

Translate this page into:
Computational kinetic study on atmospheric oxidation reaction mechanism of 1-fluoro-2-methoxypropane with OH and ClO radicals
⁎Corresponding author. marufai2424@gmail.com (Abubakar Rufai Mohammed)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
The knowledge of atmospheric chemistry has explained several reactions that occur in atmosphere regarding volatile organic compounds (VOCs). In this original paper, computational kinetic study was carried out on atmospheric oxidation reaction mechanism of 1-fluoro-2-methoxypropane (CH3CH(OCH3)CFH2) with OH/ClO radicals using DFT/M06-2X/6–311++G∗∗ method. To improve the results, DFT/M06-2X/6–311++G(2df,2p) single-point calculations were performed on the reacting species involved. The Monte Carlo search on the investigating hydrofluoroether (H3CCH(OCH3)CFH2) showed nine conformers with the lowest global minimum conformer being considered for this study. The reaction proceeded via four (4) reaction routes which led to dehydrogenation in each cases of the radical (OH/ClO) used. The total rate for dehydrogenation reaction of H3CCH(OCH3)CFH2 with OH/ClO radicals is 9.13 ∗ 103 s−1 and 1.23 ∗ 106 s−1 at 298 K which is in agreement with the available experimental rates recorded by Healthfield et al. (1998), Kazuaki Tokuhashi et al. (2000). Also, in each radical cases, thermodynamics parameters, the major and minor products’ heat of formation were computed with potential energy surfaces (PES) for the reactions constructed at absolute temperature of 298 K.
Keywords
1-fluoro-2-methoxypropane
Density functional theory
Ozone depletion
Hydrofluoroethers (HFEs)
OH & ClO radicals

1 Introduction
Atmosphere is self-cleaned, as such volatile organic compounds (VOCS) released into it usually undergo primary degradation through oxidation with atmospheric radicals e.g. OH, ClO, IO, BrO, NO3, OH2, RO2 etc. (Smook and Henne, 1950; Anderson, 1987; Philip, 1996; Gligorovski et al., 2014; Gour et al., 2016). Recent researches have shown that chlorofluorocarbons (CFCs) which are used for aerosols: deodorant sprays, paint sprays, furniture polishes and refrigerators pose direct environmental threats on ozone (O3) by diffusing into O3 layer and thus deplete it at a rate faster than it can be replaced in the atmosphere (Garfield, 1988; Hurley et al., 2004; Rohrer and Berresheim, 2006; Papadimitriou et al., 2007, Yang et al., 2007; WMO, 2010; Mendes et al., 2014; Baidya et al., 2017). As a result, international restrictions and warning was placed on the usage of CFCs as agreed upon in the Montreal Protocol (Good et al., 1998; Lelieveld et al., 2002; Ye et al., 2016; Kovacevic and Sabljic, 2017). Instead, alternative chemicals which cannot pose or with less posing ability on ozone is recommended as substantial substitutes for the CFCs (Espinosa-Garcia, 2003; Furin, 2006; Galano et al., 2010; Carr et al., 2013; Li et al., 2014). Currently, oxygenated hydrofluoroethers (HFEs) (White and Martell, 2015), have been discovered as substantial substitutes for CFCs with numerous applications ranging from refrigerators, cleaning agents, propellants, painting, solvents, pesticides, varnishes in laboratories etc. (Prather and Spivakovsky, 1990; Sako et al., 1996; Laszlo et al., 1997; Wang et al., 2009; Deka and Mishra, 2014; Mishra et al., 2014; Hashemi and Saheb, 2017). Th e presence of —O— linkage between these series increases their chemical reactivity in the atmosphere which account for the chemistry of their short lifetime and lesser atmospheric effects compared to CFCs (Gour et al., 2016; Baidya et al., 2017).
The 1-fluoro-2-methoxypropane (CH3CH(OCH3)CFH2) as an ether, is used as a cleaning agent, coolants/refrigerators, industrial intermediates, propellants etc. Due to its numerous applications, the atmospheric oxidation reaction study of this molecule (CH3CH(OCH3)CFH2) with OH/ClO radicals become very important as it will provides information on the tropospheric reactivity and degradation pathways of it in the atmosphere. Hence, it is necessary to research into the atmospheric chemistry of it for better understanding of this sample as a substantial substitute for CFCs and possibly determine more of its impacts on the environment.
Many experimental studies carried out on the related study of this kind only succeeded in providing the overall rate constants but found it very difficult to reveal the actual mechanism and the true reaction picture especially when the reaction is multi routes. However, with computational/Insilco study the detailed mechanism, thermochemistry of multi routes of reactions and overall rate constant can be determined accurately at once. This as a method is very simple, time and cost saver, accurate and very fast compares to wet laboratory method.
The major objective of this study was to make a systematic computational kinetic study on reaction of CH3CH(OCH3)CFH2 with OH/ClO radicals using simple computational techniques. And also aimed at studying the computational kinetics of dehydrogenation reaction of CH3CH(OCH3)CFH2 by OH/ClO radicals which involves four (4) dehydrogenation positions of
as shown in the chemical reactions in Eqs. (1)–(8).
2 Materials and methods
2.1 Samples used (HFEs and radicals)
1-fluoro-2-methoxypropane (HFE), OH/ClO radicals were selected from the literature and used for this computational kinetics study (Anderson, 1987; WMO, 2010; Hashemi and Saheb, 2017; Baidya et al., 2017). 2D structures were drawn with ChemDraw ultra V12.0 module and transferred to Spartan 14 v 1.1.4 where the 3D structures were obtained.
2.2 Computational procedures
In this study, Spartan 14 v 114 suite program was used for all the electronic calculations. The geometry optimization of stationary points or all chemical species involved in the reaction were carried out using density functional theory (DFT) based M06-2X method in conjunction with 6–311++G** basis set since dehydrogenation entails diffuse and polarization functions on Hydrogen (White and Martell, 2015; Hashemi and Saheb, 2017). To improve the results, DFT/M06-2X/6–311++G(2df,2p) single-point calculations were performed on the reacting species involved. This geometry optimization method/basis sets were employed due to the remarkability in their accuracy and the precise stability of the reacting species that were involved (Mishra et al., 2014; Baidya et al., 2017). The minimum energy equilibrium structure obtained at each stationary point has all real frequencies, meanwhile transition state possesses one imaginary frequency. The imaginary frequency in transition state corresponds to the coupling of stretching modes of the breaking C—H/O—C and forming O—C bonds. Transformation from the reactant to product via the transition state (TS) along the minimum energy path was confirmed with the help of intrinsic reaction coordinate (IRC) calculations at the M06-2X level of DF theory (Gour et al., 2016). IRC calculations indicate the formation of pre- and post-reaction complexes of the reactant/product molecule with the OH/ClO radicals (H2O or HOCl) in both the entry and exit of each reaction route.
The rate constants (k) for dehydrogenation involved in the reactions were derived directly from the general proposed reaction mechanisms in Eqs. (9)–(10).
The rate coefficient of this study was computed as reported by Siaka et al. (2017) as shown in Eq. (11)
The
was determined using expression (12).
Where, kB is Boltzmann’s constant, h is Planck’s constant, is change in entropy’s of transition state, is the change in enthalpy of transition state, T is absolute temperature, R is universal gas constant and k is rate constant.
The total rate coefficient of this investigation is however computed as stated by Galano et al., 2010 as expressed in Eq. (13).
The branching ratios for the dehydrogenation reaction channels, which was reported (Gour et al., 2016) to represent the individual contribution of a reaction channel toward overall reaction rate have also been determined by using an expression in Eq. (14).
The
,
as well as
are computed using Eqs. (15)–(17)
The change in enthalpy, change in entropy as well as Gibb’s free energy of each transition states were estimated using expressions (18)–(20)
3 Results and discussion
3.1 Sample conformers
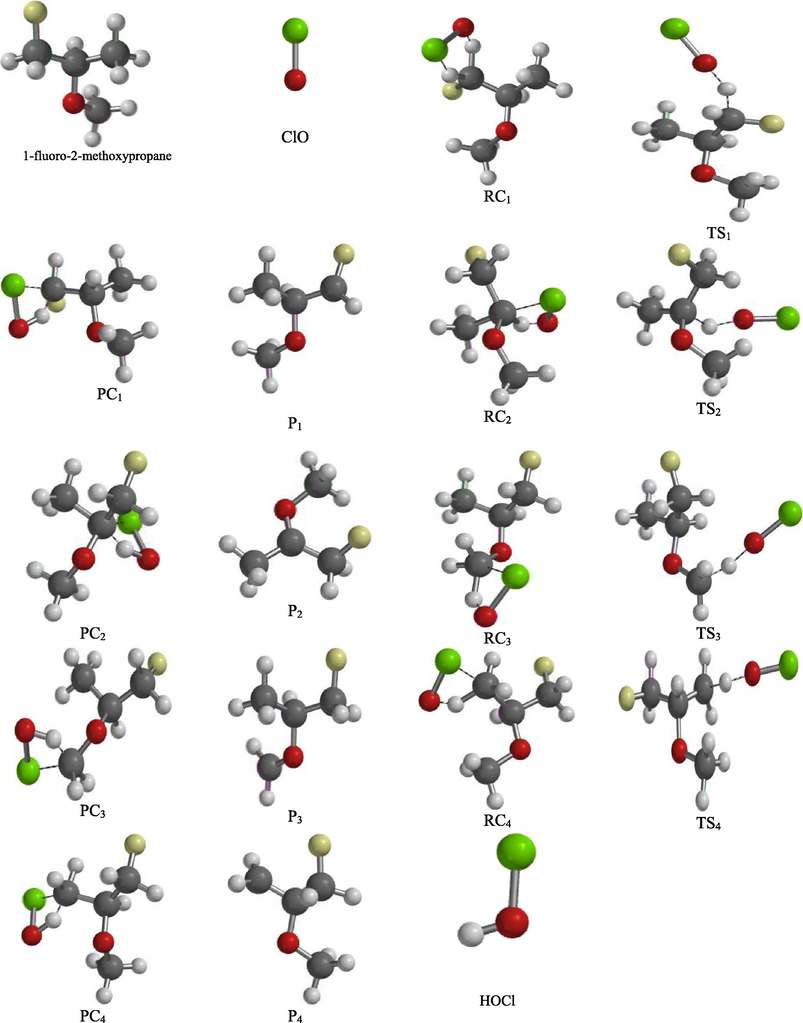
Fig. 1 shows all the nine (9) possible Conformers of 1-fluoro-2-methoxypropane molecule ranging from the lowest to highest energy conformers.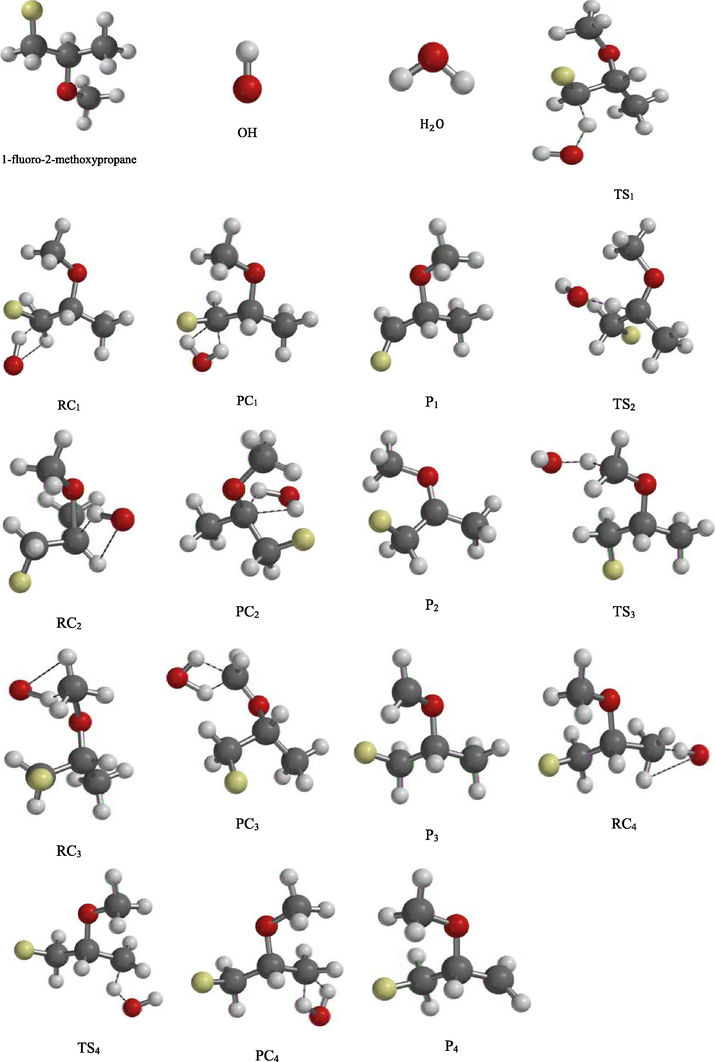
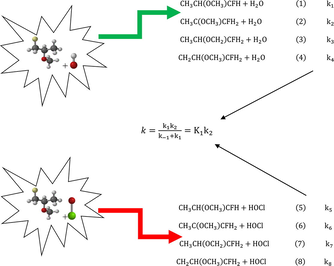
The optimized structures of reacting species with OH radical.
The optimized structures of reacting species with ClO radical.
Conformational analysis carried out on H3CCH(OCH3)CFH2 molecule revealed that it has nine (9) possible conformers as illustrated in Figs. 2 and 3. From the analysis, conformer A (88.90 kJmol−1) has the lowest energy predicted. Conformational analysis is however aimed at identifying the possible conformers of molecule H3CCH(OCH3)CFH2 and to select the most stable conformer with lowest energy predicted. Hence, higher energy conformers are not stable and were not considered for this computational study (basic rule of quantum mechanics) (Baidya et al., 2017). The resultant conformers are arranged in Fig. 2 in order of increasing in their energy stabilities, so conformer I (107.67 kJmol−1) has the highest energy predicted and it is the most unstable conformer among all as shown in the Fig. 3.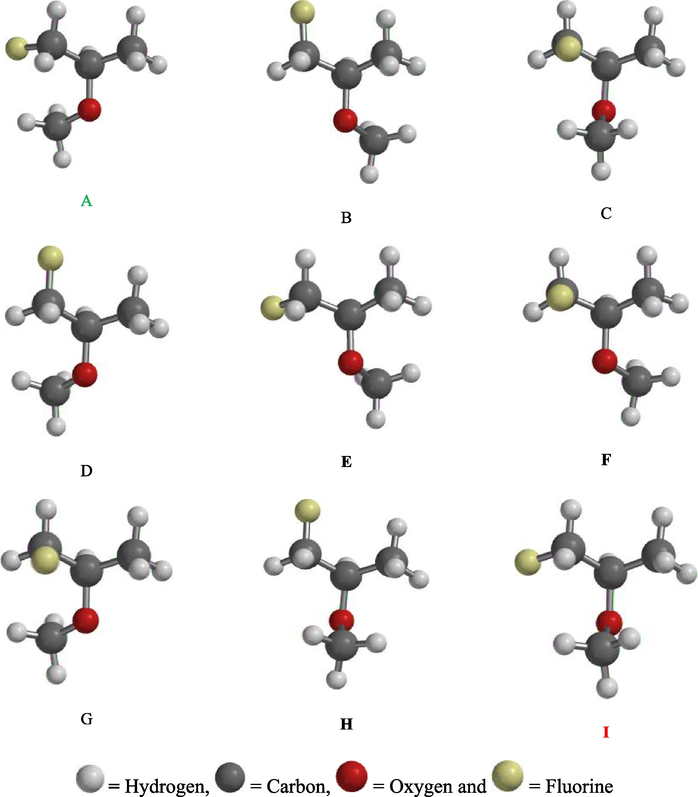
2. Nine possible Conformers of 1-fluoro-2-methoxypropane molecule.
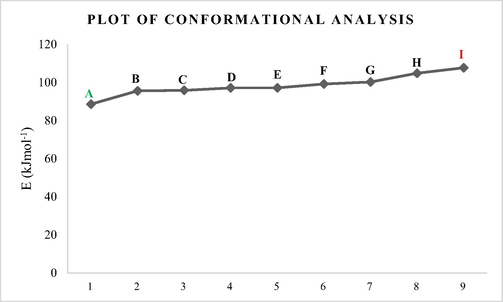
. Plot of conformational analysis results (lowest (A) and highest energy Conformers (I)).
The Tables 1a and 1b show the results on single-point calculation which was aimed at improving the reacting species energies and enthalpies (kJmol−1) using DFT/M06-2X/6–311++G(2df,2p) level. PMajor: Major product = HFE radical PMinor: Minor product = H2O or HOCl.
Reaction routes
HFEs (kJmol−1)
Radical (kJmol−1)
Reactant complexes (kJmol−1)
Transition states (kJmol−1)
Product complexes (kJmol−1)
PMajor (kJmol−1)
PMinor (kJmol−1)
R1
335.15
31.97
358.76
357.91
365.31
301.32
66.83
R2
335.15
31.97
357.81
372.25
368.77
304.58
66.83
R3
335.15
31.97
359.38
358.66
362.13
298.14
66.83
R4
335.15
31.97
360.99
345.82
365.82
301.83
66.83
Reaction routes
HFEs (kJmol−1)
Radical (kJmol−1)
Reactant complexes (kJmol−1)
Transition states (kJmol−1)
Product complexes (kJmol−1)
PMajor (kJmol−1)
PMinor (kJmol−1)
R5
335.15
13.01
349.52
344.30
353.07
301.32
45.47
R6
335.15
13.01
351.38
337.64
355.83
304.58
45.47
R7
335.15
13.01
348.34
335.52
357.40
298.14
45.47
R8
335.15
13.01
349.86
340.86
358.85
301.83
45.47
The Table 2a shows the thermodynamics calculation results of dehydrogenation reaction routes (R1–R4) that were performed at DFT/M06-2X/6–311++G(2df,2p) with OH radicals. These results indicate that reaction through R1, R2 and R4 routes are endothermic (
in nature and thermodynamically unfavorable while reaction across R3 is exothermic. However, route R2 has lowest
which accounts for high possibility of dehydrogenation to occurs through route R2 over the others. Also, route R2 has highest
and lowest
which implies that, the product of this route is thermodynamically feasible. For this reason, R2 is considered more favored than every other routes and the rate of dehydrogenation reaction along this route is faster. While Table 2b shows the thermodynamics computational results of dehydrogenation reaction routes (R5–R8) that were performed on CH3CH(OCH3)CFH2 at DFT/M06-2X/6–311++G(2df,2p) with ClO radicals. The results indicate that reaction through R6 is endothermic (
in nature and thermodynamically unfavorable while dehydrogenation along R5, R7 and R8 are exothermic. The lowest
and highest
nature of R6 accounts for its high possibility of dehydrogenation reaction. R6 with highest
and lowest
also explains that, the product from this route is more feasible. Hence, R6 is confirmed more thermodynamically favorable.
Reaction Routes
R1
1.03
800.23
802.80
32.94
R2
4.29
685.46
675.29
52.29
R3
−2.15
715.42
718.86
33.69
R4
1.54
688.70
689.60
20.85
Reaction Routes
R5
−1.37
960.10
965.45
40.24
R6
1.89
825.36
846.70
46.54
R7
−4.55
868.10
873.30
34.96
R8
−0.86
845.60
848.96
39.61
There are four possible dehydrogenation carbon sites of H3CCH(OCH3)CFH2 which are —CFH2, —OCH, —OCH3, and —CH3 groups. This resulted to eight transition states (TS1, TS2, TS3, TS4, TS5, TS6, TS7 and TS8) as the system reacted with OH/ClO radicals. For each transition state of the radicals investigated with system, the C—H bond of the dehydrogenation and newly formed bond between H and O atoms in OH/ClO radicals are observed. The intrinsic reaction coordinates (IRC) performed on all transition states at the same level were characterized by the occurrence of one imaginary frequencies identified at (840i, 1125i, 826i and 815i cm−1 for OH radical) and (420i, 350i, 515i and 410i cm−1 for ClO radical). This confirmed the existence of transition states connecting reactants and products for each route. During entrance and exit routes of this reaction study (R1–R8), pre – reactive complexes (RC1, RC2, RC3, RC4, RC5, RC6, RC7 and RC8) and post – reactive complexes (PC1, PC2, PC3, PC4, PC5, PC6, PC7 and PC8) formed have been validated as recommended by White and Martell (2015). Also, hydrogen bond formation was observed in the pre – reactive and post – reactive complexes between Oxygen atom of OH/ClO radicals with H-atom in the H3CCH(OCH3)CFH2 which result from the weak attraction force. Thus, this implies that the reaction routes can proceed through indirect mechanism.
Figs. 4a and 4b show potential energy surfaces (OH/ClO) of the reaction at DFT/M06-2X/6–311++G(2df,2p) with arbitrary ground energies of the reactants taken as zero. Route R2/R6 in Figs. 4a and 4b is the most favorable route of dehydrogenation since they are characterized by lowest energies.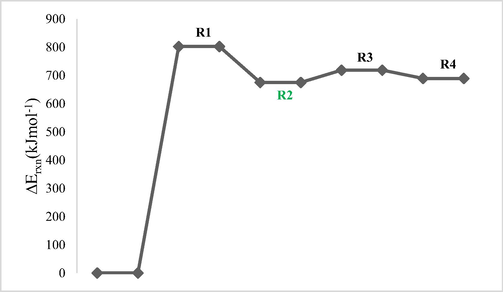
Potential energy surface of dehydrogenation reaction routes performed at DFT/M06-2X/6–311++G(2df,2p) for 1-fluoro-2-methoxypropane with OH radical.
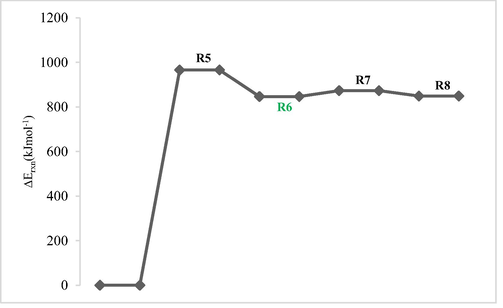
Potential energy surface of dehydrogenation reaction routes performed at DFT/M06-2X/6–311++G(2df,2p) for 1-fluoro-2-methoxypropane with ClO radical.
The Tables 3a and 3b show that reaction of OH/ClO radicals with the sample proceeded in two steps: (1) transition state formation and (2) product formation. According to the Gibb’s free energies of the two steps computed in each radical cases, the rate determining step (RDS) of each reaction channel was identified on the basis of
>
. Thus, first steps i.e. transition states formation across all reaction channels in the two Tables (3a and 3b) were the RDS. The rate determining steps are the slowest steps in the reaction between the HFE and the radical species that were investigated.
Routes
R1
−9.21
99.56
10.24
73.41
R2
5.13
87.46
−0.84
50.78
R3
−8.46
95.68
6.31
58.82
R4
−21.30
88.27
22.84
64.58
Routes
R5
−3.86
107.35
2.49
52.83
R6
−10.52
102.51
12.41
48.68
R7
−12.64
110.28
8.09
47.98
R8
−7.30
108.10
6.44
52.57
Table 4 shows the branching ratios for dehydrogenation reaction across the routes in the two radical cases which represent the individual contribution of a reaction channel toward overall reaction rate. For route one in OH radical case, the branching ratio is higher which can be attributed to Fluorine atom attach to carbon one. The total kClO value (1.23 * 106 s−1) is higher than total kOH value (9.13 * 103 s−1). Thus, the difference between kClO and kOH is 1.22 * 106 s−1. This implies that in an ideal atmospheric condition, OH radicals are expected to be higher than ClO radicals since they (OH) are the major atmospheric scavenger through which atmospheric pollutants are removed (primary degradation). However, whenever ClO radicals over shadow OH radicals at the atmosphere the possibility of ozone depletion cannot be quantified.
BROH
BRClO
kOH (s−1)
kClO, (s−1)
EaOH (kJ/mol)
EaClO (kJ/mol)
79.90
34.93
7.3 * 103
4.3 * 105
4.946
4.951
1.30
3.33
1.2 * 102
4.1 * 104
4.960
4.945
4.60
40.62
4.1 * 102
5.0 * 105
4.947
4.943
14.20
21.12
1.3 * 103
2.6 * 105
4.933
4.948
Furthermore, Tables 5 (a and b) and 6 (a and b) illustrate some significant transformation and comparisons in bond lengths variation of the reactant complexes (RC1, RC2, RC3, RC4, RC5, RC6, RC7 and RC8), transition states (TS1, TS2, TS3, TS4, TS5, TS6, TS7 and TS8), product complexes (PC1, PC2, PC3, PC4, PC5, PC6, PC7 and PC8) and products (P1, P2, P3, P4, P5, P6, P7 and P8) that were involved in the eight routes of dehydrogenation from CH3CH(OCH3)CFH2 with OH and ClO radicals.
In addition, Tables 7 (a and b) and 8 (a and b) also illustrate some significant chemical transformation and comparisons in the bond angles variation of the reactant complexes (RC1, RC2, RC3, RC4, RC5, RC6, RC7 and RC8), transition states (TS1, TS2, TS3, TS4, TS5, TS6, TS7 and TS8), product complexes (PC1, PC2, PC3, PC4, PC5, PC6, PC7 and PC8) and products (P1, P2, P3, P4, P5, P6, P7 and P8) that were involved in the eight routes of dehydrogenation from CH3CH(OCH3)CFH2 with OH and ClO radicals.
3.2 Reaction mechanism and rate of reaction
The two overall reaction mechanism proposed entail two basic steps as
By applying the steady state approximation to be above equations, we got Eq. (21)
Substitutes Eq. (21) into Eq. (22) to gives Eq. (23)
If [A] and [B] concentration is 1 M, then Eq. (23) becomes Eq. (24)
There are two chemical conditions of which one may be applicable. First is when
and second is when
. The first condition transforms Eqs. (24)–(26)
3.3 Atmospheric implications
The atmospheric lifetime of 1-fluoro-2-methoxypropane is estimated by assumption that its primary removal from atmosphere occurs through its reaction with oxidants (atmospheric radicals). In that case, it can be estimated using Eq. (27).
4 Conclusion
The potential energy surface and kinetics data for the reaction of H3CCH(OCH3)CFH2 with the OH/ClO radicals were studied for the first time using DFT/M06-2X/6–311++G(2df,2p). The most energetically stable conformer of H3CCH(OCH3)CFH2 molecule (A) was identified and the difference between the most stable and most unstable conformer is found to be 18.77 kJ/mol. The dehydrogenation reaction of H3CCH(OCH3)CFH2 with the radicals (OH/ClO) proceeded via indirect mechanism involving the formation of reactant and product complexes at entry and exit of all reaction routes in each radical cases. Eight (8) reaction routes were identified for the stable conformer’s reaction with both radicals. The results obtained showed that R2 and R6 are ideal dehydrogenation reaction routes of H3CCH(OCH3)CFH2 with OH/ClO radicals which occur at
. The total rate for dehydrogenation reaction of H3CCH(OCH3)CFH2 with OH/ClO radicals is 9.13 × 103 s−1 and1.23 × 106 s−1 at 298 K which is in agreement with the available experimental rates recorded by Kazuaki Tokuhashi et al., 2000). This work therefore, provides kinetics and thermodynamics data on atmospheric oxidation reaction of H3CCH(OCH3)CFH2 with OH/ClO which serves as precursor for further theoretical study on similar or related work. The work also revealed that ClO radicals’ reaction with H3CCH(OCH3)CFH2 can leads to ozone depletion since HOCl was among the product formed. So, higher ultraviolet radiation from sun will break off Cl from HOCl which has high tendency for ozone depletion as illustrated in the reactions (28)–(34).
Furthermore, the wet laboratory method is recommended on this same HFEs and OH/ClO radicals to further ascertain the reliability of HFEs as a substantial substitute for CFCs.
Acknowledgement
Mohammed Rufai Abubakar acknowledges the financial support of Petroleum Technology Development Fund (PTDF), Abuja, Nigeria, for providing a PTDF M.Sc. award (Award Letter No: PTDF/ED/LSS/MSc/MRA/244/17).
References
- FREE RADICALS IN THE EARTH'S ATMOSPHERE: their Measurement and Interpretation. Ann. Rev. Phys. Chem.. 1987;38:489-520.
- [Google Scholar]
- Theoretical study on atmospheric chemistry of CHF2CF2CH2OH: Reaction with OH radicals, lifetime and global warming potentials. Comput. Theor. Chem.. 2017;1119:1-9.
- [Google Scholar]
- Experimental and theoretical study of the kinetics and mechanism of the reaction of OH radicals with dimethyl ether. J. Phys. Chem. A.. 2013;117:11142-11154.
- [Google Scholar]
- Theoretical studies on kinetics, mechanism and thermochemistry of gas – phase reactions of HFE – 449mec-f with OH radicals and Cl atom. J. Mol. Graph. Model.. 2014;53:23-30.
- [Google Scholar]
- Ab initio and variational transition – state theory of the CF3CF2OCH3 + OH reaction using integrated methods: mechanism and kinetics. J. Phys. Chem. A.. 2003;107:1618-1626.
- [Google Scholar]
- Synthesis and application of fluorine-containing ethers based on perfluoroolefins. Chem. Sustain. Dev.. 2006;14:303-318.
- [Google Scholar]
- Mechanism and branching ratio of hydroxyl ethers + OH gas phase reactions: relevance of h bond interactions. J. Phys. Chem. A. 2010;144:7525-7536.
- [Google Scholar]
- Ozone-Layer depletion: its consequences, the causal debate, and international cooperation. Essays Inform. Sci. Sci. Literacy Policy Eval. Essays. 1988;11:39-49.
- [Google Scholar]
- Environmental Implications of Hydroxyl Radicals (•OH) ACS (Chem. Rev.) 2014 XXXX, XXX, XXX−XXX, A – AP.
- [Google Scholar]
- Global Ozone Research and Monitoring Project – Report No. 52 Scientific Assessment of Ozone Depletion: World Meteorological Organization, 2010. Geneva Switzerland, 516 p. ISBN: 9966-7319-6-2.
- Lifetime and global warming potentials for Dimethyl ether and for fluorinated ethers: CH3OCH3 (E143a), CHF2OCHF2 (E134), CHF2OCF3 (E125) J. Geophys. Res.. 1998;103 28,186-28,186
- [Google Scholar]
- Theoretical Investigation on the kinetics and Thermochemistry of H-atom abstraction reactions of 2-chloroethyl methyl ether (CH3OCH2CH2Cl) with OH radical at 298 K. Struct. Chem.. 2016;2016:1-9.
- [Google Scholar]
- Theoretical studies on the mechanism and kinetics of the hydrogen abstraction reactions of threo- CF3CHFCHFC2F5 and erthro- CF3CHFCHFC2F5 (HFC-43-10mee) by OH radicals. Comput. Theor. Chem. 2017:1-28.
- [Google Scholar]
- Atmospheric chemistry of fluorinated alcohols: reaction with atoms and OH radicals and atmospheric lifetimes. J. Phys. Chem. A.. 2004;108:1973-1979.
- [Google Scholar]
- Rate constants for the reactions of OH radicals with CH3OCF2CHF2, CHF2OCH2CF2CHF2, CHF2OCH2CF2CF3, and CF3CH2OCF2CHF2 over the temperature range 250–430 K. J. Phys. Chem. A. 2000;104:1165-1170.
- [Google Scholar]
- Atmospheric oxidation of halogenated aromatics: comparative analysis of reaction mechanisms and reaction kinetics. Environ. Sci. Process. Impact.. 2017;19:357-369.
- [Google Scholar]
- Kinetic studies of the reactions of BrO and IO radicals. J. Geophys. Res.. 1997;102:1523-1532.
- [Google Scholar]
- Stability of tropospheric hydroxyl chemistry. J. Geophys. Res.. 2002;107(D23):4715.
- [Google Scholar]
- Development of a model for predicting hydroxyl radical reaction rate constants of organic chemicals at different temperature. Chemosphere. 2014;95:613-618.
- [Google Scholar]
- Rate constant calculations of H-atom abstraction reactions from ethers by HO2 radicals. J. Phys. Chem. A. 2014;118:1300-1308.
- [Google Scholar]
- Theoretical investigation on gas – phase reaction of CF3CH2OCH3 with OH radicals and fate of alkoxyl radicals. (CF3CH(O.)OCH3/CF3CH2OCH2O.) J. Mol. Graph. Model.. 2014;50:90-99.
- [Google Scholar]
- Kinetics study of the Reactions of Cl Atoms with CF3CH2CH2OH, CF3CF2CH2OH, CHF2CF2CH2OH and CF3CHFCF2CH2OH. J. Phys. Chem. A. 2007;111:11608-11617.
- [Google Scholar]
- Advanced Chemistry Cambridge Low Price Edition. Cambridge University Press; 1996. p. :630-631.
- Tropospheric OH and the Lifetimes of hydrochlorofluorocarbons. J. Geophys. Res.. 1990;95:18723-18729.
- [Google Scholar]
- Strong correlation between levels of Tropospheric hydroxyl radical and solar ultraviolet radiation. Nature. 2006;442:184-187.
- [Google Scholar]
- Thermodynamics and kinetics of spiro – heterocycle formation mechanism: computational study. Phys. Chem. Res.. 2017;5(3):439-446.
- [Google Scholar]
- Predicting rate constants of hydroxyl radical reactions with organic pollutants: algorithm, Validation, applicability domain and mechanistic interpretation. Atmos. Environ.. 2009;43:1131-1135.
- [Google Scholar]
- Hydrogen abstraction from fluorinated ethyl methyl ether system by OH radicals. Adv. Phys. Chem. 2015:1-10.
- [Google Scholar]
- Theoretical study of the reactions CF3CH2OCHF2 + OH/Cl and its product radicals and parent ether (CH3CH2OCH3) with OH. J. Comput. Chem.. 2007;29:550-561.
- [Google Scholar]
- Computational study of H-abstraction reactions from CH3OCH2CH2Cl/CH3CH2OCH2CH2Cl by Cl atom and OH radical and fate of alkoxy radicals. Environ. Sci. Pollut. Res.. 2016;23:23467-23484.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.jksus.2018.08.011.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




