer and Dimer), Molecular Docking, Molecular Dynamics Simulation and Hirshfeld Surface Analysis of 2-Amino-6-Methylpyridine. Polycycl. Aromat. Compd.. 2023;43(5):3910-3940.
- [CrossRef] [Google Scholar]
- Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem.. 2004;47(7):1739-1749.
- [CrossRef] [Google Scholar]
- RAF265, a dual BRAF and VEGFR2 inhibitor, prevents osteoclast formation and resorption. Therapeutic Implications. Invest. New. Drugs.. 2013;31(1):200-205.
- [CrossRef] [Google Scholar]
- Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals.. 2022;15:646.
- [CrossRef] [Google Scholar]
- H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic. Acids. Res.. 2005;33(Web Server issue), W368–71
- [CrossRef] [Google Scholar]
- Cabozantinib: a MET, RET, and VEGFR2 tyrosine kinase inhibitor. Recent. Results. Cancer. Res.. 2014;201:207-214.
- [CrossRef] [Google Scholar]
- OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory. Comput.. 2016;12(1):281-296.
- [CrossRef] [Google Scholar]
- Novel dual inhibitors targeting CDK4 and VEGFR2 synergistically suppressed cancer progression and angiogenesis. Eur J Med Chem.. 2019;181:111541
- [CrossRef] [Google Scholar]
- ZINC20-A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model.. 2020;60(12):6065-6073.
- [CrossRef] [Google Scholar]
- Concatenation of molecular docking and molecular simulation of BACE-1, γ-secretase targeted ligands: in pursuit of Alzheimer's treatment. Ann. Med.. 2021;53(1):2332-2344.
- [CrossRef] [Google Scholar]
- Spectroscopic (FTIR, FT-Raman), molecular electrostatic potential, NBO and HOMO-LUMO analysis of P-bromobenzene sulfonyl chloride based on DFT calculations. Spectrochim. Acta. a. Mol. Biomol. Spectrosc.. 2015;136 Pt B:890-899.
- [CrossRef] [Google Scholar]
- Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules. 2015;20(12):22799-22832.
- [CrossRef] [Google Scholar]
- Potential antifungal activity of novel carbohydrate derivatives validated by POM, molecular docking and molecular dynamic simulations analyses. Mol. Simul.. 2023;49:60-75.
- [CrossRef] [Google Scholar]
- pharmacophore search of the ZINC database. Nucleic. Acid. Res.. 2012.ZINCPharmer:;40(Web Server issue):W409-W414.
- [CrossRef] [Google Scholar]
- Immunoinformatics-based multi-epitope containing fused polypeptide vaccine design against visceral leishmaniasis with high immunogenicity and TLR binding. Int. J. Biol. Macromol.. 2023;253(Pt 8):127567
- [CrossRef] [Google Scholar]
- Identification of Natural Inhibitors Against SARS-CoV-2 Drugable Targets Using Molecular Docking, Molecular Dynamics Simulation, and MM-PBSA Approach. Front. Cell. Infect. Microbiol.. 2021;11:730288
- [CrossRef] [Google Scholar]
- Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep.. 2017;7:46277.
- [CrossRef] [Google Scholar]
- Apatinib promotes autophagy and apoptosis through VEGFR2/STAT3/BCL-2 signaling in osteosarcoma. Cell. Death. Dis.. 2017;8(8):e3015.
- [Google Scholar]
- Molecular interaction studies of Deguelin and its derivatives with Cyclin D1 and Cyclin E in cancer cell signaling pathway: The computational approach. Sci. Rep.. 2019;9(1):1778.
- [CrossRef] [Google Scholar]
- Molecular docking and simulation studies on SARS-CoV-2 Mpro reveals Mitoxantrone, Leucovorin, Birinapant, and Dynasore as potent drugs against COVID-19. J. Biomol. Struct. Dyn.. 2021;39(18):7294-7305.
- [CrossRef] [Google Scholar]
- Biflavonoids from Rhus succedanea as probable natural inhibitors against SARS-CoV-2: a molecular docking and molecular dynamics approach. J. Biomol. Struct. Dyn.. 2022;40(10):4376-4388.
- [CrossRef] [Google Scholar]
- In silico discovery of potent inhibitors against monkeypox's major structural proteins. J. Biomol. Struct. Dyn.. 2023;41(23):14259-14274.
- [CrossRef] [Google Scholar]
- Combining PD-1 Inhibitor with VEGF/VEGFR2 Inhibitor in Chemotherapy: Report of a Patient with End-Stage Cholangiocarcinoma and Review of Literature. Recent. Pat. Anticancer. Drug. Discov.. 2021;16(1):101-107.
- [CrossRef] [Google Scholar]
- A Bioavailability Score. J. Med. Chem.. 2005;48:3164-3170.
- [CrossRef]
- Ligand-Based Virtual Screening, Molecular Docking, and Molecular Dynamic Simulations of New β-Estrogen Receptor Activators with Potential for Pharmacological Obesity Treatment. Molecules. 2023;28:4389.
- [CrossRef] [Google Scholar]
- GC-MS profiling of Bauhinia variegata major phytoconstituents with computational identification of potential lead inhibitors of SARS-CoV-2 Mpro. Comput. Biol. Med.. 2022;147:105679
- [CrossRef] [Google Scholar]
- CaMKII inhibitor KN-93 impaired angiogenesis and aggravated cardiac remodelling and heart failure via inhibiting NOX2/mtROS/p-VEGFR2 and STAT3 pathways. J. Cell. Mol. Med.. 2022;26(2):312-325.
- [CrossRef] [Google Scholar]
- Software for molecular docking: a review. Biophys Rev.. 2017;9(2):91-102.
- [CrossRef] [Google Scholar]
- Web-Based Job Submission Interface for the GAMESS Computational Chemistry Program. J. Chem. Educ.. 2014;91:2206-2208.
- [CrossRef] [Google Scholar]
- Regorafenib in hepatocellular carcinoma: latest evidence and clinical implications. Drugs. Context.. 2018;7:212533
- [CrossRef] [Google Scholar]
- Structural insights into the binding mode of flavonols with the active site of matrix metalloproteinase-9 through molecular docking and molecular dynamic simulations studies. J. Biomol. Struct. Dyn.. 2018;36(14):3718-3739.
- [CrossRef] [Google Scholar]
- VEGFa/VEGFR2 autocrine and paracrine signaling promotes cervical carcinogenesis via β-catenin and snail. Int. J. Biochem. Cell. Biol.. 2022;142:106122
- [CrossRef] [Google Scholar]
- Combination Therapy of Ledipasvir and Itraconazole in the Treatment of COVID-19 Patients Coinfected with Black Fungus: An In Silico Statement. BioMed. Res. Int.. 2022;2022:5904261.
- [CrossRef] [Google Scholar]
- In Silico Assessment of the Role of Iridoid in the Treatment of Zika and Influenza Virus Infection. Philippine. J. Sci.. 2023;152(5):1953-1988.
- [CrossRef] [Google Scholar]
- Ligand-Based Pharmacophore Modeling, Molecular Docking, and Molecular Dynamic Studies of Dual Tyrosine Kinase Inhibitor of EGFR and VEGFR2. Int. J. Mol. Sci.. 2020;21(20):7779.
- [CrossRef] [Google Scholar]
- PharmaGist: a webserver for ligand-based pharmacophore detection. Nucleic. Acid. Res.. 2008;36(Web Server issue), W223–8
- [CrossRef] [Google Scholar]
- Cabozantinib-based combination therapy for the treatment of hepatocellular carcinoma. Gut. 2021;70(9):1746-1757.
- [CrossRef] [Google Scholar]
- Axitinib for renal cell carcinoma. Expert. Opin. Investig. Drugs.. 2008;17(9):1405. PMID: 18447599
- [Google Scholar]
- Theoretical Studies on the Molecular Properties, Toxicity, and Biological Efficacy of 21 New Chemical Entities. ACS Omega. 2021;6(38):24891-24901.
- [CrossRef] [Google Scholar]
- Electron acceptor, excitation energies, oscillatory strength, spectroscopic and solvent effects on 5-amino-4,6-dichloro-2-(propylthio) pyrimidine - anticancer agent. Chemical Physics Impact.. 2023;6:100145
- [CrossRef] [Google Scholar]
- Development of Novel VEGFR2 Inhibitors Originating from Natural Product Analogues with Antiangiogenic Impact. J. Med. Chem.. 2021;64(21):15858-15867.
- [CrossRef] [Google Scholar]
- Identification of synthetically tractable MERS-CoV main protease inhibitors using structure-based virtual screening and molecular dynamics potential of mean force (PMF) calculations. J. Biomol. Struct. Dyn. 2023:1-11. Epub ahead of print
- [CrossRef] [Google Scholar]
- AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31(2):455-461.
- [CrossRef] [Google Scholar]
- Hunting the main protease of SARS-CoV-2 by plitidepsin: Molecular docking and temperature-dependent molecular dynamics simulations. Amino Acids. 2022;54:205-213.
- [CrossRef] [Google Scholar]
- Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Frontiers. Cell. Developmental. Biol.. 2020;8:599281
- [CrossRef] [Google Scholar]
- Theoretical investigation on the non-linear optical properties, vibrational spectroscopy and frontier molecular orbital of (E)-2-cyano-3-(3-hydroxyphenyl)acrylamide molecule. Spectrochim. Acta. a. Mol. Biomol. Spectrosc.. 2015;137:321-327.
- [CrossRef] [Google Scholar]
- Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer. Ther.. 2011;10(12):2298-2308.
- [CrossRef] [Google Scholar]
- Rhamnazin, a novel inhibitor of VEGFR2 signaling with potent antiangiogenic activity and antitumor efficacy. Biochem. Biophys. Res. Commun.. 2015;458(4):913-919.
- [CrossRef] [Google Scholar]
- Targeting VEGF-A/VEGFR2 Y949 Signaling-Mediated Vascular Permeability Alleviates Hypoxic Pulmonary Hypertension. Circulation. 2022;146(24):1855-1881.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2024.103285.
Appendix A
Supplementary data
The following supporting information can be downloaded from Journal Website. Table S1. List of established VEGFR2 inhibitors. Table S2. Parameters of Molecular dynamics simulation. Table S3. Molecular docking interaction scores and interacting residues of virtually screened molecules, designed molecules and Sorafenib VEGFR2 receptor (PDB id: 2OH4). Table S4: Drug Ability profiling of virtually screened molecules, designed molecules and Sorafenib standard. Table S5. Toxicity analysis of the virtually screened VEGFR2 inhibitotrs, designed VEGFR2 inhibitors and sorafenib. Figure S1. Ramachandran Plot of VEGFR2 receptor (PDB id: 2OH4). Figure S2. 2D Chemical structures of the virtually screened and designed VEGFR2 inhibitors. Figure S3. 3D interaction image of B3, B5, B7, and Sorafenib against 2OH4. Figure S4. FMO analysis of B3, B5, B7, and Sorafenib. Figure S5. Complexed ligand GIG interacted with VEGFR2 receptor (PDB id: 2OH4). The following are the Supplementary data to this article:




