

Translate this page into:
Design & discovery of small molecule COVID-19 inhibitor via dual approach based virtual screening and molecular simulation studies
⁎Corresponding author at: Department of Biology, College of Science, Princess Nourah bint Abdulrahman University, Riyadh 13415, Saudi Arabia. nhaljarba@pnu.edu.sa (Nada H. Aljarba)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The emerged COVID-19 (SARS corona virus) pandemic leads to severe or fatal respiratory tract infections affecting millions of people worldwide since its outbreak. The situation needs the newer molecule to control the infections as the pandemic had very badly affected the health and socioeconomic conditions of human being. CoV-2 main protease is considered to be key enzyme by targeting which we can design or develop the drug candidate. The active fitting and binding of any molecule depends upon the shape and electrostatic properties of ligand complementary to the receptor site. In this study ZINC13 database, a drug like subset (13,195,609 molecules) was subjected to shape and electrostic based virtual screening (VROCS & EON software) and followed by molecular modelling studies using docking and molecular dynamics simulation. Further the drug ability of identified candidate was predicted by the SiteMap analysis. The best shape and electrostatic similarities were observed between ZINC19973962 and reference molecule. The Tamintoshape and Tanimotoelectrostatic was found to be 0.667 and 0.022 respectively. The molecule also displayed the identical binding pattern with docking score −7.964 and this interaction was further validated by the molecular dynamics simulations. The RMSD & RMSF values were found to be 1.5 Å and1.8 Å respectively suggesting the stability of complex and very low fluctuation in ligand–protein complex over the entire MD simulation run. SiteMap analysis showed the identical Dscore of reference and identified HIT that indicated the molecule ZINC19973962 would be the promising druggable candidate against COVID main protease enzyme and can be used as lead molecule for the development of anti-COVID molecule.
Keywords
SARS
COVID-19
Zinc13 database
Dynamics simulation
SiteMap analysis

1 Introduction
Severe acute respiratory syndrome (SARS) coronavirus 2 causes severe highly prevalent and deadliest diseases including middle east respiratory syndrome (MERS) and SARS in human and animals. The later outbreak was first reported in Wuhan province of China and declared as pandemic by world health organization (WHO) in March 2020. (Surveillances 2020, Zumla and Niederman 2020) Since then millions of COVID cases and death associated with COVID infection have been reported worldwide. The data indicated that the transmission rate of SARS-CoV-2 is higher (2–2.5%) and fatality rate (5%) is lower than previously reported coronaviruses like SARS coronavirus (1.7–1.9% transmissibility, 9.5% fatality) and MERS coronavirus (<1% transmissibility, 34.4% fatality) (Mahase, 2020). The major route of transmission is respiratory droplets generated during infected through person’s cough, sneezes, speaks and contact with particles contaminated with virus droplets, whereas some reports suggested that transmission may occur through digestive tract ( Zhou et al., 2020). The incubation period of virus is 1–14 days which may increase up-to 19 days in asymptomatic carriers. The period from onset of symptoms to death may range from 6 to 41 days which depends upon immunity and age of patient. Patients with one or more comorbidities like hypertension, diabetes and cardiovascular disease have more fatality rates (Mazumder et al., 2020; Zhao et al., 2020). The major symptoms of COVID-19 are difficulty breathing, cough, runny nose or symptoms associated with pneumonia. Severity of symptoms may be mild, moderate or severe depending on the clinical features. Mild infection is manifested by development of pneumonia or non-pneumonia whereas severe infection is manifested by breathlessness, acute respiratory failure or sometime multiple organ failure that led death (Lovato et al., 2020; Yin et al., 2020; Zare-Zardini et al., 2020). Clinical laboratory studies showed leucopenia and lymphopenia are the prime feature of COVID-19. In many patients abnormal myocardial zymograms was observed due elevated level of lactate dehydrogenase and creatinine kinase. Increased level of C-reactive protein was noted with high erythrocytes sedimentation rate and D-dimer were also noted (Aghagoli et al., 2020; Cui et al., 2020). Since the outbreak various research groups and companies have been tested the library of existing antiviral drugs for the promising drug candidate or lead against the SARS-COV2 virus. Various reports have been published on generated in-silico data for development of effective candidate against this enzyme (Cao et al., 2015; Tian et al., 2021), but none of them was found to be effective. Still there is tremendous demand for development of newer molecule that can cure the acute viral SARS-COV2 infection. Recently a few reports have been indicated the possible use of some compounds from natural or synthetic origin such as Kobophenol A, (Gangadevi et al., 2021) Tideglusib, Carmofur, Shikonin, Ebselen, Disulfiram and PX-12 for their possible use in SARS-CoV-2 infection (Jin et al., 2020). The Kobophenol A was reported as an ACE2 Receptor inhibitor and binds with the Spike RBD Domain of SARS-CoV-2, whereas later all were COVID main protease inhibitors. In this study, we report the dual approach of ligand and protein based virtual screening by shape and electrostatic comparative study with previously reported inhibitor and molecular modelling-based discovery of novel HITs for the possible SARS-CoV-2 inhibitors (see Fig. 1).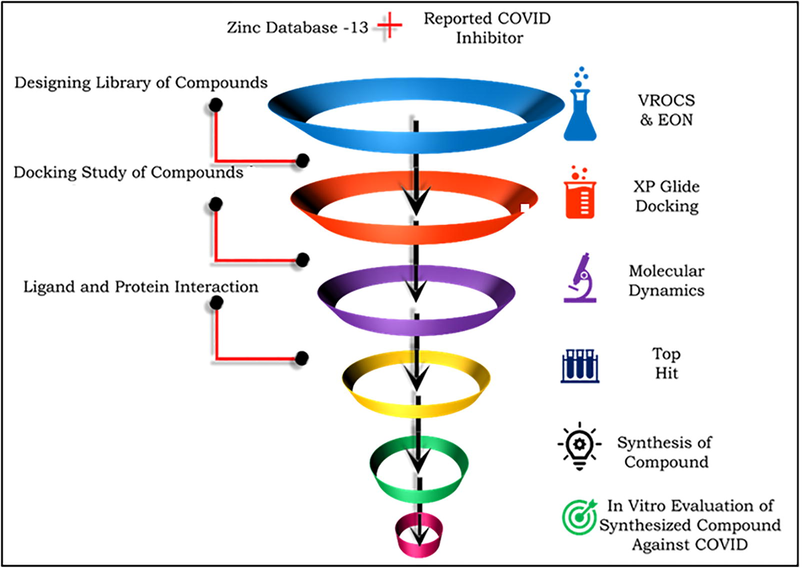
Flow chart of computational studies used for identification of HIT. The combined physico-chemical aspects of ligand (left side, highlighted as purple color) and protein (left side, highlighted in pink color) was used for in-silico data generation and analysis.
2 Material and methods
Comparative shape and electrostatic similarities indices based virtual screening of reference molecule and Zinc13 database (Irwin and Shoichet, 2005) was done by using OpenEye toolkit. (OEChem) The modeling and SiteMap analysis was performed with Maestro and SiteMap module of Schrodinger LLC suite (Schrodinger, 2011). The molecular dynamics simulation was done by using Desmond V3. The protein structure CoV-2 main protease (PDB id 6LU7) (Jin et al., 2020) was downloaded from the scientific collaboratory protein data bank (www.rcsb.org) (Berman et al., 2000). The downloaded raw protein was devoid of H bonds; some residues were missing that was updated by processing with protein preparation wizard to avoid any physicochemical constraint. All the procedures were successfully executed using above mentioned software running on ubuntu operating system 1 TB machine (Intel-R, C™ i7-8700 CPU 3.20 GHz, 3.19 GHz, 16 GB RAM 1 TB).
2.1 Shape and electrostatic study
The basis of current study was based on the comparative virtual screening of reference molecule with Zinc13 database. The reason in differentiating the topological properties is that it takes account of molecules with shape similarity by superimposition of different molecules with known inhibitors. As we know the complementary topological are the primary determinants for active site of the protein (Sastry et al., 2011; Bitencourt-Ferreira and de Azevedo, 2018; Siddique et al., 2020). Moreover, the stability of ligand–protein complex is also dependent on these interactions. Therefore, it was thought that we should consider these primary determinants at the preliminary screening of drug database. These forces are involved in electrostatic interactions and led to non-covalent bond formation between oppositely charged moieties (Morrone et al., 2016; de Ávila and de Azevedo 2018; Siddique et al., 2018). Thus, comparing shape complementary and electronic features of (Fig. 3) reference molecule with the Zinc database gave the important structural framework which realized the binding affinity and functional potency. The OpenEye ToolkitsROCS and EON algorithm were check for shape and electrostatic similarities respectively. During the shape and electrostatic screening, the molecules were screened depending on shape agreement with that of reference molecule. The obtained top HITS were used for comparative electrostatic correlations and their shape and electrostatic scores were counted for result analysis as shown in Fig. 3.
2.2 Molecular docking studies
2.2.1 Receptor grid generation
Receptor grid is set of active site residues where molecule should get bind and modulate the activity of enzyme of protein. ‘Receptor Grid Generation’ wizard in Glide was used for grid generation with default parameters for partial cut-off (0.25) and scaling factor (1.0) without any force. The site was determined by literature reported and centroid of the selected residues was used for generation of grid.
2.2.2 Docking
Ligprep utility in Glide module used for generation of various conformers, isomers of screened ligand. A highest number of 32 tautomers and stereoisomers were produced. OPLS 2005 force field was used for energy minimization and ligands were also desalted. The generated gird file with active site residues was used for docking of ligand using Glide software. The default 0.8 scaling factor (vdW) and 0.15 potential charge cut-off from were set. The analysis of results XP pose viewer utility was used by importing xpz file.
2.3 Molecular dynamics simulations
MD simulation study was used to check the ligand–protein complex at target site in physiological conditions. It was done by using Desmond V3 module running workstation. The docked protein–ligand complex was merged and used for generation for orthorhombic simulation box by the system builder utility of desmond module. It was built with Simple Point-Charge (SPC) explicit water model and the 10 Å distance was maintained between the solvent surfaces with protein surface. This solvated system was neutralized and 0.15 M concentration of physiological salt was maintained. This equilibrated system was used to MD simulations studies. The MD simulation was performed at 310.15 K temperatures over a 100 ns with constant temperature, constant pressure (1.0 bar). The CMS generated after the successful MD run was used for analysis of results using the simulation interaction analysis utility. The MD trajectory was written with 1000 frames during the entire simulation run but only initial protein backbone frames were aligned to understand the stability of ligand–protein complex. The RMSD, RMSF and interaction plots were used to understand the stability of complex.
2.4 SiteMap analysis
Single binding site region was evaluated by Sitemap tool of Schrödinger. All the molecules were separately tested for calculation of Dscore, hydrophilicity, hydrophobicity, H bond donor and acceptor properties. These computed properties are used to calculate the site score and Dscore. Depending on these scores, the druggability of target was also predicted. The binding site of Covid main protease (CYP1B1) was validated by comparing scores to all the molecules. (Halgren, 2007; Halgren, 2009).
2.5 In-silico ADME prediction
ADME drug likeliness properties are the crucial part of new drug development procedure as many molecules are withdrawn from the market due to their poor pharmacokinetic profiles. We used QikProp v3, Schrödinger, 2005 for prediction of ADME properties. To calculate the in-vitro and in-vivo drug likeliness properties, we considered various parameters such as H bond donor and acceptor, human oral absorption, molecular weight, Lipinski’s rule of five and predicted aqueous solubility as these are the important determinants of how the body going to react with any external molecule. If all these came under the identical or normal values, then we consider the identified HIT to be taken into further stages of drug development processes.
3 Results & discussion
HTVS based on the shape and electrostatic complementary of reference molecule (Ebselen) with Zinc13 (13, 195, 609) was performed. The best scoring top 15,000 molecules were further subjected to molecular modeling studies via XP mode in glide. The top 10 molecules were analyzed and top identifies molecule was used for molecular dynamics, SiteMap analysis and in-silico ADME calculation for drug likeliness properties (Fig. 2).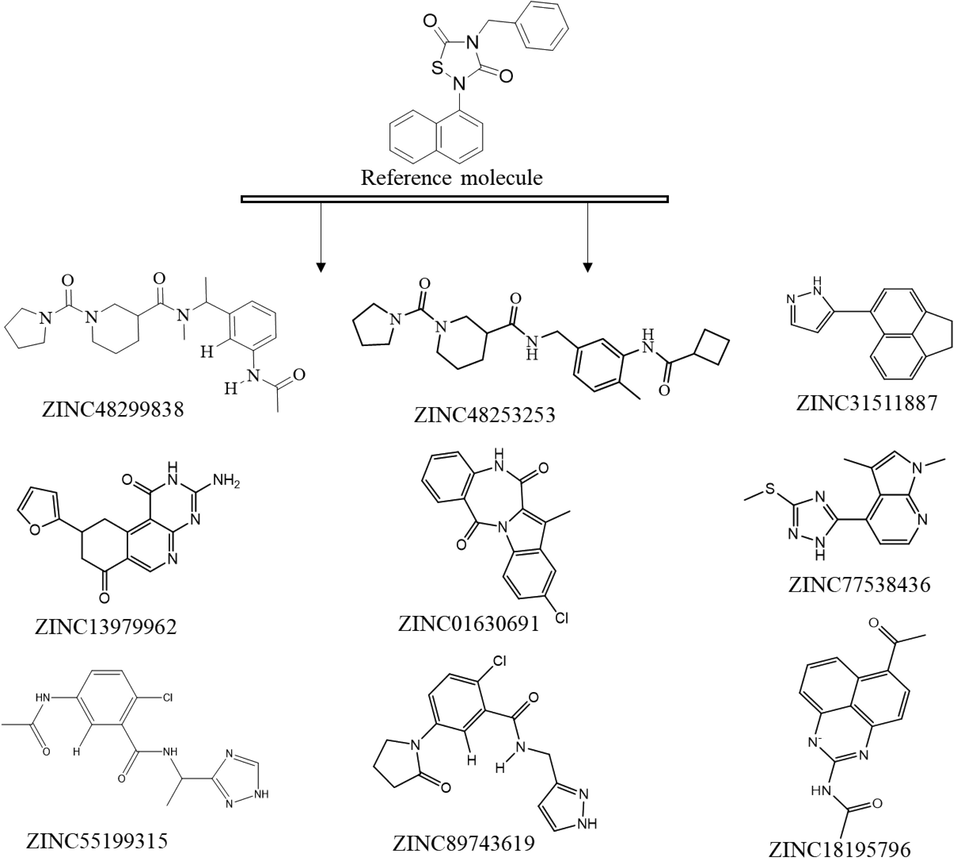
displayed the best scoring identified top 10 HITS via virtual screening.
3.1 Shape and electrostatic screening
The shape and electrostatic properties of any molecule are the primary topological determinants that govern the active fitting and binding at the catalytic site of protein. In the present study the top HIT was displayed the good structural similarities with reference molecule. Fig. 3B and C displayed the shape and electrostatic potential of the ZINC19973962 compared to reference molecule whereas Fig. 3A showed the complete overlapping of reference molecule with identified HIT. The Tanimoto shape and electrostaticscores of ZINC19973962 with reference molecule was found to be 0.667 and 0.22 respectively. The identified top HIT ZINC19973962 was chemically different from the reference molecule but it displayed the good shape and electrostatic complementary with reference molecule and these values suggested that ZINC19973962 could actively bind with the amino acid residues present at protein catalytic site. Based on the active binding and fitting of HITs at the active pocket it was considered that these molecules would be potential inhibitors of COVID main protease enzyme. But generated in-silico data do not confirm the actual in vitro or in vivo biological activity. The identified best scoring top 1500 molecules from preliminary shape and electrostatic screening was led to further in silico studies. Molecular docking studies of these molecules were performed against the COVID main protease enzyme using 6LU7 pdb file.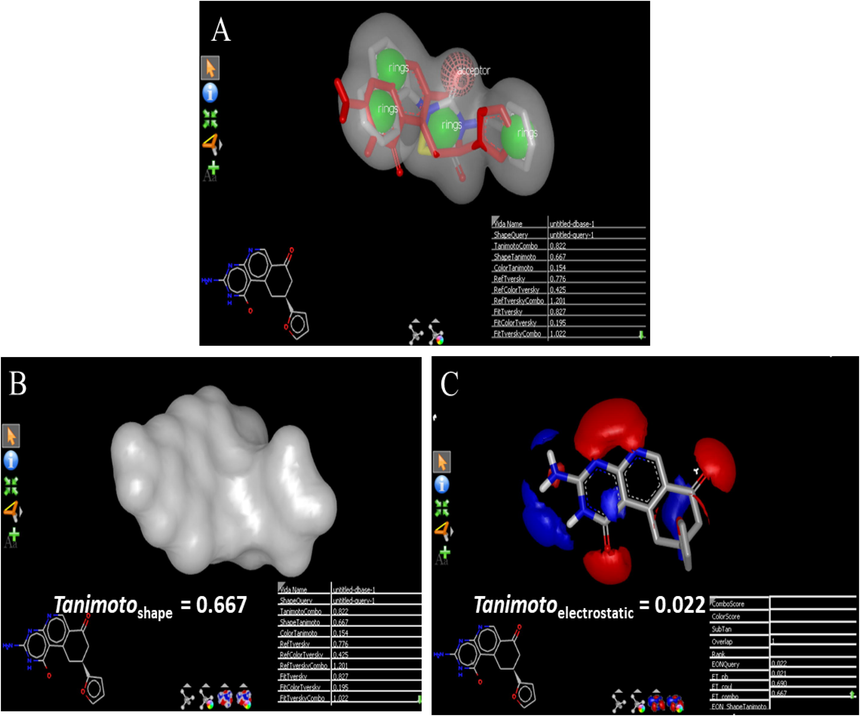
A: Overlapping of identified HITS ZINC19973962 (red color) on reference molecule (silver color) B: The display of shape coefficient of ZINC19973962 in neutral form with that of reference molecule. C: Electrostatic potential of ZINC19973962 red color & blue color represents electronegative and electronegative areas respectively.
3.2 Molecular modeling studies
Before the run of docking studies, the software needs to be validated and it was simply done by extracting out the internal ligand and again redocking at its actual place without changing its states or generating any conformers. The obtained root means square deviation value 3.365 Å suggested the software is ready to use for study. The top 1500 HITs obtained from the preliminary screening were docked at the active site of COVID main protease. The key interaction between molecule and active site residues was determined by extra precision docking. From the docking studies compound with id ZINC19973962 displayed the good docking score of −7.9 and was identified as the best scoring molecule from the top 1500 molecules. The negative sign in the score indicates about the energy release of exothermic reaction occurring when the ligand is going combined with protein. The Fig. 4A represents the molecular surface diagram of docked drug-protein complex. Fig. 4B displayed the overlay of reference molecule and ZINC19973962 with key interactions present Fig. 4C for reference and identified HIT. The analysis of docking results revealed that the ZINC19973962 and reference molecule bound with the active pocket residues in similar fashion. The interactions were also found to be quite similar between these two molecules. It was hypothesized that these electrostatic interactions would lead to the same biological behavior of ZINC19973962. However, it requires the experimental data to prove the proposed hypothesis. The molecule ZINC19973962 displayed the hydrophobic interaction with Phe140, Leu141, Cyc145, Met165, Val42, Met49 and Leu27. These interactions were stabilized by 3 polar H bond formed with Thr26, Gly143 and Gln189. The polar interaction with His164, Gln189, Thr25, Thr26, Asg142 and His41 was also observed as shown in the Table 1. The comparative analysis of reference and ZINC19973962 interactions was shown the maximum active site residues were conserved in both the docked drug-protein complex. These interactions determine the stability of drug-protein complex which give an idea about the potential biological activity of any molecule.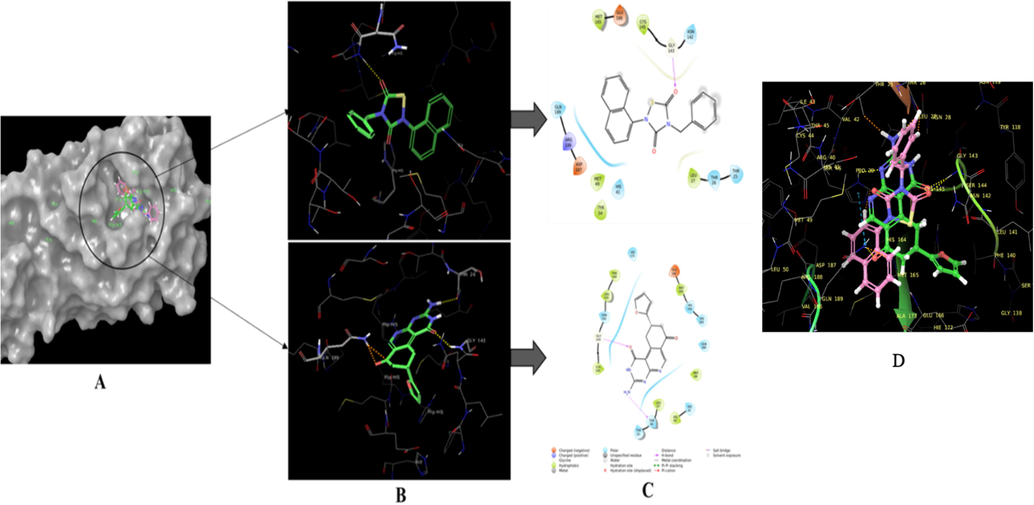
B:3D interactive diagram of ZINC19973962 at the active site of Dengue protease enzyme. The molecule displayed the two H Bonding and hydrophobic interaction with active site residues considered as key interaction for possible inhibitory activity of corona virus; C: 2D diagram of identified HIT at the active site of corona virus (PDB id 6LU7). D: Superimposed structure of reference (pink color) and identified molecules (Green color) at the active pocket of COVID main protease.
Parameter
Docking Score
Interaction
Residues
COV-Reference
−4.605
H-Bond
GLY-143
Hydrophobic
LEU-141, CYS-145, TYR-54, MET-165, PRO-52, MET-49, LEU-27, VAL-42, CYS-44
Zinc3962
−7.964
H-Bond
HIS-41, GLU-166
Hydrophobic
PHE-140, LEU-141, MET-165, MET-49, VAL-42, LEU-27, CYS-44, CYS-145
3.3 Molecular dynamics studies
The two combined methods (MM/PBSA) Poisson-Boltzmann surface area continuum solvation and molecular mechanical energies have been proven for the prediction of accurate binding free energies and its usefulness in drug discovery and development. (Genheden and Ryde, 2015; Siddique et al., 2021) In present study the same hypothesis was used to check the accurate binding of Zinc at the active site of COVID main protease. The complex stability was predicted based on the RMSD and RMSF calculation during the entire run of MD trajectories. Fig. 5 displayed the RMSD value of Zinc protein complex over 100 ns run. The complex was stable over the entire 100 ns run. The energies involved in the ligand–protein complex were mainly H bonding, hydrophobic and Venderwal interactions as shown in Fig. 7. The active site residues Gly143, Ser144, Leu141 & Gln189 shown the H bonds and remain conserved as they were also responsible for the maintaining the stability during the docking. The additional π-π interactions with His163 and hydrophobic interactions with Leu141& Met165 suggested the good stability of the complex. The RMSF value thus obtained (Fig. 6) was suggested about the very low fluctuations of molecule form the protein over entire MD trajectories (see Fig. 8).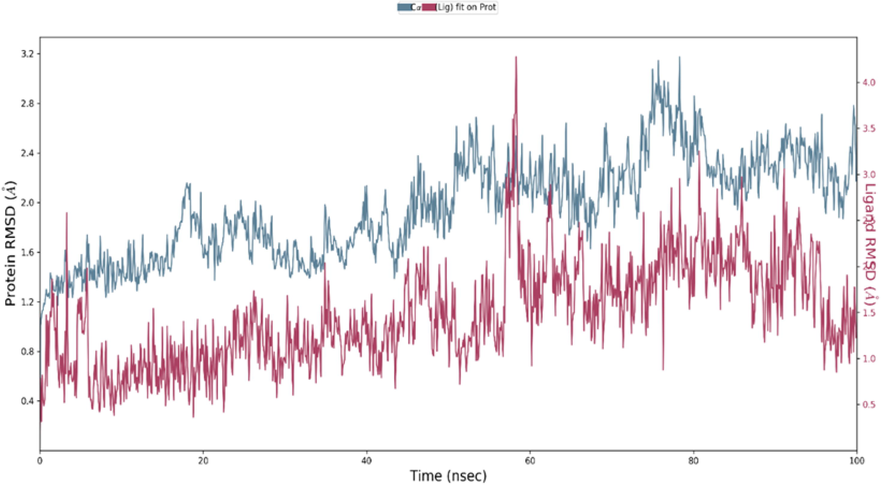
Structural changes over the entire trajectory run of 100 ns upon binding of ligand at the active site of Corona virus (PDB id: 6LU7).
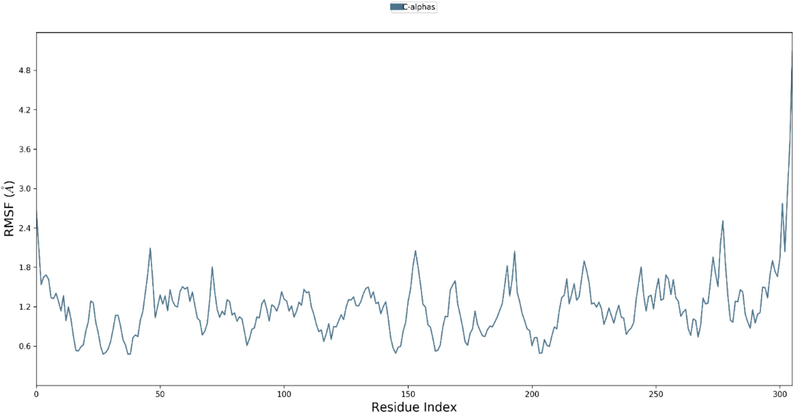
Fluctuations of residues over the entire trajectory run of 100 ns upon binding of ligand at the active site of Corona virus (PDB id: 6LU7).
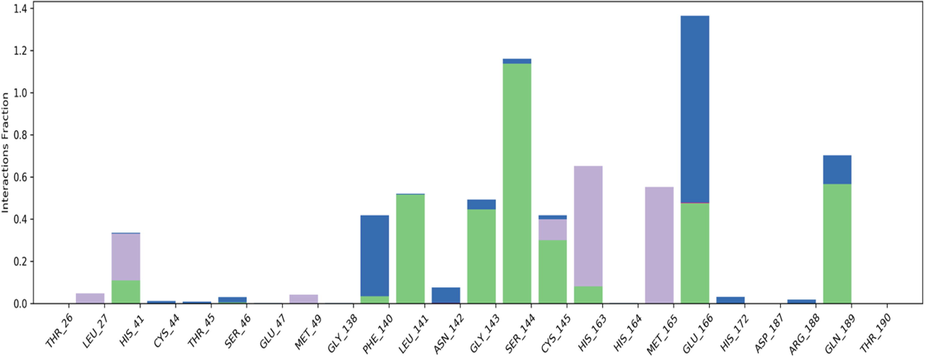
Different interactions in the molecular recognition of the active site of Corona virus (PDB id: 6LU7) (Green color: H Bond, magenta color: hydrophobic interactions, Blue color: water bridges).
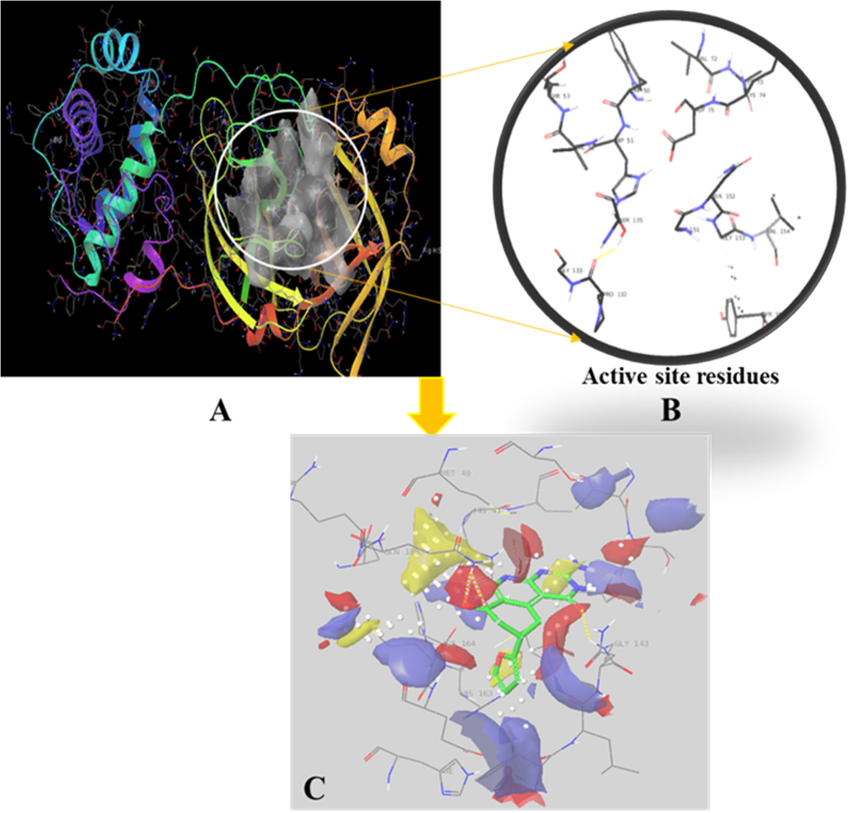
Binding mode analysis of ZINC19973962 at active site of Dengue protease (PDB id 2FOM). The various color contours represent the presence of active site residue that could possibly had the different interactions such as blue and red color displayed H-bond donor and H-bond acceptor whereas yellow color shows the hydrophobic interaction.
3.4 Site map analysis
The reference and ZINC19973962 molecules were used for SiteMap analysis to check the drug ability of identified HIT. It was considered that identical scores and values could assure the potential biological activity of identified HIT. The drug ability of molecule can be assessed by various parameters involved in the Site Map analysis like D-score, sites score, hydrophobic, hydrophilic, H-bond donor and acceptor etc. The Fig. 4b displayed the active site residues and 3C indicates the site map analysis of HIT. The different colored contours represent various parameter of SiteMap analysis as described in figure legend. The obtained values for both compounds were identical and all were very close proximity to average values as listed in Table 2. The obtained values of identified HIT and reference molecule were found to be similar hence we can say that the ZINC19973962 would be the promising druggable candidate against the COVID main protease and can be act as a lead molecule for development of anti COVID molecule.
Site Properties
Score (reference)
ZINC19973962
Sitescore
0.28
1.016
Size
97
121
DScore
1.070
1.065
Site Vol
262.7
255.5
Exposure
0.571
0.531
Enclosure
0.700
0.664
Contacts
0.964
0.901
Phobic
0.936
0.858
Philic
0.845
0.838
Donor/Acceptor
1.097
1.093
3.5 ADME properties
The pharmacokinetic properties of any drug candidate are a very crucial aspect that should be taken in consideration in the drug discovery and development process. The reason behind this is many promising and strong druggable candidate was failed in the later stages of drug discovery processes due to poor pharmacokinetic profiling. This causes huge loss of time and cost of the drug development process that ultimately add the cost of the project. Hence, if we can check or predict the in-silico pharmacokinetic properties of HIT or lead molecule which is supposed to be taken into further stages of drug discovery process to avoid the later stage failure of drug discovery process. Here we also checked the in-silico pharmacokinetic profile of identified HIT i.e. ZINC19973962. Different physicochemical properties that are going to play a major role in human body or behavior of body onto the drug candidate were determined by using qikprop utility of Schrodinger tool. The various physicochemical parameters of drug candidate like molecular weight, LogP, metabolism, logBB human oral absorption and polar surface area was predicted. For a molecule to be orally active, the violation of Lipniski’s rule of five was checked. The results obtained were listed in Table 3. The obtained values were suggested that the molecule was not violated the Lipniski’s rule of five hence there is chance that molecule may be orally active. The obtained LogBB value indicated that the molecule will not cross the blood brain barrier and not be able to produce any CNS related toxicities. Thus, the obtained values suggested the fate of ZINC19973962 i.e. absorption, distribution, metabolism and excretion. By this it was postulated that the identified HIT can absorb orally and reached to specific target in unaltered form to produce the desired effect.
ADME Properties
Reference
ZINC19973962
Molecular Weight
334.3
296.2
HB Donor
0
3
HB Acceptor
3
7
LogPO/W
4.225
0.298
logBB
−0.141
−1.689
#Metabolism
1
5
% Human oral Absorption
100
60.404
PSA
125.94
56.14
Ro5 Violation
0
0
Ro3 Violation
0
0
4 Conclusion
Many promising drug candidates were failed in later stages of the drug development due to lesser credibility of in-silico data. Hence, the more validated in-silico data is needed to consider any molecule as a suitable lead that can be taken into further stages of drug development. In the present study combined ligand and protein based physicochemical parameters were considered for HTVS based design and discovery of small molecule inhibitors of covid-19 main protease enzyme. Then top scoring 15,000 molecules were subjected to molecular docking and dynamics simulation studies. Considering the fact that these two-sided physicochemical aspects and validated in-silico results and chances of lesser failure of later stage drug development process. The results obtained based on these studies were analyzed and ZINC19973962 was identified as top scoring HIT after the successful completion of computational studies. Based on topological similarity indices with reference molecule, identical binding pattern, stability of ligand–protein and drug ability the identified molecule would be the promising HIT and can used as a lead molecule for further development of small molecular inhibitors against SARS COVID main protease enzyme.
5 Availability of data and materials
The data generated or analyzed in this article are online publicly available without request.
6 Authors' contributions
Nada H. Aljarba was performed the electrostatic study; Saad Alkahtani was performed the molecular docking studies. Mashael Bin-Meferij was acquired and analyzed the data. Nada H. Aljarba and Mashael Bin-Meferij were performed dynamics simulations and SiteMap analysis. Md Saquib Hasnain and Saad Alkahtani were involved in the conception and design of the study, data interpretation, and critically revised the manuscript.
Acknowledgement
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number (PNU-DRI-Targeted-20-031).
Conflict of interest
The authors declare no conflicts of interest associated with this manuscript.
References
- Cardiac involvement in COVID-19 patients: risk factors, predictors, and complications: A review. J. Card. Surg.. 2020;35(6):1302-1305.
- [Google Scholar]
- Development of a machine-learning model to predict Gibbs free energy of binding for protein-ligand complexes. Biophys. Chem.. 2018;240:63-69.
- [Google Scholar]
- A screen of the NIH Clinical Collection small molecule library identifies potential anti-coronavirus drugs. Antiviral Res.. 2015;114:1-10.
- [Google Scholar]
- A 55-day-old female infant infected with 2019 novel coronavirus disease: presenting with pneumonia, liver injury, and heart damage. J. Infect. Dis.. 2020;221(11):1775-1781.
- [Google Scholar]
- Development of machine learning models to predict inhibition of 3-dehydroquinate dehydratase. Chem. Biol. Drug Des.. 2018;92(2):1468-1474.
- [Google Scholar]
- Kobophenol a inhibits binding of host ace2 receptor with spike rbd domain of sars-cov-2, a lead compound for blocking covid-19. J. Phys. Chem. Lett.. 2021;12(7):1793-1802.
- [Google Scholar]
- The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov.. 2015;10(5):449-461.
- [Google Scholar]
- New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des.. 2007;69(2):146-148.
- [Google Scholar]
- Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model.. 2009;49(2):377-389.
- [Google Scholar]
- ZINC− a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model.. 2005;45(1):177-182.
- [Google Scholar]
- Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289-293.
- [Google Scholar]
- Upper airway symptoms in coronavirus disease 2019 (COVID-19) Am. J. Otolaryngol.. 2020;41(3):102474.
- [CrossRef] [Google Scholar]
- Coronavirus: covid-19 has killed more people than SARS and MERS combined, despite lower case fatality rate. Br. Med. J. Publishing Group 2020
- [Google Scholar]
- Geriatric care during public health emergencies: lessons learned from novel corona virus disease (COVID-19) pandemic. J. Gerontol. Social Work. 2020;63(4):257-258.
- [Google Scholar]
- SAnDReS a computational tool for statistical analysis of docking results and development of scoring functions. Comb. Chem. High Throughput Screening. 2016;19(10)
- [CrossRef] [Google Scholar]
- OEChem, T. “version 2.0. 0. OpenEye Scientific Software, Santa Fe, NM.” There is no corresponding record for this reference.
- Rapid shape-based ligand alignment and virtual screening method based on atom/feature-pair similarities and volume overlap scoring. J. Chem. Inf. Model.. 2011;51(10):2455-2466.
- [Google Scholar]
- Schrodinger Software Suite. New York: Schrödinger LLC; 2011.
- Comparative Computational Studies on Selective CytochromeP450 1B1 Inhibitors. Int. J. Bioautomation. 2020;24(3):213.
- [Google Scholar]
- Comparative shape and electrostatic study of highly potent and selective CYP1B1 inhibitor: assessment of active site of CYP1B1 by Binding Mode Analysis Using Site Map Tool. Indian J. Pharmaceut. Educ. Res.. 2018;52(1):159-165.
- [Google Scholar]
- Non-carboxylic acid inhibitors of aldose reductase based on N-substituted thiazolidinedione derivatives. Eur. J. Med. Chem.. 2021;113630
- [Google Scholar]
- The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19)—China, 2020. China CDC Weekly. 2020;2(8):113-122.
- [Google Scholar]
- An update review of emerging small-molecule therapeutic options for COVID-19. Biomed. Pharmacother.. 2021;111313
- [Google Scholar]
- Concomitant neurological symptoms observed in a patient diagnosed with coronavirus disease 2019. J. Med. Virol.. 2020;92(10):1782-1784.
- [Google Scholar]
- Coronavirus disease 2019 (COVID-19) in children: prevalence, diagnosis, clinical symptoms, and treatment. Int. J. General Med.. 2020;13:477.
- [Google Scholar]
- Advances in the relationship between coronavirus infection and cardiovascular diseases. Biomed. Pharmacother.. 2020;127:110230
- [Google Scholar]
- Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054-1062.
- [Google Scholar]
- Zumla, A. and M. S. Niederman (2020). “The explosive epidemic outbreak of novel coronavirus disease 2019 (COVID-19) and the persistent threat of respiratory tract infectious diseases to global health security.” Curr. Opin. Pulmonary Med.




