

Translate this page into:
Computational studies on potential new anti-Covid-19 agents with a multi-target mode of action
⁎Corresponding authors. ranjank_mohapatra@yahoo.com (Ranjan K. Mohapatra), azam_res@yahoo.com (Mohammad Azam), ajaz.alam2@gmail.com (Azaj Ansari), mraval@yahoo.com (Mukesh K. Raval)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A compound that could inhibit multiple targets associated with SARS-CoV-2 infection would prove to be a drug of choice against the virus. Human receptor-ACE2, receptor binding domain (RBD) of SARS-CoV-2 S-protein, Papain-like protein of SARS-CoV-2 (PLpro), reverse transcriptase of SARS-CoV-2 (RdRp) were chosen for in silico study. A set of previously synthesized compounds (1–5) were docked into the active sites of the targets. Based on the docking score, ligand efficiency, binding free energy, and dissociation constants for a definite conformational position of the ligand, inhibitory potentials of the compounds were measured. The stability of the protein–ligand (P-L) complex was validated in silico by using molecular dynamics simulations using the YASARA suit. Moreover, the pharmacokinetic properties, FMO and NBO analysis were performed for ranking the potentiality of the compounds as drug. The geometry optimizations and electronic structures were investigated using DFT. As per the study, compound-5 has the best binding affinity against all four targets. Moreover, compounds 1, 3 and 5 are less toxic and can be considered for oral consumption.
Keywords
Molecular electrostatic potential
Frontiers molecular orbital
Natural bond orbital
Molecular docking
Molecular dynamics
Pharmacokinetics
Drug-likeness prediction
SARS-CoV-2

1 Introduction
Covid-19 caused by SARS-CoV-2, was first reported from China on December 2019. Since then, the virus has been rapidly spreading around the whole world and has become a global pandemic (Mohapatra et al., 2020; Mohapatra et al., 2021a, 2021b). Initial infection control measures (e.g. quarantine, lockdown, and the use of masks) implemented to tackle the high transmission rate of the virus proved beneficial. These steps, however, are not sufficient on their own to control the spread of this rapidly-mutating virus. SARS-CoV-2 variants, including B.1.351 (SA), B.1.1.7 (UK), B.1.427, B.1.429 (USA), B.1.617.2 (India), and B.1.1.529 (South Africa, Botswana) are mainly transmitted through respiratory droplets (Sah et al., 2021; Mohapatra et al., 2022a). However, hospital-associated, airborne and fecal-oral transmissions and transmissions from contaminated objects/surfaces/fomites were also reported (Morawska and Cao, 2020; Mohapatra et al., 2021c). SARS-CoV-2 and its variants of concern infects the respiratory tract, liver, kidney, gut, heart, nervous system and may lead to multiple organ damage. A number of countries have already deployed a rapid vaccination programmes with their own vaccines such as AstraZeneca (UK), Covishield/Covaxin (India), Pfizer/Comirnaty (Germany), Moderna/Novavax (USA), CanSino/Sinovac/BBIBP-CorV (China) and Sputnik V (Russia) (Madkaikar et al., 2021; Bhuyan, 2021). However, the recently emerged Omicron variant reduces the protective effect of vaccination and showed immune-evasive property and it is currently the dominant strain of SARS-CoV-2 around the globe (Mohapatra et al., 2022b). It remains to be seen if such vaccines show efficacy against all potential SARS-CoV-2 variants (Callaway, 2021). The recent discovery of two new antivirals targeting the SARS-CoV-2 virus represents a promising strategy to control this pandemic (Mohapatra et al., 2022a; WHO, 2022; Soriano et al., 2022) and it is essential that more efforts are focused on developing such drugs. Computational approaches have been extensively used to date to identify new chemical scaffolds that could potentially inhibit key biological targets involved in the pathogenicity of SARS-CoV-2 (Mohapatra et al., 2021d; Sahu et al., 2021). In the present study, we carried out frontiers molecular orbital (FMO), natural bond orbital (NBO), molecular electrostatic potential (MEP) mapping, molecular docking, molecular dynamics (MD) simulation, pharmacokinetics and drug-likeness prediction studies on five compounds (1–5) previously synthesized by our group (Sahu et al., 2021) (Fig. S1) in an effort to evaluate their interactions, and drug-like potential, with the human ACE2 receptor, as well as the RBD spike protein, the papain-like (PL) protein and the reverse transcriptase of SARS-CoV-2.
2 Methods
2.1 Molecular electrostatic potential (MEP) mapping studies
Compounds 1–5 were prepared using the Gaussian 09 software package as previously reported (Mohapatra et al., 2021e). Calculations were performed using the B3LYP-D2 (Grimme, 2006)/6-31G** basis set in the Gaussian 16 program suite (Fig. S2) (Marenich et al., 2009). The electrostatic potential distribution for each molecule was measured by using the following equation (Takayanagi et al., 1996); where, ρ explains both the nuclear and electronic charge of a molecule, with the integration performed over the molecular volume of the same molecule, and r’ is the atomic position relative to same origin.
All molecular electrostatic potentials were colour-graded, with red and blue corresponding to the most negative and most positive potential, respectively, and green and yellow corresponding to a potential halfway between the two extremes.
2.2 Frontier molecular orbitals (FMO) studies
The TD-DFT (time-dependent density functional theory) calculations were reported by using Gaussian 09 platform (Sahu et al., 2021) with B3LYP method and 6–31 + G (d, p) basis set to identify the chemical reactivity of compounds, by calculating EHOMO and ELUMO energies and the [EHOMO - ELUMO] energy difference (band gap). This also helped to characterise global reactivity descriptors including chemical hardness (η), electrophilicity index (ω), electronic chemical potential (μ), and electronegativity (χ) for all compounds (Sahu et al., 2021).
2.3 Natural bond orbital (NBO) studies
Natural bond orbital (NBO) analysis was conducted in order to explore the bonding aspects related to compounds 1–5, using a set of anti-bond, localized bond, and Rydberg extra valence orbitals (Mohamed et al., 2018; Monika et al., 2021). NBO studies were used to analyse donor–acceptor interactions for all compounds using a second-order perturbation approach (Fock matrix). The strength of the stabilization energy E(2) associated with electron delocalisation among donor (i) and acceptor (j) NBO was deduced using the following equation: where, qi denotes donor orbital occupancy, F(i, j) implies the off diagonal NBO Fock matrix element and εj and εi are the diagonal elements, High stabilization energy E(2) is indicative of a strong interaction between the electrons of the acceptor–donor NBO, suggesting the presence of a higher conjugation system (Sumrra et al., 2018).
2.4 Molecular docking study
The 3D crystal structures of the human ACE2 receptor (PDB ID:1R42), the SARS-CoV-2 RBD S-protein (PDB ID:6MOJ Chain B), Papain-Like protein (PLpro) (PDB ID:7CJD Chain A) and reverse transcriptase (RdRp) (PDB ID:6YYT Chain A) were obtained from Protein Data Bank (https://www.rcsb.org/pdb) and used as biological targets for the molecular docking study (Xu et al., 2020; Xu et al., 2004; Gao et al., 2021; Hillen et al., 2020). All proteins were prepared by removing water molecules, ions, and heteroatoms, with grid maps created around the respective binding sites using AutoDock Tools and Auto Grid programs. Prior to the docking, the 2D and 3D structures of all ligands were drawn using HyperChem v.8.0 and optimized geometrically using the MM + force field (Hocquet and Langgård, 1998). The ligands and the protein active site residues were allowed to be flexible during the docking process, which was conducted using AutoDock Vina (Trott and Olson, 2010). The active site was chosen around 5 Å of the ligan-protein interaction region. The docking grid dimension was taken to be 25 × 25 × 25 Å and the default grid point is taken to be 0.375 Å. The best pose (with lowest docking score) was used for further studies. All protein–ligand interactions were visualized using Biovia Discovery Studio Visualizer v.16.1.0. Ligand efficiencies (LE) were calculated for each compound (Hopkins et al., 2014) by using the following equation; where, HA is the number of heavy (non-hydrogen) atoms, pKd is the negative logarithm to the base 10 of the dissociation constant and pIC50 is the negative logarithm to the base 10 of the half-maximal inhibitory concentration.
2.4.1 Calculation of binding free energy (ΔGbind) by MM/PBSA (GBSA)
Protein – ligand binding energy is calculated by MM/PB(GB)SA methods using farPPI- a webserver (Wang et al., 2019). The formula to compute the MM/PB(GB)SA binding free energy is given by where, ΔGbind is the difference between bound-state (Gcomplex) and free-state (Greceptor + Gligand) and called total binding free energy. Using the AmberTools17, seven different MM/PB(GB)SA calculation procedures (PB1, PB3, PB4, GB1, GB2, GB5, and GB6) are done.
2.5 Molecular dynamics (MD) simulation studies
With the help of YASARA molecular modeling v.18.4.24, the stability of the protein–ligand (P-L) complexes was determined (Krieger and Vriend, 2014). The P-L complexes were put in a cubic simulation box (density 0.997 g L−1, TIP3P water model, NaCl 0.9 as counter ions) in aqueous medium, keeping at least a 5 Å buffer space around each complex. The systems were energy-minimized using the AMBER14 force field with a steepest gradient approach (100 cycles). The MD simulation was carried out for 40 ns with a frame capture every 25 ps and trajectories were generated from the data captured.
2.6 Pharmacokinetic, drug-likeness and toxicity prediction studies
The pharmacokinetic properties of compounds 1–5 were predicted using the online admetSAR v1/v2 and SwissADME software. The Lipinski’s properties were tabulated by using physicochemical data obtained from PubChem database. Drug-likeness filters (Ghose, Veber, Muegge and Egan) were also determined to enhance the predictions (Mohapatra et al., 2021b). Toxicity of the compounds was predicted by using ProTox II online server (Banerjee et al., 2018).
3 Results and discussion
3.1 MEP mapping studies
The molecular electrostatic potential maps obtained for compound 1–5 are shown in Fig. 1. All compounds displayed reactive centres with the most negative potential predicted at oxygen atoms. Compounds 1–3 showed a more negative electrostatic potential (with darker red oxygen atoms) compared to compounds 4–5. MEP mapping studies are used to predict the chemical reactivity of molecules, with high electron density regions (prone to electrophilic attack) and low electron density regions (prone to nucleophilic attack) showing a negative and a positive electrostatic potential, respectively (Yadav et al., 2021).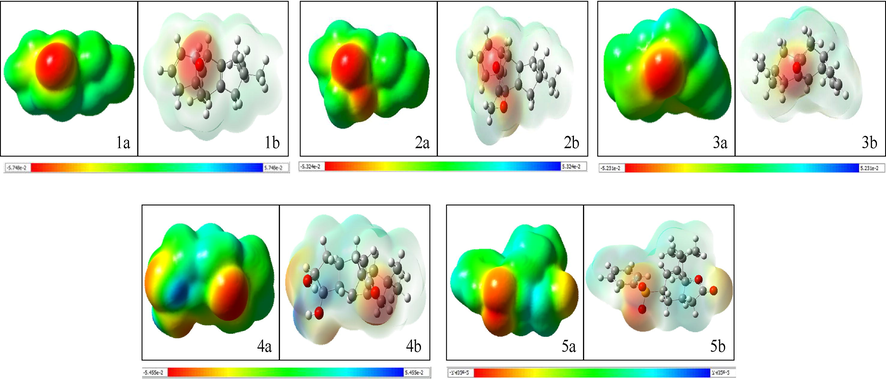
Molecular electrostatic potential (MEP) maps obtained for compounds 1–5 (a: denotes transparent display and b denotes solid display).
3.2 FMO studies
The electronic transitions taking place from the HOMO to LUMO orbitals of compounds 1–5 are shown in Fig. 2. Compound 1 displayed the highest EHOMO-ELUMO gap (5.349 eV) in comparison with 2 and 5 which had the lowest gaps. As a lower transition gap indicates a higher reactivity/lower stability (Choudhary et al., 2013), the energy difference values obtained suggest that compounds 2 and 5 show the greatest reactivity amongst all compounds. The values obtained for the electronic chemical potential (μ), electronegativity (χ), chemical hardness (η), and electrophilicity index (ω) are presented in Table 1. Compound 1 showed the highest value for chemical hardness and electrophilicity, while compound 5 showed the highest value for chemical potential and electronegativity. These results confirmed that compound 1 was the most stable amongst all compounds as high chemical hardness and low electrophilicity are directly linked to high stability (Choudhary et al., 2013).![[EHOMO − ELUMO] energy band gaps obtained for compounds 1–5 (value in electron volt).](/content/185/2022/34/5/img/10.1016_j.jksus.2022.102086-fig2.png)
[EHOMO − ELUMO] energy band gaps obtained for compounds 1–5 (value in electron volt).
Compounds
EHOMO (eV)
ELUMO (eV)
ELUMO-EHOMO (eV)
Chemical hardness (ɳ)
Chemical potential (μ)
Electronegativity (χ)
Electrophilicity (ω)
1
−6.046
−0.696
5.349
2.326
−6.394
6.394
3.830
2
−6.097
−1.118
4.979
1.930
−6.657
6.657
2.004
3
−6.195
−1.112
5.083
1.985
−6.752
6.752
2.193
4
−6.405
−1.161
5.243
2.040
−6.986
6.986
1.946
5
−6.783
−1.817
4.966
1.574
−7.692
7.692
3.115
3.3 NBO studies
The NBO plots corresponding to the carbonyl (C⚌O) bond of compound 1–5 are shown in Fig. 3. The examination of the NBO plot obtained for compound 1 revealed that pz orbital of both the carbon and the oxygen atoms were involved in σ-bond formation with the carbon and oxygen atom showing a 34.95 % and 65.05 % orbital contribution, respectively. The py orbital is further involved in the formation of a π-bond with the carbon and the oxygen atom showing a 32.71 % and 67.29 % orbital contribution, respectively. The orbital contributions obtained for all compounds are listed in Table 2, while the corresponding natural electronic configurations of all carbonyl bonds are listed in Table 3.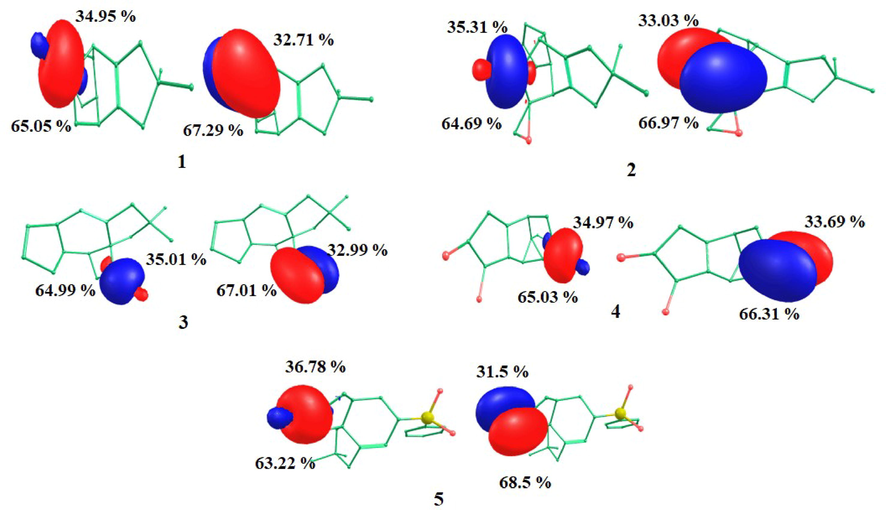
Natural Bond Orbital (NBO) plots obtained for compounds 1–5.
Compounds
σ bond orbital contribution (%)
π bond orbital contribution (%)
Carbon
Oxygen
Carbon
Oxygen
1
34.95
65.05
32.71
67.29
2
35.31
64.69
33.03
66.97
3
35.01
64.99
32.99
67.01
4
34.97
65.03
33.69
66.31
5
36.78
63.22
31.50
68.50
Compounds
Natural electronic configuration
Carbon
Oxygen
1
[core]2S(0.45)2p(1.23) 3p(0.01)
[core]2S(0.85)2p(2.41)
2
[core]2S(0.45)2p(1.23) 3p(0.01)
[core]2S(0.85)2p(2.40)
3
[core]2S(0.44)2p(1.24) 3p(0.01)
[core]2S(0.85)2p(2.41)
4
[core]2S(0.44)2p(1.25) 3p(0.01)
[core]2S(0.85)2p(2.41)
5
[core]2S(0.41)2p(1.20)
[core]2S(0.85)2p(2.41) 3p(0.01)
The total Lewis and non-Lewis contributions for compound 1 and all other compounds (2–5) are illustrated in Fig. 3 and Fig. S3, respectively. Compound 1 exhibited a Lewis type structure (99.931%) with a non-Lewis (Valence-2.12% and Rydberg-0.16%) contribution. Compound 2 had a Lewis type structure (98.905%) with a non-Lewis (Valence-0.931% and Rydberg-0.164%) contribution. Compound 3 exhibited a Lewis type structure (98.928%) with a non-Lewis (Valence-0.917% and Rydberg-0.155%) contribution. Compound 4 had a Lewis type structure (98.959%) with a non-Lewis (Valence-0.888% and Rydberg-0.154%) contribution. Compound 5 exhibited a Lewis type structure (98.144%) with a non-Lewis (Valence-1.6215% and Rydberg-0.2337%) contribution.
The electron donor–acceptor orbitals and interaction stabilization energy values obtained for all compounds are illustrated in Table 4. Strong intramolecular interactions were predicted to occur in all compounds. Compound 1–5 were stabilized predominantly by n to σ* transitions. This included n(O17) → σ*(C12-C16), n(O17) → σ*(C13-C14) (10.80 kcal/mol each) for 1; n (O17) → σ*(C12-C13), n(O17) → σ*(C13-C14) (10.46 and 11.94 kcal/mol, respectively) for 2; n(O15) → σ*(C4-C13), n(O15) → σ*(C13-C14) (11.66 and 10.46 kcal/mol, respectively) for 3; n(O14) → σ*(C1-C2), n(O14) → σ*(C2-C3) (10.26 and 11.35 kcal/mol, respectively) for 4; n(O1) → σ*(C2-O13), n(O13) → σ*(O1-C2), n(O13) → σ*(C2-C3) (22.26, 16.04, 9.08 kcal/mol, respectively) for 5. The latter showed additional π(C–C) and antibonding π* interactions, including π(C26-C28) → π*(C25-C27), π(C26-C28) → π(C29-C30), πC29-C30) → π*(C26-C28), πC25-C27) → π*(C29-C30) (12.07, 9.99, 10.03, 11.3 kcal/mol, respectively). This indicated that it showed better conjugation and greater stability compared to other compounds. These π → π* and n → π*interactions have remarkable impact on crystal packing and charge transfer interactions (Sumrra et al., 2018). The bending angles corresponding to C⚌O bonds (i.e. angle of deviation from the direction of the line connecting the two nuclei centres) for all compounds are reported in Table S1.
Donor NBO(i)
Acceptor NBO(j)
E(2a)kcal/mol
E(j)-E(i)b(a.u.)
F(i,j)c(a.u.)
Compound 1
n(O17)
σ*(C12-C16)
10.80
0.60
0.103
n(O17)
σ*(C13-C14)
10.80
0.60
0.103
σ(C13-O17)
σ* (C11-O14)
0.33
1.38
0.027
π(C13-O17) *
σ*(C4-C12)
0.57
0.75
0.026
π(C13-C17)
σ*(C12-C16)
0.74
0.68
0.028
π(C13-O17)
σ *(C14-H35)
0.58
0.71
0.026
π (C13-O17)
σ*(C14-H37)
0.59
0.72
0.026
Compound 2
n(O17)
σ*(C12-C13)
10.46
0.60
0.102
n(O17)
σ*(C13-C14)
11.94
0.61
0.108
σ(C13-O17)
σ *(C11-O14)
0.38
1.38
0.029
π(C13-O17)
σ *(C4-C12)
0.56
0.75
0.026
π(C13-O17)
σ*(C14-C36)
0.32
0.66
0.018
π(C13-O17)
σ *(C14-O37)
1.44
0.63
0.038
Compound 3
n(O15)
σ*(C4-C13)
11.66
0.62
0.108
n(O15)
σ*(C13-C14)
10.46
0.63
0.104
π(C13-O15)
σ*(C3-C4)
0.70
0.71
0.028
π (C13-O15)
σ*(C4-C5)
0.73
0.76
0.030
π(C13-O15)
σ*(C14-H18)
0.62
0.72
0.027
π(C13-O15)
σ*(C14-H19)
0.78
0.71
0.030
Compound 4
n(O14)
σ*(C1-C2)
10.26
0.62
0.102
n(O14)
σ*(C2-C3)
11.35
0.63
0.107
σ(C2-O14)
σ*(C1-C5)
0.30
1.41
0.026
σ(C2-O14)
σ *C3-C4
0.26
1.34
0.024
π(C2-O14)
σ*(C1 H21)
0.63
0.71
0.027
π(C2-O14)
σ*(C1-H22)
0.76
0.71
0.029
π(C2-O14)
σ*(C3-C7)
1.51
0.66
0.040
π(C2-O14)
σ*(C3-C10)
0.43
0.71
0.022
Compound 5
π(C26-C28)
π*(C25-C27)
12.07
0.26
0.071
π*(C29-C30)
9.99
0.27
0.066
π(C29-C30)
π*(C26-C28)
10.03
0.28
0.067
π(C25-C27)
π*(C29-C30
11.03
0.28
0.070
n(O1)
π*(C2-O13)
22.56
0.34
0.111
n(O13)
π*(O1-C2)
16.04
0.60
0.125
n(O13)
π*C2-C3
9.08
0.60
0.095
σ(C2-O13)
σ*(O1-C5)
0.97
1.36
0.046
σ(C2-O13)
σ*(C2-C3)
0.32
1.41
0.027
π(C2-O13)
π*(C2-O13)
0.45
0.38
0.017
π (C2-O13)
σ*(C3-H45)
0.44
0.70
0.022
π(C2-O13)
σ*(C3-H46)
0.70
0.70
0.028
3.4 Molecular docking studies
The docking scores (binding energy, BE), ligand efficiency (LE), and Ki values obtained for the best conformational positions of 1–5 with ACE2, RBD, PLpro, RdRp, and detailed intermolecular interactions of each ligand with the target proteins are presented in Tables S2–S9, and depicted in Fig. 4. Compounds 4 and 5 displayed the best results against PLpro, ACE2, and RdRp, while compounds 1, 2, 5 displayed best results against RBD. The BE for compound 5 against ACE2, RBD, PLpro, RdRp found to be −7.549, −7.392, −8.031 and −7.883 kcal/mole respectively which showed better interaction and stability of the ligand against the targets. The Lys441 and Gln442 of ACE2; Gly339 and Ser371 of RBD; Tyr273, Asp164 and Pro248 of PLpro; and Gly327 and Pro328 of RdRp forms H-bond with the ligand at the active site. The results obtained for the binding free energy (ΔGbind) of compound 5 with all target proteins using MM/PBSA (GBSA) are presented in Table S10.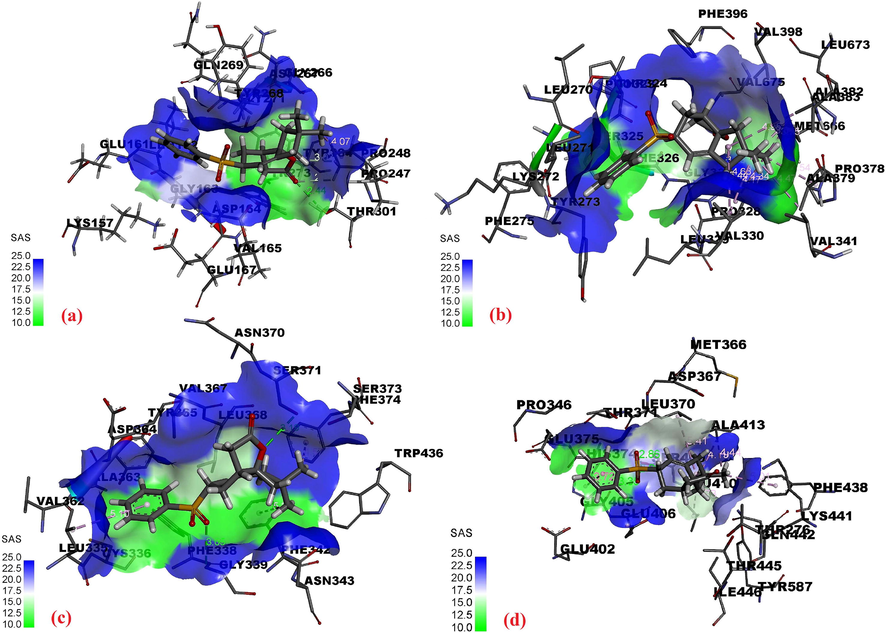
Intermolecular interactions of compound 5 with (a) PLpro, (b) RdRp, (c) RBD, (d) ACE2.
3.5 MD simulation studies
During MD simulation studies, two important trajectories - ligand movement and ligand conformation − were analyzed and expressed in terms of RMSD in Å. All protein–ligand complexes analysed revealed RMSD values ≤3.0 Å (Fig. 5a). The trajectories of RMSD of Cα of the backbone of P-L complexes are shown in Fig. 5b. The RMSD values obtained for ACE2-5 and RBD-5 were <2.0 Å while PLpro-5 had a value >2 Å. The average RMSDs of the ligand movement trajectories of ACE2-5, RBD-5, PLpro-5 and RdRp-5 were 3.753, 6.038, 4.009 and 6.186, respectively. These values (>3 Å) suggest a re-orientation of the ligands in each protein cavity. Structural investigation shows the ligands are well within the cavity of the active site. The radius of gyration for all complexes remained within <1 Å of their average values, which suggests that the P-L complexes stayed compact throughout the simulation (Fig. 6). The trajectories of radius of gyration of the P-L complexes of all the ligands suggest compact conformations, reflecting the stability of the complexes (Mohapatra et al., 2021f).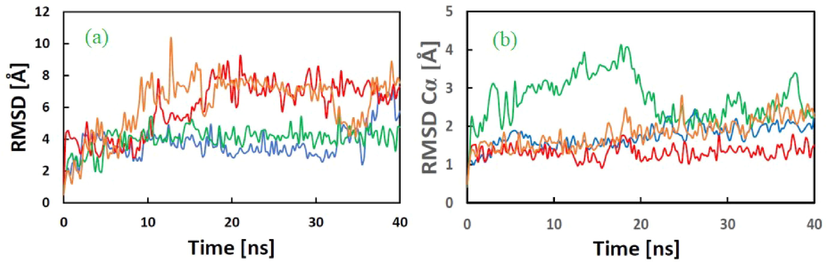
(a) RMSD in Å of the ligand movement of ACE2-5 (blue, avg. 3.753), RBD-5 (red, avg. 6.038), PLpro-5 (green, avg. 4.009), and RdRp-5 (brown, avg. 6.186) complexes during the MD simulations. (b) RMSD in Å of protein–ligand Cα trajectory of ACE2-5 (blue, avg. 1.728), RBD-5 (red, avg.1.319), PLpro-5 (green, avg. 2.715), and RdRp-5 (brown, avg. 1.809) complexes during the MD simulations.
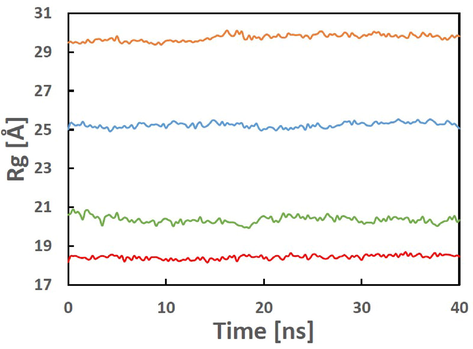
Radius of gyration (Rg) in Å of ACE2-5 (blue, avg. 25.236), RBD-5 (red, avg. 18.435), PLpro-5 (green, avg. 18.435), and RdRp-5 (brown, 29.74) complexes during the MD simulations.
3.6 Pharmacokinetic, drug-likeness and toxicity parameters
The pharmacokinetic and drug-likeness parameters for the ligands (Fig. S4) were calculated and are reported in Tables S11–S16. All investigated compounds obeyed the rules for oral drug-likeness, including the Lipinski’s rule of five (Lipinski, 2000; Lipinski, 2004). In the case of compounds 1–5, the molar refractivity was <100. The topological polar surface area (TPSA) for these compounds was < 70 Å2, indicating their potentiality as favourable drug molecules (Dariya and Nagaraju, 2020). Their number of rotatable bonds (≤2) suggested flexibility. These compounds were soluble and highly absorbable in gastrointestinal (GI) tract. Their synthetic accessibility value was nearly 5, indicating the feasibility of their synthesis (Mohapatra et al., 2021b). The toxicity parameters including LD50 and toxicity class of the five compounds are listed in Table S13. Compounds 2 and 4 with lower LD50 values of 750 and 1000 mg/kg predicted to be more toxic. Compounds 1, 3 and 5 having LD50 values of 5000 mg/kg, 4600 mg/kg and 2000 mg/kg are less toxic and can be considered for oral consumption.
4 Conclusion
Analysis of the interactions of compounds 1–5 against the human angiotensin receptor (ACE2) as well as the receptor binding domain (RBD) spike protein, the papain-like (PL) protein and the reverse transcriptase of SARS-CoV-2 identified compound-5 has the best binding ligand against all four targets. This was evidenced by the formation of a number of key intermolecular interactions with the target proteins, high negative values of docking scores and of free binding energy values, and low calculated values of Ki binding constants. The stability of each protein-compound 5 complex was validated by the results obtained following MD simulation studies. These results, along with the favourable pharmacokinetic properties predictions, suggest that compound 5 may be a good candidate for the future design of novel drugs. Moreover, compounds 1, 3 and 5 are less toxic with LD50 values of 5000 mg/kg, 4600 mg/kg and 2000 mg/kg respectively and can be considered for oral consumption. Further studies are necessary to fully explore the potential of compound 5 in the fight against Covid-19.
Acknowledgements
The authors acknowledge the financial support through Researchers Supporting Project number (RSP-2021/147), King Saud University, Riyadh, Saudi Arabia. All authors are thankful to their respective Institutions for the support they received.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Banerjee, P., Eckert, A.O., Schrey, A.K., Preissner, R., 2018. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 46, W257–W263, https://doi.org/10.1093/nar/gky318
- India begins SARS-COV-2 vaccination amid trial allegations. Lancet. 2021;397(10271):264.
- [Google Scholar]
- Could new COVID variants undermine vaccines? Labs scramble to find out. Nature. 2021;589(7841):177-178.
- [Google Scholar]
- Comparative vibrational spectroscopic studies, HOMO–LUMO and NBO analysis of N-(phenyl)-2,2-dichloroacetamide, N-(2-chloro phenyl)-2,2-dichloroacetamide and N-(4-chloro phenyl)-2,2-dichloroacetamide based on density functional theory. Comput. Theor. Chem.. 2013;1016:8-21.
- [Google Scholar]
- Understanding novel COVID-19: Its impact on organ failure and risk assessment for diabetic and cancer patients. Cytokine Growth Factor Rev.. 2020;53:43-52.
- [Google Scholar]
- Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B. 2021;11(1):237-245.
- [Google Scholar]
- Semiempirical GGA-type density functional constructed with a long-range dispersion correction. ComputChem.. 2006;27(15):1787-1799.
- [Google Scholar]
- The role of ligand efficiency matrics in drug discovery. Nat. Rev. Drug Discov.. 2014;13:105-121.
- [Google Scholar]
- YASARA view – molecular graphics for all devices – from smartphones to workstations. Bioinformatics. 2014;30:2981-2982.
- [Google Scholar]
- Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000;44(1):235-249.
- [Google Scholar]
- Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today. Technol.. 2004;1(4):337-341.
- [Google Scholar]
- Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 2009;113(18):6378-6396.
- [Google Scholar]
- Design, synthesis, molecular modeling, and biological evaluation of novel α-aminophosphonates based quinazolinone moiety as potential anticancer agents: DFT, NBO and vibrational studies. J. Mol. Struct.. 2018;1173:128-141.
- [Google Scholar]
- Challenges in controlling COVID-19 in migrants in Odisha, India. Diab. Metab. Synd.: Clin. Res. Rev.. 2020;14(6):1593-1594.
- [Google Scholar]
- Repurposing benzimidazole and benzothiazole derivatives as potential inhibitors of SARS-CoV-2: DFT, QSAR, molecular docking, molecular dynamics simulation, and in-silico pharmacokinetic and toxicity studies. J. King Saud Univ. – Sci.. 2021;33(8):101637
- [Google Scholar]
- Infection capability of SARS-CoV-2 on different surfaces. Egypt. J. Basic Appl. Sci.. 2021;8(1):75-80.
- [Google Scholar]
- Mohapatra, R.K., El-ajaily, M.M., Alassbaly, F.S., Sarangi, A.K., Das, D., Maihub, A.A., Ben-Gweirif, S.F., Mahal, A., Suleiman, M., Perekhoda, L., Azam, M., Al-Noor, T.H., 2021e. DFT, anticancer, antioxidant and molecular docking investigations of some ternary Ni(II) complexes with 2-[(E)-[4-(dimethylamino) phenyl]methyleneamino]phenol. Chem. Papers 75, 1005–1019.
- Mohapatra, P.K., Chopdar, K.S., Dash, G.C., , A.K., Raval, M.K., 2021f. In silico screening and covalent binding of phytochemicals of Ocimum sanctum against SARS-CoV-2 (COVID 19) main protease, J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2021.2007170
- Computational investigations of three main drugs and their comparison with synthesized compounds as potent inhibitors of SARS-CoV-2 main protease (Mpro): DFT, QSAR, molecular docking, and in silico toxicity analysis. J. King Saud Univ. – Sci.. 2021;33(2):101315.
- [Google Scholar]
- SARS-CoV-2 and its variants of concern including Omicron: a never ending pandemic. Chem. Biol. Drug Des. 2022
- [CrossRef] [Google Scholar]
- Challenges of the Omicron (B.1.1.529) Variant and Its Lineages: A Global Perspective. ChemBioChem. 2022;23(9)
- [CrossRef] [Google Scholar]
- Electronic structures, bonding, and spin state energetics of biomimetic mononuclear and bridged dinuclear iron complexes: a computational examination. Struct. Chem.. 2021;32(4):1473-1488.
- [Google Scholar]
- Airborne transmission of SARS-CoV-2: The world should face the reality. Environ. Int.. 2020;139:105730
- [Google Scholar]
- The SARS-COV-2 vaccination campaign in Nepal, emerging UK variant and futuristic vaccination strategies to combat the ongoing pandemic. Travel Med. Infect. Dis. 2021
- [CrossRef] [Google Scholar]
- An efficient synthesis towards the core of Crinipellin: TD-DFT and docking studies. J. Saudi Chem. Soc.. 2021;25(2):101193
- [Google Scholar]
- Oral antivirals for the prevention and treatment of SARS-CoV-2 infection. AIDS Rev.. 2022;0(0)
- [CrossRef] [Google Scholar]
- Synthesis, crystal structure, spectral and DFT studies of potent isatin derived metal complexes. J. Mol. Struct.. 2018;1166:110-120.
- [Google Scholar]
- Studies on picrate. VIII. Crystal and molecular structures of aromatic amine picrates: aniline, N-methylaniline, N, N-dimethylaniline and o-, m- and p-phenylenediamine picrates. Chem. Pharm. Bull.. 1996;44(12):2199-2204.
- [Google Scholar]
- AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [Google Scholar]
- farPPI: a webserver for accurate prediction of protein-ligand binding structures for small-molecule PPI inhibitors by MM/PB(GB)SA methods. Bioinformatics. 2019;35(10):1777-1779.
- [Google Scholar]
- WHO, WHO recommends two new drugs to treat COVID-19, 14 January 2022. https://www.who.int/news/item/14-01-2022-who-recommends-two-new-drugs-to-treat-covid-19 (accessed on: 06-02-2022).
- Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J. Biol. Chem.. 2004;279(47):49414-49419.
- [Google Scholar]
- Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2020
- [CrossRef] [Google Scholar]
- Electronic structures, bonding and energetics of non-heme mono and dinuclear iron-TPA complexes: a computational exploration. Struct. Chem.. 2021;32(5):2007-2018.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102086.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




