

Translate this page into:
Design, synthesis, in vitro antiproliferative effect and in situ molecular docking studies of a series of new benzoquinoline derivatives
⁎Corresponding authors. quahmed@iium.edu.my (Qamar Uddin Ahmed), zaz@ums.edu.my (Zainul Amiruddin Zakaria),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Quinoline derivatives have been reported to possess multi-therapeutic potential owing to the manifestations of different pharmacological effects. The current research work describes about the design and synthesis of a series of novel benzoquinoline analogues with an objective to evaluate their antiproliferative structure–activity relationship against colon, breast and hepatocellular cancers. Upon synthesis, all derivatives’ chemical structures were elucidated through FTIR, 1HNMR and 13CNMR spectroscopic analysis. All derivatives were investigated for their in vitro anti-proliferative property against three different cancer cell lines (viz., colon carcinoma HT29, Caucasian breast adenocarcinoma MCF7, hepatocellular carcinoma HepG2) and a normal non-transformed human foreskin fibroblast Hs27 cell line. All derivatives demonstrated varied degrees of strong anticancer effect against all of the cell lines with the 2-Amino-4-(4-nitrophenyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile (CNMP, 2) exhibited the most potent antiproliferative effect viz. LC50 21.23 μM for breast, 8.24 μM for colon, and 26.15 μM for the hepatocellular, respectively. Molecular docking studies against all the the target crystal structures of cancer proteins (1HK7, 3EQM, 3IG7 and 4FM9) revealed significant binding affinities via hydrophobic and H-bonding interactions with all the compounds in conformity with the wet lab results. CNMP showed the highest binding energy of −7.55 in the HT29, −6.9 (both in MCF7 HepG2) kcal/mol. Based on the results obtained from wet lab and dry lab experiments, it can be proposed that CNMP might prove to be a potential lead structure for the design and synthesis of more potent anticancer candidates.
Keywords
Quinolines
Benzoquinoline derivatives
Cytotoxicity
Molecular docking
Computational studies

1 Introduction
Although there have been numerous anticancer compounds synthesized and consumed frequently, nevertheless, the upsurge in the cancer rate is recurrent in every country and its prevention has certainly become one of the most significant public health problems and a prodigious challenge of the 21st century. It has been suggested that about 40 percent of the cancer incidences might be controlled if the vulnerability to cancer risk factors including diet, nutrition and physical activity could be decreased and monitored (Sung et al., 2020). Majority of the synthesized anticancer drugs have been reported to be inefficacious due to untoward toxicities associated with them, Hence, this has made it necessary to put a great demand and efforts in producing more safe and efficacious drugs to counteract this detrimental disease (Tang et al., 2020).
The quinolone (molecular weight: 129.16) is a nitrogenous based heterocyclic compound with molecular formula C9H7N. It can react with acids to form salts and presents both nucleophilic and electrophilic substitutions (Marella et al., 2013). Quinoline and its derivatives are well-known diverse heterocyclic compounds with a variety of reported pharmacological activities which include anticancer, antibacterial, antifungal, anti-inflammatory, antiplasmodial, antimalarial among others. A huge number of different quinoline derivatives had earlier been synthesized mainly through chemical modification of quinoline which led to the synthesis of different promising candidates against various types of cancer and other diseases (Hussaini, 2016; Jain et al., 2016).
Based on the above quinoline features, medicinal chemists have devoted their time for developing various methods which include employment of organometallic reagents such as CuCN, LiCl, BF3, THF among others for the synthesis of different types of bioactive quinoline derivatives with the help of new computer simulation tools (Kumar et al., 2009). For instance Katarzyna et al. synthesized indolo[2,3-b]quinoline derivatives with a guanidine group that showed high selective anticancer effect as highly introducers of apoptosis through DNA-methyl green assay (Sidoryk et al., 2015; Nqoro et al., 2017). The progression in drug research in the new modern era of advanced technology of computational processes that includes molecular docking, quantitative structure activity relationship among others, make drug synthesis much more effective, economical and less time consuming (Surabhi and Singh, 2018).
Molecular docking is a computational approach that evaluates the binding affinity of the docked molecules with the receptors based on the scoring functions of mathematical algorithms, while the QSAR model generates new compounds with better predicted pharmacological efficacy which can serve as future effective drug candidates. These approaches are considered very effective tools in an in silico drug design that can either be used separately or simultaneously (Surabhi and Singh, 2018).
Indeed, the well-known most tedious efforts and cost of searching new drug candidates have been highly reduced since the emergence of different computational based chemistry methods catering the drug rationale of this purpose, for example QSAR, 3D QSAR, virtual screenings among others that collectively develop an explanatory hypothesis not only in drug synthesis, however on its mechanism of actions as well (Vilar et al., 2008). Nevertheless, several aspects of these in silico programs need to be constantly reviewed and revised due to the disparity in results when different software are used, and this is a very critical in the paradigm of drug discovery, which when overcomes will delineate a fruitful outcome in drug discovery with maximum achievements in the medicinal chemistry field (Fischer et al., 2021). Hence, in the current study, we illustrate herein the synthesis of a new series of benzoquinoline derivatives and anticancer activity evaluation at in vitro against three distinct cancer cell lines (viz., colon carcinoma HT29, Caucasian breast adenocarcinoma MCF7 and hepatocellular carcinoma HepG2). A molecular docking study implying breast protein cancer (PDB code = 1HK7 and PDB code = 3EQM), colon protein cancer (PDB code = 3IG7) and hepatocellular carcinoma (PDB code = 4FM9) was carried out to observe the binding mode of new benzoquinoline derivatives possessing in vitro anticancer effect on the active site of 1HK7, 3EQM, 3IG7 and 4FM9, respectively.
2 Results
2.1 Chemistry
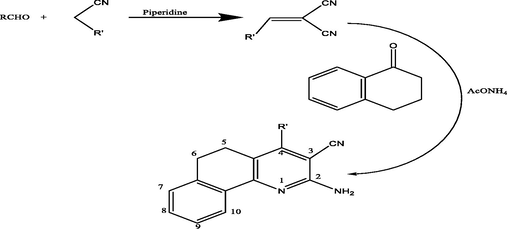
Synthesis of benzoquinoline derivatives: 1. R = 4-CH3C6H4 (CMMP 1), 2. R = 4-NO2C6H4 (CNMP 2), 3. R = 4-(CH3)2NC6H4 (CPMP 3), 4. R = 2-Theinyl (CRMP 4), 5. R = 1-Naphthyl (CTMP 5).
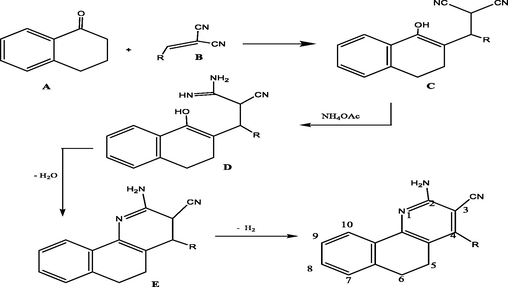
Reaction mechanism for the synthesis of benzoquinoline derivatives.
![Structures of 2-amino-4-subistituted-5,6-dihydrobenzo[h]quinoline-3 carbonitrile derivatives.](/content/185/2022/34/4/img/10.1016_j.jksus.2022.102003-fig3.png)
Structures of 2-amino-4-subistituted-5,6-dihydrobenzo[h]quinoline-3 carbonitrile derivatives.
2.2 Anticancer activity
2.2.1 In vitro MTT cytotoxicity of assay of 2-amino-4-subistituted-5,6 dihydrobenzo[h]quinoline-3-carbonitriles
S/No
Compound Codes
Human colon carcinoma HT29
Human hepatocellular carcinoma HePG2
Human breast cancer MCF 7
1
CMMP (1)
42.64
78.5
20.56
2
CNMP (2)
21.23
26.15
8.24
3
CPMP (3)
85.84
–
b-
4
CRMP (4)
74.5
–
54.45
5
CTMP (5)
30.12
25.20
10.12
Ref.
Doxorubicin c
40.0
3.0
4.0
2.3 Molecular docking
2.3.1 Breast cancer
.![3D docking, site view & 2D of the CMMP (1) (2-Amino-4-(p-tolyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile) and 3EQM protein cancer of breast cancer protein.](/content/185/2022/34/4/img/10.1016_j.jksus.2022.102003-fig4.png)
3D docking, site view & 2D of the CMMP (1) (2-Amino-4-(p-tolyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile) and 3EQM protein cancer of breast cancer protein.
mol
S
rmsd_refine
E_conf
E_place
E_score1
E_refine
E_score2
CMMP (1)
−7.24
0.78
−28.29
−77.10
−10.40
−27.16
−7.24
−7.03
0.98
−28.90
−83.32
−9.94
−28.83
−7.03
−6.88
1.01
–32.85
−73.07
−10.73
−34.00
−6.88
−6.87
2.32
−38.88
−97.65
−10.36
−35.17
−6.87
−6.82
1.29
−36.64
−87.97
−9.93
−29.06
−6.82
CNMP (2)
−7.55
0.73
−0.55
−79.68
−10.61
−36.75
−7.55
−7.54
0.86
9.13
−79.41
−11.17
−27.95
−7.54
−7.53
1.33
3.78
−81.73
−10.84
−36.80
−7.53
−7.46
1.32
−1.90
−101.67
−10.74
−36.30
−7.46
−7.41
1.16
1.33
−96.69
−10.72
−37.65
−7.41
CPMP
(3)−6.61
0.74
−15.18
−70.33
−9.50
−26.14
−6.61
−6.48
0.95
−20.08
−86.56
−9.12
−30.38
−6.48
−6.47
1.02
−20.15
−77.36
−8.94
−31.02
−6.47
−6.41
4.98
−20.99
−69.87
−10.30
−29.63
−6.41
−6.37
2.33
−18.56
−69.25
−9.11
−26.41
−6.37
CRMP
(4)−6.86
1.05
−18.54
−81.17
−9.65
−30.58
−6.86
−6.80
0.90
−16.94
−77.32
−9.67
−28.40
−6.80
−6.76
0.90
−21.62
−71.69
−9.81
−25.78
−6.76
−6.73
1.30
–22.96
−82.51
−10.06
–32.29
−6.73
−6.70
1.18
−1.72
−83.66
−9.39
−35.69
−6.70
CTMP
(5)−7.41
1.40
−19.25
−68.66
−10.38
–32.46
−7.41
−7.34
0.70
–22.89
−90.73
−10.57
−28.94
−7.34
−7.27
3.05
−25.14
−74.92
−9.98
−39.27
−7.27
−7.17
1.11
−16.44
−73.64
−9.89
–33.62
−7.17
−7.15
1.71
–23.67
−99.32
−11.01
−34.25
−7.15
.
Ligand
Receptor
Interaction
Distance E
(kcal/mol)
CMMP (1)
N 41
CA MET 303
H-acceptor
3.59
−0.9
6-ring
N ALA 438
pi-H
4.23
−1.1
CNMP (2)
O 37
NE1 TRP 141
H-acceptor
3.39
−1.4
O 37
NH2 ARG 145
H-acceptor
3.14
−1.3
O 38
NH2 ARG 115
H-acceptor
3.08
−1.1
O 38
NE ARG 435
H-acceptor
2.97
−2.9
O 38
NH2 ARG 435
H-acceptor
3.17
−1.9
CPMP (3)
N 36
SD MET 311
H-donor
4
−0.6
CRMP (4)
N 24
SD MET 311
H-donor
3.54
−1.7
6-ring
CA GLY 439
pi-H
4.33
−0.7
CTMP (5)
N 23
O ARG 435
H-donor
2.98
−1.7
2.3.2 Colon cancer
![3D docking, site view & 2D of the CMMP (1) (2-Amino-4-(p-tolyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile) and 3IG7 protein cancer of colon-cancer protein.](/content/185/2022/34/4/img/10.1016_j.jksus.2022.102003-fig5.png)
3D docking, site view & 2D of the CMMP (1) (2-Amino-4-(p-tolyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile) and 3IG7 protein cancer of colon-cancer protein.
2.3.3 Hepato cancer
![3D docking, site view & 2D of the CMMP (1) (2-Amino-4-(p-tolyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile) and 4FM9 protein cancer of hepato-cancer protein.](/content/185/2022/34/4/img/10.1016_j.jksus.2022.102003-fig6.png)
3D docking, site view & 2D of the CMMP (1) (2-Amino-4-(p-tolyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile) and 4FM9 protein cancer of hepato-cancer protein.
3 Discussion
α-Tetralone upon reacting with the arylidene malononitriles (derivatives 1–5) in ammonium acetate’s presence afforded 2-amino-4-substituted-5,6-dihydrobenzo[h]quinoline-3-carbonitriles in good yields (). The formation of the 2-aminotetraquinoline derivatives, could well be described according to the mechanism in which the reaction was initiated by active hydrogen addition of compound A to the ethylenic double bond of compound B to give compound C. Ammonia (from ammonium acetate) was added to the nitrile group in compound C which afforded compound D which underwent dehydration by eleminiting a molecule of water to yield compound E which eventually underwent an auto-oxidation to produce the desirable final compound in the form of final product.
The IR spectra of the 2-aminotetraquinolines exhibited absorption bands within the range of 2205–2220 cm−1 and 3324–3482 cm−1 for the –CN and –NH2 groups, respectively. Their structures were more tentatively confirmed through 1H NMR spectral results which displayed alongside the aromatic protons, two different signals as multiplets at δ2.29–2.35 and 2.59–2.68 ppm which were attributable to the H-5 and H-6, respectively. 13C NMR spectral data further supported the structures of the compounds that also displayed the expected number of aromatic carbons. Moreover, two different signals observed at δ 23.69–24.14 and 27.44–27.94 were attributable to C-5 and C-6, respectively.
3.1 Antiproliferative effect
Three different human tumor cell lines that include colon carcinoma HT29, hepatocellular carcinoma HePG2, and Caucasian breast adenocarcinoma MCF7 were taken into consideration and used to investigate an in vitro antiproliferative effect (cytotoxic activity) of the all synthesised derivatives (Fig. 1) viz. 2-amino-4-(p-tolyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile (CMMP, 1), 2-Amino-4-(4-nitrophenyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile (CNMP, 2), 2-Amino-4-(dimethylamino)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile(CPMP,3),2-Amino-4-(thiophen-3-yl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile(CRMP,4) and 2-Amino-4-(naphthalen-1-yl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile(CTMP,5) through MTT assay(https://www.dojindo.eu.com/Protocol/Dojindo-Cell-Proliferation-Protocol.pdf, 2019). Table 1 demonstrates the LC50 (μM) values (concentration of the test benozquinoline derivatives that were responsible to lead the death of 50% of the cells in 24 h (lethal dose)). The obtained data showed that all the different cancer cell lines which were taken into account displayed dissimilar degree of sensitivity activities against all of the above mentioned benzoquinoline based derivatives that were evaluated in this study. In the colon cancer cell line (HT29), compounds 2 and 5 (viz. LC50 21.23 and 30.12 μM, respectively) revealed distinctive sensitivity in comparison to the referenced anticancer drug, doxorubicin (LC50 40.0 μM). Meanwhile, the compound 2 superseded the doxorubicin (LC50 40.0 μM) activity, however compounds 1, 3 and 4 (LC50 42.64, 85.84 and 74.7 μM, respectively) displayed moderate cytotoxic effect towards the HT29 with compound 2 being the most potent among them. However, the MCF 7 (human breast cancer cell line) showed a significant sensitivity towards four of the investigated structure analogues and their LC50 ranged 8.24–54.45 μM, in comparison to doxorubicin (LC50 4.0 μM). Between all the examined compounds, the highest activity was shown by compounds 2 and 5 (LC50 8.24 and 10.12 μM, respectively). While the hepatocellular carcinoma HepG2 was found to be the slightest sensitive as the growth of this cell line was impacted by the three structure analogues of benzoquinoline only. Moreover, compounds 2 and 5 were found to show a potent growth inhibition potential as evidenced from their small LC50 values (26.15 and 25.20 μM, respectively), that represented around 40–60% of the activity of doxorubicin (LC50 4.0 μM). This implied that the compounds 1, 3 and 4 had a good cytotoxic effect against the all cell lines used in this study. The compound 2 was found to be the most active member in the current research work with distinct efficacy towards both the MCF 7 (human breast cancer) (about 50% of the activity of doxorubicin; LC50 8.24 versus 4.0 μM, respectively) and HT29 (the colon carcinoma) (almost two-fold as active as doxorubicin; LC50 21.23 versus 40 μM, respectively). Existence of two cyano functional groups on the double bonded carbons (olefinic) especially nitro group (being the most active substituent) appeared to structurally influence the cytotoxic activity of the coded compound 2 among others (Larsson et al., 2020).
3.2 Molecular docking
Molecular docking is one of the proficient ways to calculate as well as surmise the kind of interaction and binding sites with the interacting molecules. MOE modeling program was employed in order to visualise and calculate the binding sites and their docking scores of the coded compounds viz. CMMP (1), CNMP (2), CPMP (3), CRMP (4) and CTMP (5) with each of the three different cancer protein enzymes coded as 3EQM for human breast, 3IG7 for colon and 4FM9 for hepatocellular carcinoma cell lines, respectively. Among the all energies calculated, the least binding energy revealed the highest activity which was observed by the ranking poses generated by scoring functions which are given in Tables 2, S4 and S6, respectively. The best score was obtained by the 2 in all the three cancer proteins and the scores were −7.55, −6.90 and −6.90, respectively. List of hydrogen bonds between all the synthesized compounds figure with coenzymes of the chosen proteins are respectively presented in Tables 3, S5 and S7. The best fitted poses adopted by the compounds docked into enzymes viz. 3EQM, 3IG7 and 4FM9 are shown in Figs. 2, 3 and 4, respectively (Meng et al., 2011; Pantsar and Poso, 2018).
Molecular Operating Environment (MOE) is one of the molecular docking routines that is employed in order to recognize an accurate docking studies between the compounds and the target proteins. The docking pose and types of interaction were aligned and agreed with the experimental LD50 of these compounds against the three cancer proteins (Figs. 2-4; S1-S6). For the 3EQM protein “human breast cancer”, the compound 2 showed the high docking score through hydrogen bonding between two oxygen atoms of the nitro group towards Trp 141, Arg 145, Arg115 and Arg 435 amino acid residues. The hydrogen bond varied from 2.97 to 3.39 A° and the energy stabilization by −1.1 to −2.9 K cal. While in the case of 3IG7 protein “colon cancer”, the compound 2 showed the high docking score by the hydrogen accepting ability of one of the oxygen atoms of nitro group towards Lys 33 amino acid residue with hydrogen bond “3.26 A°” and energy stabilization by −3.3 K cal., nitrogen atom of the same nitro group demonstrated hydrogen bond “3.53 Ao and energy stabilization by −2.1 K cal. with Leu 83 amino acid residue as well as hydrogen –π-stacking with 6-membered ring of Gly 11 “3.99 A° and −0.9 K cal.”. On the other hand, it was found that the 2 exhibited the high docking score according to its interaction with 4FM9 protein “hepatocellular carcinoma cell lines” through one of the oxygens of nitro group with Arg 727 residue as a hydrogen bond viz. 3.01 A° and −2.80 K cal., respectively (Ferreira et al., 2015; Singh et al., 2019). These hydrogen bonding, ionic and hydrophobic bonding interactions with the key amino acid residues of the identified binding pockets based on the above output, stabilized the structure of the target receptor. All the docked pose with the least binding energy has the highest affinity, thereby, is considered the best docked conformation. Moreover, analysis of these docked ligands with these proteins brought into focus a very vital interaction operating at the molecular level, this is due to the fact that the docking program (MOE) was able to reproduce experimentally observed binding modes, as in this findings, it identifies the particular target-ligand conformation (Pantsar and Poso, 2018; Boittier et al., 2020).
4 Materials and methods
4.1 Instrumentation
Gallenkamp device was used to determine the melting point of all benzoquinoline derivatives. Bruker DPX-400 FT NMR spectrometer was used to determine the proton and carbon 13 spectra. Tetramethylsilane (TMS) and DMSO‑d6 were used as an internal standard and solvent, respectively. The chemical shifts were measured as δ in ppm and splitting patterns were labelled as q for quartet, m for multiplet, d for doublet and s for singlet, respectively. Shimadzu FT-IR 8400S spectrophotometer was used to measure the infrared spectra by following the KBr pellet method. 2400 Perkin Elmer Series 2 analyzer was used to carry out the elemental analysis and the observed values were found to vary with the practical values within the range of ± 0.4%. All synthetic reactions were observed through TLC on silica gel-protected aluminum sheets (Type 60 F254, Merck) and the UV-lamp at λ 254 was used to detect the spots of the final products on the TLC plate. The three kinds of distinct cancer cell lines namely breast (MCF-7), colon (HT29) and hepatocellular carcinoma were directly procured from the Sigma-Aldrich Co. (St Louis, MO, USA).
4.2 General method for the synthesis of arylidene derivatives
A combination of the suitable carbonyl compounds i.e. aldehydes (10 mmol), malononitrile i.e. propanedinitrile (10 mmol) and piperidine (azinane) (catalytic amount of it) in ethyl alcohol (25 mL) were agitated for about sixty minutes at room temperature (25 °C). Afterwards, the reaction mixture was then poured onto water (200 mL) and left undisturbed overnight. Subsequently, the solid product as precipitate was obtained through filtration process, washed carefully with plenty of H2O then completely dried. Later, it was recrystallized with the suitable organic solvents to obtain in pure form (Qandalee et al., 2013).
4.3 General procedure for synthesis of 2-Amino-4-subistituted-5,6-dihydrobenzo[h]-quinoline-3-carbonitrile.
A combination of arylidene derivative (Hussaini, 2016), 1-tetralone (1.46 g, 10 mmol), 1 (10 mmol) and excess ammonium acetate (6.2 g, 80 mmol) in an ethanol (30 mL) (absolute) was heated for about 4–6 h under reflux. The completion of reaction was closely examined by the TLC plates run in different binary solvent systems. After comprehensive conversion (with the TLC indication), the separated solid material during heating was cautiously obtained through filtration process and recrystallized from a mixture of ethyl alcohol and a few drops of DMF as needles shaped fine crystals (Asiri et al., 2011).
2-amino-4-(p-tolyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile (1) [CMMP (R = 4-CH3C6H4)] 1: Recrystallized from DMF/ethanol as needles (3.2 g, 82%), m.p. 150–152 °C. ν max (cm−1, KBr): 2205 (CN); 3332, 3482 (NH2). 1HNMR [400 MHz, DMSO‑d6, δ (ppm)]: δ 2.36 (s, 3H, CH3); 2.49 (m, 2H, H-5), 2.70 (m, 2H, H-6), 7.28–8.20 (m, 10H, Ar H + NH2). 13CNMR [150 MHz, DMSO‑d6, δ (ppm)]: 21.35 (CH3), 23.72 (C-5), 27.94 (C-6); 114.87 (CN); 86.41, 125.55, 126.84, 127.42, 127.41, 128.20, 129.82, 129.83, 130.10, 132.46, 134.25, 139.22, 150.23, 159.82, 160.23 (ArC). Anal% Calcd for C21H17N3: C, 81.00; H, 5.50; N, 13.49. Found: C, 80.98; H, 5.53; N, 13.51.
2-Amino-4-(4-nitrophenyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile (2) [CNMP (R = 4-NO2C6H4)] 2: Recrystallized from ethanol as needles (3.2 g, 79%), m.p. 190–192 °C. ν max (cm−1, KBr): 2228 (CN), 3238 (NH2).1HNMR [400 MHz, DMSO‑d6, δ (ppm)]: 2.51 (m, 2H, H-5); 2.73 (m, 2H, H-6); 6.81–8.05 (m, 10H, Ar H + NH2), 7.98 (s, 1H, NH). 13CNMR [150 MHz, DMSO‑d6, δ (ppm)]: 23.69 (C-5), 27.79 (C-6), 112.53 (CN), 115.47, 116.73, 121.76, 125.20, 125.30, 126.40, 128.50, 130.60, 139.23, 147.37, 147.59, 159.85, 161.95, 162.73 (ArC). Anal.% Calcd for C20H14N4O2: C, 70.17; H, 4.12; N, 16.37. Found: C, 70.21; H, 4.01; N, 16.51.
2-Amino-4-(dimethylamino)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile (3) [CPMP (R = 4-(CH3)2NC6H4)] 3: Recrystallized from ethanol as needles (3.6 g, 82%), m.p. 178–180 °C. ν max (cm−1, KBr): 2209 (CN); 3362, 3478 (NH2). 1HNMR [400 MHz, DMSO‑d6, δ (ppm)]: 3.10 (s, 6H, CH3); 2.54 (m, 2H, H-5), 2.71 (m, 2H, H-6), 6.85–8.17 (m,10H, Ar H + NH2). 13CNMR [150 MHz, DMSO‑d6, δ (ppm)]: 41.38 (CH3), 24.14 (C-5), 27.85 (C-6), 113.55 (CN), 85.8, 112.41, 112.52, 122.80, 125.42, 126.93, 127.52, 128.46, 128.7, 130.78, 139.52, 150.22, 155.3, 159.91, 160.35 (ArC). Anal.% Calcd for C16H16N4: C, 72.70; H, 6.10; N, 21.20. Found: C, 70.68; H, 6.13; N, 21.22.
2-Amino-4-(thiophen-3-yl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile (4) [CRMP (R = 2-Theinyl)] 4: Recrystallized from ethanol as needles (3.1 g, 77%), m.p. 175–176 °C. ν max (cm−1, KBr): 2212 (CN); 3355, 3432 (NH2). 1HNMR [400 MHz, DMSO‑d6, δ (ppm)]: 2.57 (m, 2H, H-5),2.75 (m, 2H, H-6), 7.22–8.17 (m, 9H, Ar H + NH2). 13CNMR [150 MHz, DMSO‑d6, δ (ppm)]: 24.02 (C-5), 27.44 (C-6), 113.8 (CN), 85.22, 126.84, 126.82, 127.91, 128.54, 128.44, 128.62, 128.90, 130.75, 134.22, 139.63, 139.80, 146.92, 159.78, 160.23 (ArC). Anal.% Calcd for C18H13N3S: C, 71.26; H, 4.32; N, 13.85. Found: C, 71.23; H, 4.35; N, 13.82.
2-Amino-4-(naphthalen-1-yl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile (5) [CTMP (R = 1-Naphthyl)] 5: Recrystallized from ethanol as needles (3.4 g, 80%), m.p. 220–222 °C. ν max (cm−1, KBr): 2220 (CN); 3333, 3480 (NH2). 1HNMR [400 MHz, DMSO‑d6, δ (ppm)]: 2.56 (m, 2H, H-5), 2.74 (m, 2H, H-6); 7.30–8.10 (m,13H, Ar H + NH2). 13CNMR [150 MHz, DMSO‑d6, δ (ppm)]: 24.05, 27.82 (CH2), 113.70 (CN), 85.54, 124.22, 125.54, 125.71, 126.62, 126.64, 127.50, 128.38, 128.41, 128.62, 128.92, 130.40, 132.79, 134.25, 134.98, 138.89, 139.3, 150.35, 159.62, 161.29 (ArC). Anal.% Calcd for C24H17N3: C, 82.97; H, 4.93; N, 12.10. Found: C, 82.95; H, 4.96; N, 12.13.
4.4 In vitro cytotoxicity investigation via MTT assay
All the synthesized derivatives of benzoquinoline were tested for their in vitro cytotoxic effect by following an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) method (Cell Counting Kit-8 (WST-8 based), Dojindo Molecular Technologies, Inc. Cat.# CK04-01) (Denizot and Lang, 1986). Three distinct human cancer cell lines, viz., colon carcinoma HT29, Caucasian breast adenocarcinoma MCF7 and hepatocellular carcinoma HepG2 as well as a normal non transformed human foreskin fibroblast Hs27 cell line were used for abovementioned cytotoxicity investigation. The in vitro cytotoxicity investigation through MTT assay was carried out with slight modification. Briefly, all experiments were carefully carried out in a laminar flow cabinet biosafety class II (Baker, SG403INT, Stanford, ME, USA) level particularly within a sterile region. Cells were cultured in batches for a specific period of 10 days and then carefully seeded at 10 × 103 cells/well (concentration) in a freshly prepared growth medium. This process was carried in a 96-well microtiter plastic plate for twenty four hours under 5% CO2 with the help of a water jacketed CO2 incubator (Sheldon, TC2323, Cornelius, OR, USA) at 37 °C. Media was cautiously extracted, freshly prepared medium without serum was then transferred, and cells (negative control) were subjected to incubation only. Leter, they were treated with various strengths of all the synthesized quinoline derivatives investigated at 100–50-25–12.5–6.25–3.125–1.56–0.78 μg/mL. All the synthesized quinoline derivatives were dissolved in DMSO which was taken as a vehicle. The final concentration of the DMSO solution was not increased more than 0.2% for any cell preparations. DMEM was used for MCF 7 cell line and RPMI 1640 medium was used for HT29 and HepG2 cell lines suspensions and then 1% antibiotic–antimycotic mixture (10,000 μg/mL streptomycin sulphate, 10,000 IU/mL penicillin potassium and 25 μg/mL amphotericin B), and 1% L-glutamine were also added in 96-well flat bottom microplate at 37 °C under 5% CO2. After incubation process for 24 h, aspiration of the medium was carried out and then 40 μL of MTT reagent (2.5 μg/mL) was vigilantly transferred to every well and then plate was further kept for incubation for another four hours at 37 °C under 5% CO2. Afterwards, 10% sodium dodecyl sulphate (SDS) (200 μL) in deionized water was separately/individually transferred to every well to terminate the reaction by dissolving the crystals formed and then plate was again put for incubation all night at 37 °C. At 595 nm, the absorbance was measured using microplate multiwall reader (Bio-Rad Laboratories Inc., model 3350, Hercules, California, USA). 620 nm was used as a reference wavelength in this experiment. The values are displayed in Table 1 as LC50 (μM) (toxic amount that was responsible to kill 50% cells in the period of 24 h) for all the synthesized quinoline based derivatives investigated in this study (Sylvester, 2011; Larsson et al., 2020).
4.5 Molecular docking methodology
All the scores obtained in the molecular docking process were calculated and characterized through the special software package known as Molecular Operating Environment (MOE). Initially, the 3D structures of all the synthetic compounds tested were built with the help of Chem3D ultra 12.0 software [Molecular Modeling and Analysis; Cambridge Soft Corporation] and subsequently all the compounds were evaluated for energy minimization through MOPAC and finally kept as MDL MolFile (*.mol). Protein Data Bank (https://www.rcsb.org/pdb/ welcome.do) was used to recover and generate the target crystal structures of breast protein cancer (PDB code = 1HK7 and PDB code = 3EQM), colon protein cancer (PDB code = 3IG7) and hepatocellular carcinoma (PDB code = 4FM9) (Mashat et al., 2019). For the optimization process, all bound cofactors and waters ligands were disconnected from the protein structure and the hydrogen atoms were finally attached to it. The active sites were sequestered and taken as dummies atoms. MMFF94x force field was used to apportion all the parameters and charges (Abdellattif et al., 2018). Upon generation of the alpha-site spheres through the use of MOE’s site finder module, the molecules structural model was docked on the surface of the interior of the cancer protein through the MOE’s DOCK module. For executing all the calculations, intel(R) core(TM)i7, 3.8 GHz based machine running MS Windows 10 as the operating system was used. London dG scoring function was used to execute the Dock scoring in MOE software and two unrelated refinement methods were further used to upgrade. Auto rotatable bonds were subsequently permitted for the top five binding poses that were targeted for the analysis to attain the best possible score (Althagafi et al., 2019). The database browser was then employed in order to compare the docking poses to the ligand in the co-crystallized structure as well as to acquire RMSD of the docking pose. Later, the binding free energy and hydrogen bonds between the synthesized molecules and amino acid residues in the receptors were calculated in order to rank the binding affinity of the molecules to the protein molecules taken into account. Hydrogen bond length below 3.5 A° was used to ascertain the intensity of the hydrogen bonding between the synthesized quinoline derivatives and amino acids in the receptors. Moreover, RMSD of the synthesized quinoline derivatives’ position was further matched to the docking pose employed in ranking. Both the types of interactions as well as the RMSD of the ligand (native) within the receptor’s structure were taken as standard docked model (Abdel-Rhman et al., 2019).
4.6 Statistical analysis
A statistical significance was confirmed among the samples in question and the cells with vehicle (negative control) using independent t-test by SPSS 20 program.
5 Conclusion
The quinoline-3-carbonitrile derivatives were synthesized in an excellent yield in this research study. The antitumor activity results indicated that compound CNMP (2) bearing para-nitro substituted phenyl ring and CTMP (5) bearing naphthyl substitution possess the overall tumor growth inhibition sequentially both in vitro and in silico. The anticancer property results against breast (MCF-7) and colon (HT29) carcinoma cell lines showed that presence of nitro substitution and naphthyl rings play a major role in activity. The derivative CNMP revealed a very good growth inhibition of cancer cells with IC50 value very close to the standard drugs as presented in all the cell lines. The computational studies revealed that CNMP possessed the maximum binding affinity with the target protein of both breast and colon cancer proteins with binding energies −7.55, −6.90 Kcal/mol, respectively. While the CTMP revealed a better cytotoxicity against the hepatocellular carcinoma cell lines. CNMP and CTMP were found to be the most potent compared to the others. On the basis of the above facts of both in silico and wet lab outcomes, 2-Amino-4-(4-nitrophenyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile (CNMP, 2) and 2-Amino-4-(naphthalen-1-yl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile (CTMP, 5) may be proposed to serve as lead structures for the design and synthesis of more potent anticancer drugs.
6 Data availability statement
All data generated in this study can be provided upon request.
CRediT authorship contribution statement
Abdulrashid Umar: Investigation, Writing – original draft. Hassan M. Faidallah: Conceptualization, Methodology, Validation, Formal analysis, Resources, Writing – review & editing, Supervision, Project administration, Funding acquisition. Qamar Uddin Ahmed: Conceptualization, Writing – original draft, Writing – review & editing. Khalid.A. Alamry: Methodology. Sayeed Mukhtar: Writing – review & editing. Meshari A. Alsharif: Writing – review & editing. Syed Najmul Hejaz Azmi: Writing – review & editing. Humaira Parveen: Writing – review & editing. Zainul Amiruddin Zakaria: Writing – review & editing. Mostafa A. Hussien: Methodology, Software, Resources.
Acknowledgements
Authors are thankful to the Department of Chemistry, Faculty of Science, King Abdulaziz University, KSA for providing all the essential facilities, chemicals and reagents through internal research fund to accomplish this research. Faculty of Pharmacy, King Abdulaziz University, KSA and Alexandria University, Egypt is also duly acknowledged for providing all the necessary facilities to carry out an in vitro anticancer evaluation.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.. 2020;2021
- [CrossRef] [Google Scholar]
- Design and synthesis of novel quinoline derivatives bearing oxadiazole, isoxazoline, triazolothiadiazole, triazolothiadiazine, and piperazine moieties. J. Heterocycl. Chem.. 2020;57(6):2330-2338.
- [Google Scholar]
- Therapeutic significance of quinolines: a patent review (2013–2015) Expert Opin. Ther. Pat.. 2016;26(10):1201-1221.
- [Google Scholar]
- Comprehensive review on current developments of quinoline-based anticancer agents. Arab J. Chem.. 2016;12(8):4920-4946.
- [Google Scholar]
- Biological activities of quinoline derivatives. Mini Rev. Med. Chem.. 2009;9:1648-1654.
- [Google Scholar]
- The synthesis of indolo [2, 3-b] quinoline derivatives with a guanidine group: Highly selective cytotoxic agents. Eur. J. Med. Chem.. 2015;105:208-219.
- [Google Scholar]
- Quinoline-based hybrid compounds with antimalarial activity. Molecules. 2017;22(12):2268.
- [Google Scholar]
- Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem.. 2008;8(18):1555-1572.
- [Google Scholar]
- A novel ligand of the translationally controlled tumor protein (TCTP) identified by virtual drug screening for cancer differentiation therapy. Invest. New Drugs. 2021;39(4):914-927.
- [Google Scholar]
- Synthesis of quinoline derivatives from the reaction of aminobenzophenones and acetylenic esters in the presence of SnO2 nanoparticles. Int. Nano Lett.. 2013;3:49.
- [Google Scholar]
- 2-Amino-4-(4-chlorophenyl)-5,6-dihydrobenzo[h]quinoline-3-carbonitrile–3-amino-1-(4-chlorophenyl)-9,10-dihydrophenanthrene-2,4-dicarbonitrile (1/4) Acta Cryst.. 2011;E67:o2874
- [Google Scholar]
- Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. lmmunol. Methods. 1986;89(2):271-277.
- [Google Scholar]
- Optimization of the tetrazolium dye (MTT) colorimetric assay for cellular growth and viability. Methods Mol. Biol.. 2011;716:157-168.
- [Google Scholar]
- Optimization of cell viability assays to improve replicability and reproducibility of cancer drug sensitivity screens. Sci. Rep.. 2020;10(1):5798.
- [Google Scholar]
- Synthesis, structures, DNA-binding and anticancer activities of some copper(I)-phosphine complexes. Polyhedron. 2019;158(2):164-172.
- [Google Scholar]
- New approaches of 4-aryl-2-hydrazinothiazole derivatives synthesis, molecular docking, and biological evaluations. Int. J. Pharm. Sci. Res.. 2018;9(12):5060-5078.
- [Google Scholar]
- Characterization of new Pt(IV)–thiazole complexes: Analytical, spectral, molecular modeling and molecular docking studies and applications in two opposing pathways. Appl. Organomet. Chem.. 2019;33(9):1-18.
- [Google Scholar]
- Synthesis, characterization, molecular docking and cytotoxicity studies on N-benzyl-2-isonicotinoylhydrazine-1-carbothioamide and its metal complexes. J. Mol. Struct.. 2019;1196:417-428.
- [Google Scholar]
- Available online: https://www.dojindo.eu.com/Protocol/Dojindo-Cell-Proliferation-Protocol.pdf (accessed on 17 October 2019).
- Molecular Docking: Apoerful approach for structure based drug discovery. Curr. Comput. Aided Drug Des.. 2011;7(2):146-157.
- [Google Scholar]
- Molecular docking and structure-based drug design strategies. Molecules. 2015;20(7):13384-13421.
- [Google Scholar]
- Rigorous sampling of docking poses unveils binding hypothesis for the halogenated ligands of L-type amino acid transporter 1 (LAT1) Sci. Rep.. 2019;9(1):15061.
- [CrossRef] [Google Scholar]
- Assessing molecular docking tools to guide targeted drug discovery of CD38 inhibitors. Int. J. Mol. Sci.. 2020;21(15):5183.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102003.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




