

Translate this page into:
Computational insight into the spectroscopic and molecular docking analysis of estrogen receptor with ligand 2,3-dimethyl-N[2-(hydroxy)benzylidene] aniline
⁎Corresponding authors. draprabakaran@veltech.edu.in (A. Prabakaran), issaoui_noureddine@yahoo.fr (Noureddine ISSAOUI)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
A comprehensive approach encompassing a blend of spectral techniques and Density Functional Theory (DFT) was employed to elucidate the electronic, vibrational, structural, and nonlinear optical properties of 2,3-dimethyl-N[2-(Hydroxy)benzylidene] aniline (DNHBA). The investigation of this chemical compound involved a meticulous examination using spectroscopic techniques (FTIR, FT-Raman). The DFT calculations utilized the B3LYP method in conjunction with 6-311G (d, p) basis sets. The findings were substantiated by comparing the estimated vibrational frequencies and molecule geometry with experimental data. Using the Time Dependent −Density Functional Theory (TD-DFT) method to calculate the energy band gap of the compound at different solvents. We looked into frontier molecular orbitals (FMO) Molecular electrostatic potential (MEP) and natural bond analysis (NBO) to determine the compound’s kinetic stability and chemical reactivity. Hirshfeld surface (HS) analysis calculated the O–H..O, H-H and intermolecular interaction energy of the compound. The electron density distribution, interactions, and excitation within the title molecule are illustrated by performing topological analyses (RDG, ELF and LOL) using the Multiwfn software. In addition to these assessments, molecular dynamics provides an understanding of the system's dynamic development over a predefined duration during which atoms and molecules are permitted to interact. Utilizing the molecular docking approach allows us to energize the atomic-level interplay between small molecules and proteins within the binding sites of target proteins. This approach enhances our comprehension of vital biochemical processes associated with the behavior of small molecule. The intricacy and hazards associated with drug discovery and development processes have increased dramatically, leading to higher drug research costs.
Keywords
Density functional theory
Computational chemistry
Molecular dynamics
Molecular docking

1 Introduction
Organic materials with extremely non-linear optical responses, or NLOs, have attracted a lot of attention in the last several decades. Optical frequency doublers, rapid processing of information, photonics, high-density optical disc data storage, telecommunication, and optoelectronics are just a few of the areas in which these materials may find use (Zhang et al., 2007; Munn and Ironside, 1993). The goal has been to create and synthesis novel materials with enhanced NLO qualities for application (Mohd et al., 2015; Muhammad et al., 2013; Senthil Kannan et al., 2020). Even though inorganic crystals may produce light using second harmonic generation (SHG), they have disadvantages, including being difficult to synthesis and modify (In Chung and Kanatzidis, 2014). On the other hand, due to their dispersed π-electron clouds and non-symmetric charge transfer mechanism, which results in large-scale nonlinearities, organic materials are now receiving a lot of attention. They provide benefits for device applications, including molecular engineering and synthetic flexibility. It is known that organic nonlinear crystals have higher laser damage thresholds, lower dielectric constants, fast reaction times, and a large nonlinear optical (NLO) response across a wide frequency range (Zhang et al., 2008). Hence, an examination of organic nonlinear optical (NLO) molecules, particularly crystals, serves to connect fundamental research with practical applications. There are several varieties of organic crystals, including BDMABA, CCBA, CDMA, MDMABA, CMOBA, and BCBA (Leela et al., 2009; Subashini et al., 2011). Because they may link and mesomerize molecules across extended distances, derivatives of Schiff base crystals have been thoroughly researched as potential linear and second-order NLO materials. DFT research and spectroscopic analysis have not yet been the subject of a thorough literature review.
The primary goal of this study is to conduct a computational and experimental examination of the compound derived from a schiff base DNHBA. Density Functional Theory (DFT) is an inexpensive quantum chemical computation method that has a strong record of producing accurate molecule structure predictions. To assess the chemical reactivity of the substance, MEP and quantum theory of atoms in molecules (QTAIM) analyses are conducted. Employing molecular docking and molecular dynamic stimulations improves our understanding of the interactions between DNHBA and biomolecules. These computational techniques enable the investigation of binding modes and dynamic behavior, providing insights into potential applications in the realm of biochemistry.
The current study compares the estimated results obtained using the DFT (B3LYP) technique to the experimental data for the DNHBA. The purpose is to investigate the impact of DFT, TD- DFT, and 6-311G (d,p) basis sets on the structure and vibrational spectra of the title compound. Computational chemistry combines theoretical chemistry approaches embedded in computer programs to calculate the structures and properties of molecules, groups of molecules, and solids. The current version of DFT has numerous limitations, including too many approximations, failures for systems with strong correlations; too flow for liquids, etc. The computational chemist has to decide which DFT method to apply for a certain application.
2 Computational studies
In this study, quantum chemistry simulations were carried out using Gauss View 5.0 (Dennington et al., 2009) and Gaussian 09 W software (Frisch et al., 2009). The molecular structure was optimized using a specific basic set, namely (6-311G(d,p)); this basis set was essential to the computational strategy. To determine the potential energy distribution in terms of wavenumbers, the VEDA4 software (Michal, 2004) was utilized. Analyzing the compound's vibrational frequencies and modes was part of this DFT calculations were employed to ascertain the vibrational frequencies in the realm of organic molecules. To account for electron correlation and anharmonicity, a scaling factor of 0.961 was employed to adjust the computed frequencies. The compound’s electronic properties were examined in different solvents. Various tests were performed on the compound, such FMO, ultraviolet (UV), and MEP studies. The FMO analysis helped us understand how the electric charges inside the molecule moved, and the MEP analysis predicted reactions that would happen between electrons and protons, nucleophiles and electrons, and hydrogen bonds (Issaoui et al., 2014). The study also utilized a natural bond orbital (NBO) study to explore electronic arrangement and bonding attributes (Daghar et al., 2021). We obtained local reactivity descriptors using the Fukui function, which informed us of areas that electrophiles and nucleophiles might attack. Fukui and NBO analyses were utilized to gain insights into molecular relationships and molecular receptive areas, respectively. Additionally, Hirshfeld surface analysis was conducted to analysis the molecular surface area, providing information about molecular packing and interactions. Lastly, multiwave function analysis was performed for topological studies, which involved the analysis of electron density distributions.
3 Results and discussion
3.1 Molecular geometry
DNHBA was analyzed using energy minimization to determine its most stable geometry, with an energy value of −710.783547 Hartree. This procedure was completed using Gaussian 09 W, and Gauss View 5 was used to arrange the calculation's input. Fig. 1 depicts the compound's optimized structure in the gaseous phase. This compound consists of two components 2,3-dimethylaniline (C16-C29/N15) and salicylaldehyde (C15-C6/O11). The dihedral angle between these two components is measured at 46.690 (experimental) and 42.410 (theoretical). Table S1 furnishes bond lengths, bond angles, and corresponding experimental data (Tahir et al., 2010) for the compound. The predicted range for C–C bond lengths is 1.38–1.409 Å, while in this work, the experimental range lies between 1.36 and 1.394 Å. The experimental C–H bond length varies from 0.93-0.96 Å, while the theoretical range is 1.08 to 1.099 Å. The O–H bond has a length of 0.82 Å, which is the shortest among all the bonds. It has been shown that the bond energy rises with decreasing bond length, increasing the molecule's stability. As theoretical calculations are commonly performed on isolated molecules in the gaseous state, there may be a small discrepancy between the theoretical and actual results.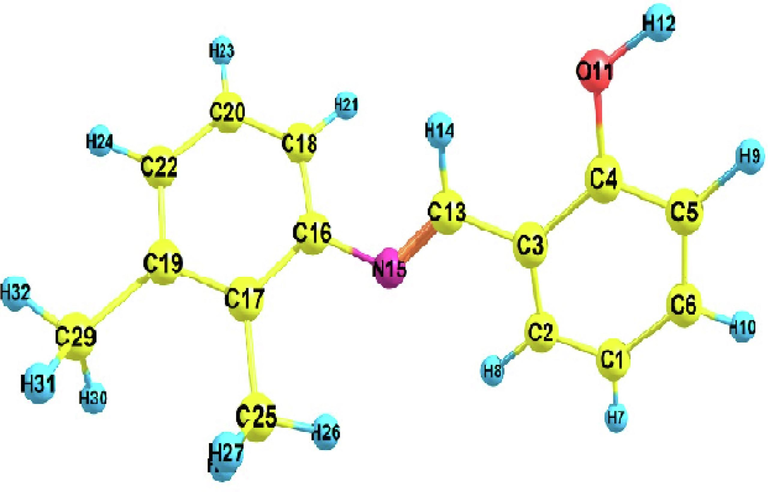
Optimized molecular structure of DNHBA.
3.2 Vibrational assignments
The functional group of the given chemical has been found by using FT-IR and Raman spectroscopy for both theoretical and experimental analysis. Under the symmetry of C1 within the point group, the compound DNHBA contains 90 vibrational modes and 32 atoms. The detailed PED has been used in this investigation to assign all 90 typical vibrational modes. The predicted, computed, and observed spectra (Balachandar et al., 2021) of DNHBA are shown in Fig. S1 and S2. Table S2 lists the basic vibrational assignments and their corresponding intensities for the compound under consideration. The frequencies of the functional groups C–H, O–H, and CH3 have been found to be in the range of 3100–3000 cm−1, 3600–3400 cm−1, and 3000–2800 cm−1, correspondingly (Charanya et al., 2023; Muthu and Prabakaran, 2015). In this investigation, the stretching frequencies associated with the functional groups were observed at 3061 and 3059 cm−1 (C–H), 3681 cm−1 (O–H) (Rekik et al., 2007; Issaoui et al., 3AD), and 2941 and 2895 cm−1 (CH3) (Kazachenko et al., 2023). These values agree well with the FTIR and FT-Raman results shown in Table S2. In the 1600–1200 cm−1 range, vibrations linked to C–N and C–C stretching were detected (Dhakshinamurthy Divya et al., 2023). The recorded frequencies of C-N at 1612 cm−1 (FTIR and FT-Raman) and C–C at 1570 cm−1 (FTIR and FT-Raman) align well with the predicted frequencies for C-N (1614 cm−1) and C–C (1581 cm−1). The calculated bending vibrations of C–H, C–C, and C-N were contrasted with the corresponding experimental values, as detailed in Table S2.
3.3 Hirshfeld surface (HS) analysis
Highly reliable structural (HS) analysis is a trustworthy method for exploring the intermolecular interactions that occur within the crystal structures of molecules. We used the HS analysis tool in the Crystal Explorer 17 (Spackman et al., 2021) package to figure out the HS map and make 2D fingerprint plots for our study of the compound DNHBA. The dnorm surface, which normalizes the function of di and de, exhibits specific red-encircled regions that denote interactions with neighboring molecules, particularly those involving oxygen and nitrogen atoms. The 3D-HS plot in Fig. 2a clearly shows the O–H…O interactions, which are shown as large red spots on the HS. The triangular red and blue areas, on the other hand, show the π-π stacking interactions. Fig. 2b illustrates 2D fingerprint plots showcasing the HS characteristics of the compound. Notably, these plots display pseudo-symmetry wings along the lower right and upper left sides of the di and de diagonal axes. For reference, the complete fingerprint outline is depicted in gray. The largest surface area within the fingerprint, marked as “H..H. contact,” is the most prevalent, constituting 57.2 % of the total HS. 30.2 % of the total hyperfine structure (HS) is represented by the inverse of the “H..C/C..H contact”, symmetrical, pointed protrusions with de + de. The “O.H./H.O. interaction” appears as the next significant region, contributing 8.3 % of the overall HS. All other interactions are observed at percentages less than 8 %, as depicted in Fig. 2b.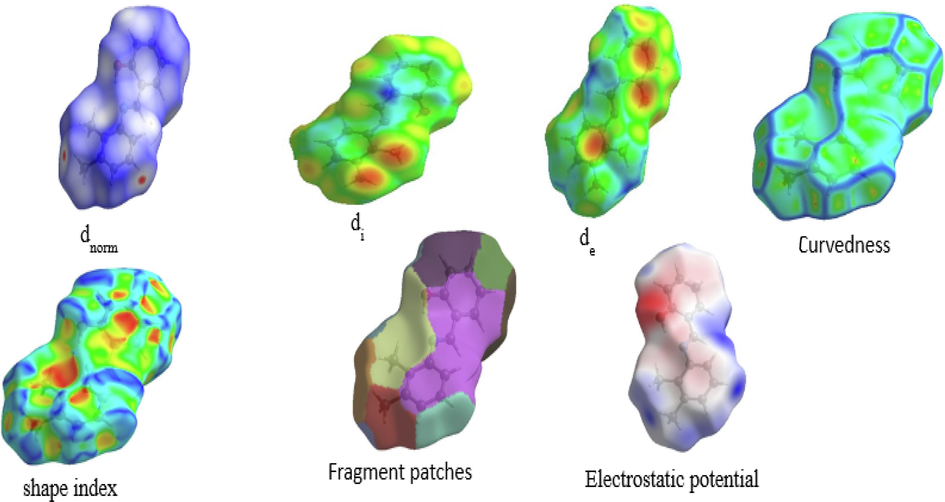
Hirshfeld surface analysis of dnorm, di, de, Shape index, Curvedness, Fragment patches, Electrostatic potential.
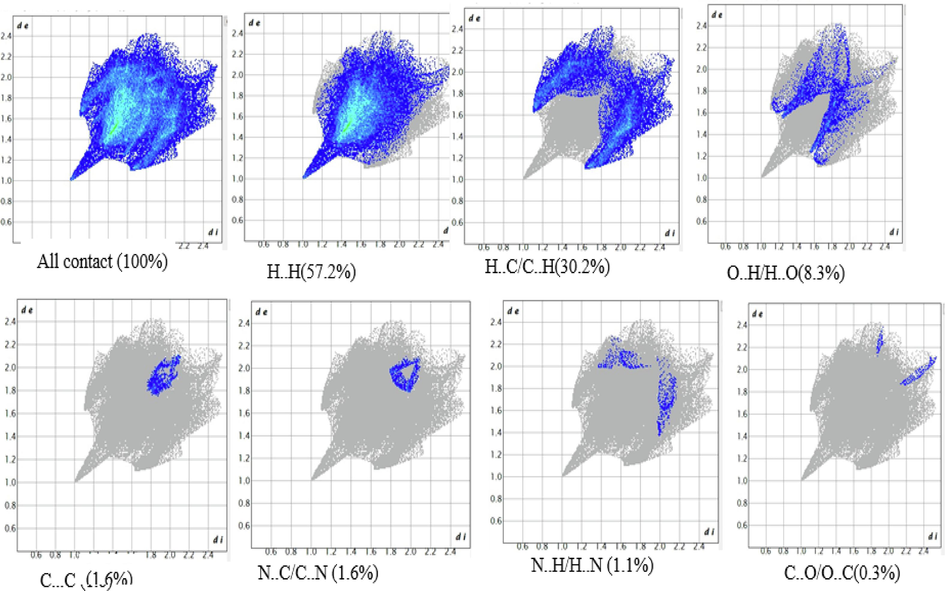
Fingerprint plots of the compound by highlighting unique interaction of each compound.
3.3.1 Interaction energy and energy framework
The Crystal Explorer uses a pairwise method to figure out the intermolecular interaction energy. We calculated the molecular interaction energy using the monomer wave function at the B3LYP/6-311G (d,p) level. This approach allows us to obtain precise values for electrostatic, dispersion, polarization, and repulsion energies, as outlined in Table 1. The calculation of the total interaction energy, shown in Fig. S3, and the calculations in our energy model show how important it is for electrostatic compounds and dispersion forces to play in the crystal's interactions. In Fig. S4, we introduced energy frameworks for a cluster of molecules within 2x1x2 unit cells of the compound, with the dispersion energy represented in green and the total energy in blue. A molecular crystal has a network of hydrogen bonds that connect its molecules. The intermolecular interaction energies between different pairs of molecules make it possible to figure out the lattice energy of a crystal. The B3LYP/6-311G (d,p) energy model yields findings that are as accurate as those derived from dispersion-corrected periodic density functional theory.
N
Sym op
R
E’ele
E’pol
E’dis
E’rep
E’tot
2
−x,y + 1/2,-z + 1/2
11.47
−1.9
−0.2
−8.0
9.6
−3.2
2
x + 1/2,-y + 1/2,-z
5.63
−5.4
0.0
−43.8
20.3
−31.3
2
−x + 1/2,-y,z + 1/2
8.28
−4.6
−1.1
−20.6
10.0
−17.4
2
x,y,z
13.06
0.4
−0.2
−7.2
0.0
−5.9
2
−x,y + 1/2,-z + 1/2
7.19
−5.5
−1.1
−18.9
8.5
−17.8
2
x + 1/2,-y + 1/2,-z
11.07
−0.5
−0.2
−7.0
1.6
−5.7
2
−x + 1/2,-y,z + 1/2
8.33
−4.2
−0.6
−22.9
13.3
−16.7
3.4 QTAIM analysis
3.4.1 AIM analysis
The QTAIM (Matta and Boyd, 2007) is instrumental in discerning within both intramolecular and intermolecular connections. The chemical structure graph topological structure was acquired with the aid of the Multiwfn 3.8 software (Lu and Chen, 2012). Tables S2 and Table 2 show a full breakdown of the DNHBA parameters, including both BCPs and RCPs (Bond and Ring Critical Points) that were found in the substance that was being studied. Fig. S5 illustrates the atomic arrangement of DNHBA visually, which helps to understand how the RCPs, bond routes, and BCPs are spread out. BCPs N15 to H26 are very important in this molecular framework because they help form intramolecular hydrogen bonds on the molecular surface during optimization. The BCPs in question have a positive ∇2ρ(r) value (0.0634 a.u.), ellipticity (1.8308), Eint energy (−48.548 K cal/mol), and the lowest ρ(r) value (0.0156 a.u.). The electron density ρ(r) ranges from 0.3011 to 0.3119 a.u., while ∇2ρ(r) values vary between −0.8139 and −0.8803. These results suggest the existence of a covalent character in the aromatic loop. Also, our AIM analysis shows that O11-H12 has the highest electron density, with ρ(r) = 0.4064 a.u. and ∇2ρ(r) = -2.53 a.u. This is because it has the most non-covalent hydrogen bonds within its molecules, which are caused by those specific BCPs.
RCP
Atom Count
ρRCP
∇2ρRCP
V(a.u)
G(a.u)
H(a.u)
MOD
(V)/GAtoms
1
6
0.0208
0.15717
−0.024
0.03154
0.0078
0.7542
C1- C2 − C3 − C4 – C5-C6
2
5
0.0154
0.07376
−0.011
0.0149
0.0035
0.7659
N15-C16-C17-C25-H26
3
6
0.0207
0.15577
−0.024
0.03128
0.0077
0.7554
C16-C17-C19-C22-C20-C18
Besides, we identified three RCPs (ring critical points) denoted as R1, R2, and R3. These RCPs exhibit positive ∇2ρ(r) values, specifically 0.1571 a.u. as well as 0.1557 a.u. concerning aromatic RCPs in addition to 0.073 a.u. for non-aromatic ring RCPs, along with ρ(r) values of 0.02085 and 0.02072 a.u. in favor of the aromatic ring and 0.0154 a.u. for the non-aromatic ring. The RDG-NCI investigation confirms the presence of a strong repulsive steric effect inside these aromatic rings because R1 and R2 exhibit higher values for ∇2ρ(r) and ρ(r) in comparison to the other RCPs.
3.4.2 RDG-NCI analysis
RDG is used to analyze the spatial distribution of electrons in a molecule. It was computed using the Multiwfn program and visualized with the VDM (Becke and Edgecombe, 1990) program. Fig. 3(a) analyzes weak interactions using circular slabs of dark green, green, and red, interspersed among blue atoms. These color-coded systems can identify interactions when there is a significant, strong steric repulsion that affects the target molecule, which is in the red color region. The blue region (H32, H8, H21, and H14) represents the strong attraction between two hydrogens, and the green peaks near −0.01 a.u. signify enhanced stability due to van der Waals forces(Hammami et al., 2021). Fig. 3(b) illustrates the pronounced steric repulsion effects within the core of the aromatic ring of the specified molecule.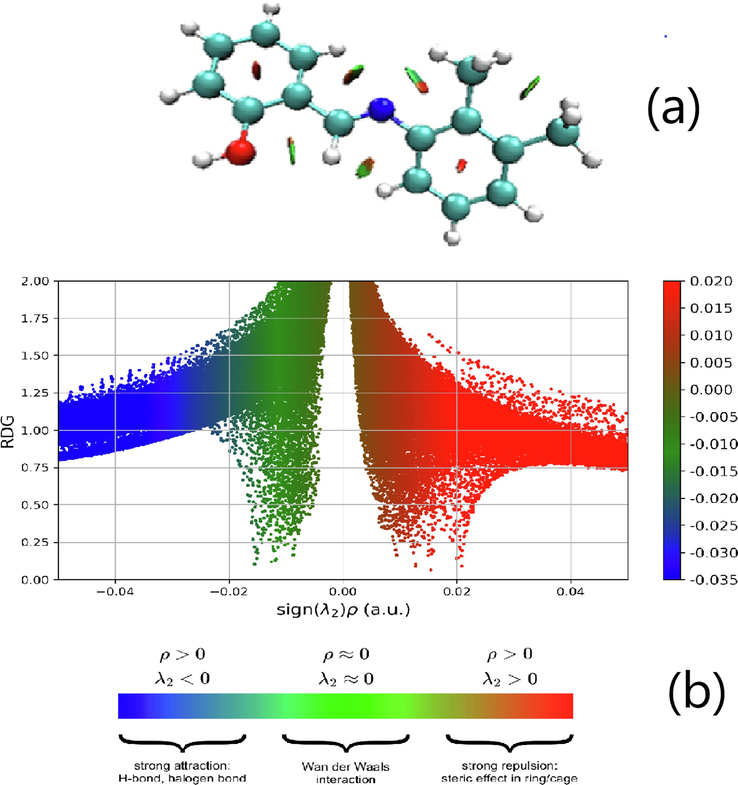
Reduced density gradient (a) Isosurface density,(b) Scatteredgraphs of DNHBA.
3.4.3 Electron localization function and localized orbital Locator analysis
In quantum chemistry, figuring out the ELF (Electron Localization Function) and LOL (Localized Orbital Locator) gives us details about the structure and spread of electron density in a molecule. It helps in understanding lone pairs, atomic shells, and chemical bonds with strong Pauli repulsion (Silvi and Savin, 1994). Fig. 4(a–d) shows the ELF and LOL diagrams of the DNHBA, which use different colors to indicate the degree of electron localization or delocalization(Medimagh et al., 2023). In this compound, hydrogen atoms (H30, H28, H26, and H14) show a rich red color, which represents a high degree of electron localization, while carbon atoms (C19, C17, C13, C4, C5, and C6) have a more blue region, which shows a high degree of electron delocalization Fig. 4(a and b). The white region is positioned at the center of the red region, which represents more electron density in the area exceeding the color scale. This can be observed in Fig. 4(c and d). Additionally, there is a notable overlap in electron density between the carbon atoms C10-C17 and C4-C5 in the red region.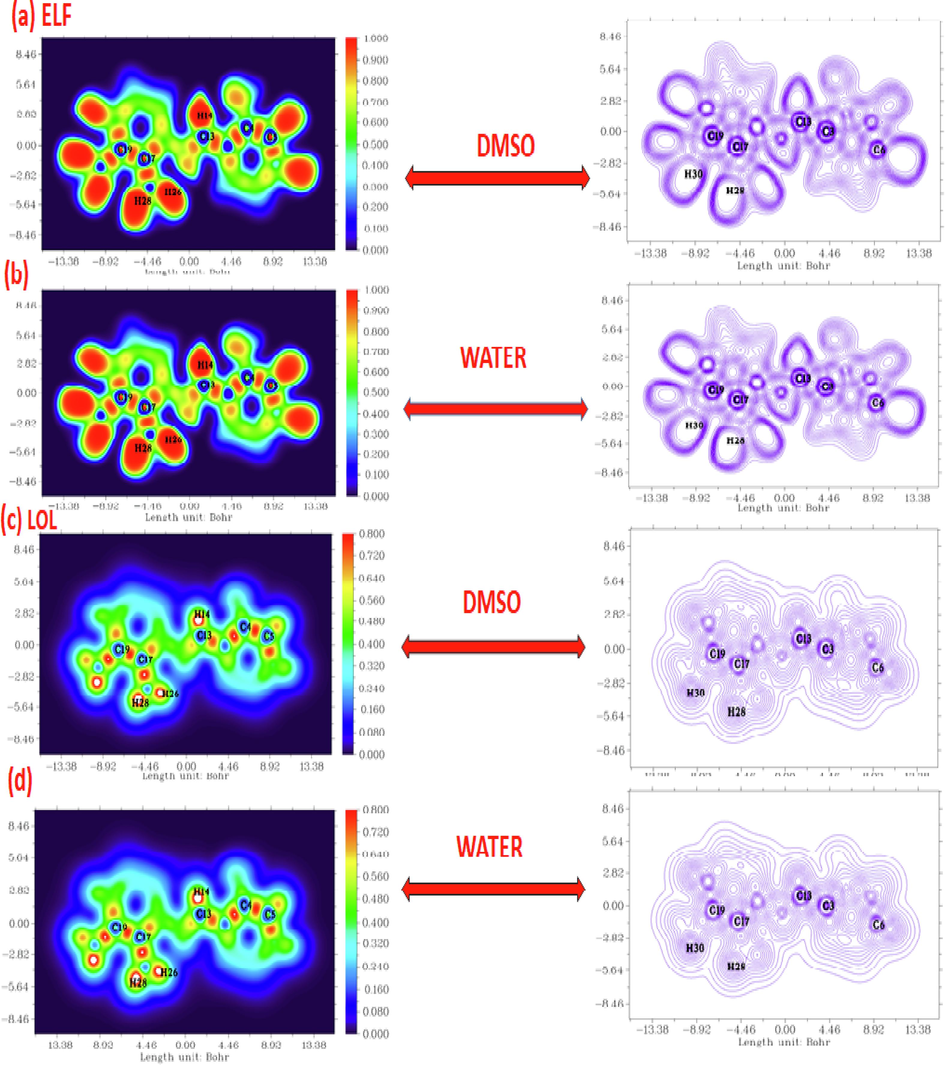
Plane color filled for the Electron Localization Function and Localized Orbital Locator in DMSO and Water of DNHBA.
3.5 Electrostatic potential maps
Molecular electrostatic potential refers to the distribution of electrostatic charges within the molecules. MEP is the three-dimensional molecules; it helps us to visualize the charge distribution, charge-related properties, shape of the molecule, and also the behavior of the complex molecules. The red color with the negative indicates the electrophilic attack, the blue color with the positive indicates the nucleophilic attack, and the zero potential region is indicated by the green color. In Fig. 6a and 6b, the potential rises sequentially from red to orange, yellow < green < blue. And the color code mapped over between −6.496 × 10-2 eV (red) and 6.496 × 10-2 eV. Finally, we concluded that oxygen (O11) and nitrogen (N15) atoms display the negative electrostatic potential, while hydrogen (H7, H8, H9, H10, H12, and H14) display the positive electrostatic potential. These locations provide details on potential areas of interaction between the drug and other molecules (Mhadhbi et al., 2022). It was expected that the site exhibiting the highest reactivity would function as a susceptible target for both electrophilic and nucleophilic attacks.
3.6 Uv–visible spectra
The UV absorption patterns of the DNHBA molecule were computed at different solvents in the TD B3LYP/6-311G level and are shown in Fig. S7. To derive the energies and oscillator strengths of electronic transitions, TD-DFT considers a molecule to be subjected to a time-dependent disturbance induced by an oscillating electrical field of incident light. TD-DFT calculations apply the linear response assumption, which assumes that changes in electron density are proportional to changes in the external field. Three separate peaks can be seen in the spectra at 347.6 nm, 303.8 nm, and 296.9 nm. These peaks in absorption signify the transition of electrons between π and π*. The oscillator strength and primary contribution are computed and displayed in Table S4., which is discussed in the FMO and Fukui function analyses.
3.6.1 Frontier molecular orbital
The HOMO and LUMO play vital roles in quantum chemistry, forming the basis of frontier molecular orbital theories. These orbitals actively participate in chemical processes, aiding the understanding of biological reactions and atomic interactions. Molecular orbital theory (Mishma et al., 2023) is instrumental in evaluating the characteristics of organic compounds, including their optical, electrical, and biological properties (Mishma et al., 2023). In a molecule, the energies of LUMO and HOMO are linked to ionization potential and electron affinity, respectively. The charge density within the methyl and methylene groups is associated with HOMO, while the charge distribution related to the carbonyl group attached to the pyridine ring is termed LUMO (Gatfaoui et al., 2017). Most HOMOs and LUMOs exhibit π-anti-bonding properties. The HOMO and LUMO energy gaps in the molecule, measured in DMSO and water, are 4.421 and 4.2219 eV, respectively, and plotted in the Eg diagram as shown in Fig. S8. Chemical hardness (
) and softness (S), chemical potential (
), and electrophilicity index (
) are some of the reactivity descriptors that are used to find Koopman's (Koopmans, 1934) approximation theorem. A molecule's chemical stability and reactivity are determined by the energy differential between its HOMO and LUMO orbitals. Finding the chemical hardness for the molecule's HOMO-LUMO energy is as follows:
Electronegativity
, which is given by, is what makes an atom able to get an electron.
Chemical potential
Electrophilicity is a species' ability to pull electrons to it. After getting an extra charge, it gives a measure of how stable the system's energy is, which is given as
Table 3, which shows the value of the above parameter. The compound's thermal resilience is indicated by its ionization energy (I), which is determined to be 6.073 eV. The value of electron affinity, 1.85 eV, is smaller than the energy of ionization, implying that electrons are reasonably attracted to one another. In addition, stability and chemical potential are closely connected. It is determined that the chemical potential is − 3.958 eV, and the electronegativity index is 3.958 eV. The calculated energy gap values in water and DMSO are less than 5 eV, which means this compound is a good bioactive material (Romani et al., 2020).
Parameter
Solvent
Water eV)
Dmso (eV)
EHOMO (I)(eV)
6.073
6.0686
ELUMO (A)(eV)
1.8511
1.8474
Energy Gap(eV)
4.2219
4.22
chemical hardness(η)
2.1109
2.11
Softness(S)
0.2368
0.236
chemical potential (μ)
−3.962
−3.958
Electronegativity(χ)
3.9625
3.958
Electrophilicity index (ω)
3.7182
3.7045
3.7 Fukui function analysis
In Chemistry, the distribution of electrons in a molecule is analysed using fukui function. Mulliken population analysis is used to identify radial, electrophilic and nucleophilic attack. The indices regarding Fukui functions at the atomic level can be calculated using a straightforward method provided by Yang and Mortier (Yang and Mortier, 1986) that is based on Mulliken population analysis (MPA). This method can be expressed as
Electrophilic and nucleophilic assaults at a certain site are distinguished by their respective signs by the dual descriptor . Electrophilic bombardment is favored if is less than zero, whereas nucleophilic assault is favored if is greater than zero (Parr and Yang, 1985). Condensed fukui function values for the DNHBA are displayed in Fig. S9 and listed in Table S5.
3.8 NBO/NLMO analysis
NBO (natural bond orbital) scrutiny adeptly examines bonding within and between molecules, supplying a practical groundwork for exploring charge transfer and conjugative interaction within molecular systems (Weinhold and Landis, 2005; Reed et al., 1998). Yet another positive attribute of the NBO method is its capability to offer substantial revelations into interaction within occupied and virtual orbital this proficiency notably enhances the examination of both intra and intermolecular interactions.
Table S6. Showcases the perturbation energies linked to notably donor–acceptor interactions. The more substantial the E(2) value, the more pronounced is the interaction between donor of electrons and receptors of electrons. Into DNHBA, the interactions between the N1-C8 and its corresponding antibonding of C9-C10, hold higher E(2) value roughly 174 kcal/mol. The additional notable interactions that contribute more robust enhancement of the structure’s stability involve C6-C7 and the antibonding of C2-C3 are the highest E (2) value around 196 kcal/mol.
The assessment of localized molecular orbital (NLMO) underwent and it reveals the composition of bonding in a molecule which originates from orbitals situated on distinct atoms. Deriving NLMOs from NBOs offers immediate understanding into the characteristics of the molecular orbital localized ‘‘delocalization tails’’ (Gnanasambandan et al., 2014). The provided table highlights the noteworthy NLMO occupancy, the percentage obtained from the parent NBO, and the atomic hybrid contributions of DNHBA. These computations were conducted at the B3LYP level, employing the 6-311G(d,p) basic set.
The NLMO associated with C2-C3 exhibits the highest degree of delocalization, with contributions solely from the localized C6-C7, and N1-C8 parent NBO ranging from 89 %to 93 %. The broader delocalization tails imply a potent extension into the regions of proximate bonding and antibonding. This spreading out of the electrons is also evident in the energy provided by the perturbation theory in the Table S6. Astabilization energy of the molecule increases correspondingly the hybrid contribution atoms delocalization tail decreases.
3.9 Molecular dynamics protein with ligand interaction
In molecular dynamics, the Gromacs (GUI) 2022.5 version command-line program is employed for the analysis and exploration of the arrangement of gases, liquids, as well as solids. Pertaining to molecular dynamics often makes use of Newton's law and equations from classical mechanics. Molecular dynamics usually makes use of Newton's law and equations from classical mechanics. The molecular dynamics simulation and energy minimization operate in the same manner. The input file is prepared for mdrun to run by Grompp. Molecular dynamics simulations also require an mdp file for setup parameters. In molecular dynamics, all of the mdrun options-aside from −x, which generates a trajectory file are used for energy reduction. One use of MD simulations in protein–ligand docking is the computation of binding free energy (Astuti et al., 2011). Accurate estimations of the protein–ligand binding affinity can be obtained using long simulations and complex sampling procedures, such as improved sampling approaches. These binding-free energies can then be used to rank and prioritize potential drug options. We described the output files of Polar energy, RMSD, RMSF, Hydrogen bond interaction, Radius of gyration (total and around axes), and Protein and ligand interaction energy as all the graph in Fig. 5 for further understanding of Molecular dynamics results (Trott and Olson, 2009). We used XM-grace software for graph XVG file creation purposes. The minimum energy, middle energy, and final energy of Potential energy of the solvated system, Radius of Gyration, Root mean square deviation (RMSD), Root Mean Square Fluctuation (RMSF), Hydrogen Bond, and Interaction between protein and ligand values are mentioned in Table 4, it is been summed up by concluding that MD simulation is essential to protein–ligand docking because it calculates binding free energy with accuracy and offers comprehensive with respect to the dynamic nature of interaction involving the protein and ligand. With the ability to investigate and refine possible drug candidates more accurately and efficiently, it has emerged as an essential tool in the drug development process.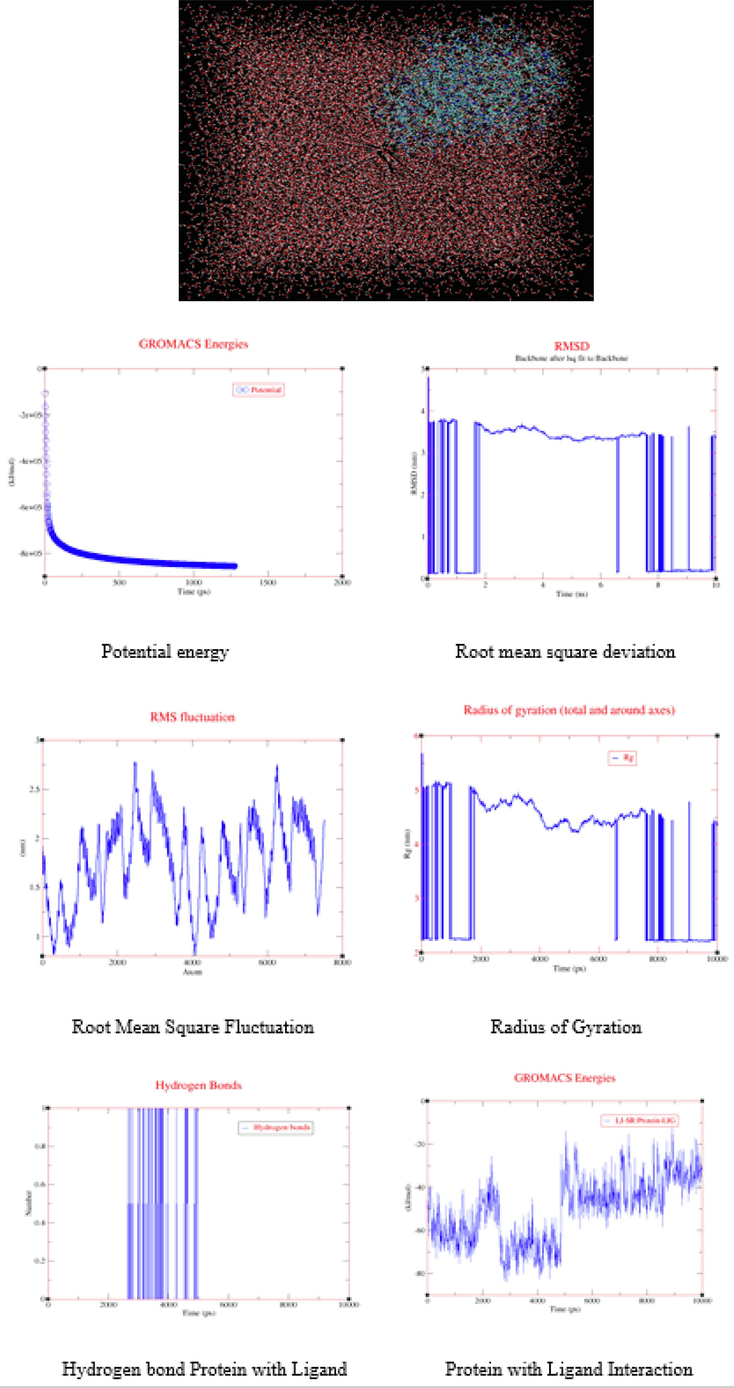
Protein with ligand in water solvent, Molecular dynamics simulation results XM-graphs.
Molecular Dynamics output Data
Energy minimization (potential Energy)
XM-Graph
Energy
ST. Value
Mid Value
End Value
X- axis
Time (ps)
0.000000
612.000000
1275.000000
Y-axis
(kJ/mol)
−106970.359375
−834631.812500
−855515.687500
Root-mean-square deviation
X- axis
Time (ns)
0.0000000
5.0000000
10.0000000
Y-axis
RMSD (nm)
0.0572647
3.3258679
3.3666923
Root-mean-square fluctuation
X- axis
Atom
1
3187
7517
Y-axis
nm
1.9206
1.1966
2.1928
Radius of gyration
X- axis
Time (ps)
0.00000
4720
10,000
Y-axis
Rg (nm)
2.21795
4.35705
4.34491
Y-axis
Rg\sX\N
1.81765
4.17504
4.16415
Y-axis
Rg\sY\N
1.95219
1.86125
1.85801
Y-axis
Rg\sZ\N
1.65036
4.13191
4.11874
Hydrogen Bond Interaction
X- axis
Time (ps)
0.0000000
4970
10,000
Y-axis
Number
0.0000000
0.0000000
1
Y-axis
Pairs within 0.35 nm
0.0000000
2
0.0000000
Protein and Ligand Interaction (LJ-SR: Protein-LIG)
X- axis
Time (ps)
0.00000
4750.000000
10000.000000
Y-axis
(kJ/mol)
−54.137070
−41.228001
−33.075733
3.10 Molecular docking of estrogen receptor with ligand
The process of molecular docking uses the atomic-level binding processes of big or small molecules interacting with protein receptors to calculate a ligand's affinity for a protein. This work used Auto Dock with Vina tools (Andrew et al., 2002) as the molecular docking-DSV analysis and visualization technique. Molecular docking chooses the protein receptor as estrogen (PDB-id 1L2I) (Organisms-Homo sapiens, Expression System- Escherichia coli, resolution − 1.95 Å) and ligand is “2, 3-dimethyl-N-[2-(hydroxy) benzylidene] aniline” (Balachandar et al., 2021). The nuclear hormone receptor for estrogen in target tissues, the steroid hormones, and their receptors control the expression of eukaryotic genes and have an impact on cellular proliferation and differentiation. Protein “chain B” interacted with ALA:491, HIS:488, and ALA:312 amino groups performed as Hydrophobic Alkyl and Pi-Alkyl and Pi-Sigma. The van der Walls of protein groups GLN: B:500, GLN: A:498, LEU: B:497, LEU: A:497, THR: B: 485, LYS: B:481, and HIS: A:501 present in docking further details shown in Fig. 6a. The Electrostatic charge of the Pi-Anion group is ASP: B:484 interacted with the ligand (Uma Maheswari et al., 2023). The hydrogen bond surface donor color is violet and the acceptor is green color and SAS surface blue color range of 25.0,22.5,20 and 17.5, and the green color range of 15.0,12.5,10.0 with these two solid surfaces shown white color is neutral Fig. 6b and surface wire mesh volume 61.361, surface area 294,954 probe radius 1.4 and position of surface is x = 24,4603, y = 8,25319, z = 21,0328 and transparency level 0.75. and ligand N1 is the acceptor and H1 is the donor. Docking best Binding affinity was found at −7.8 kcal/mol, UFF value 266.03 kcal/mol, docking rmsd lower value and upper value described in Table S7.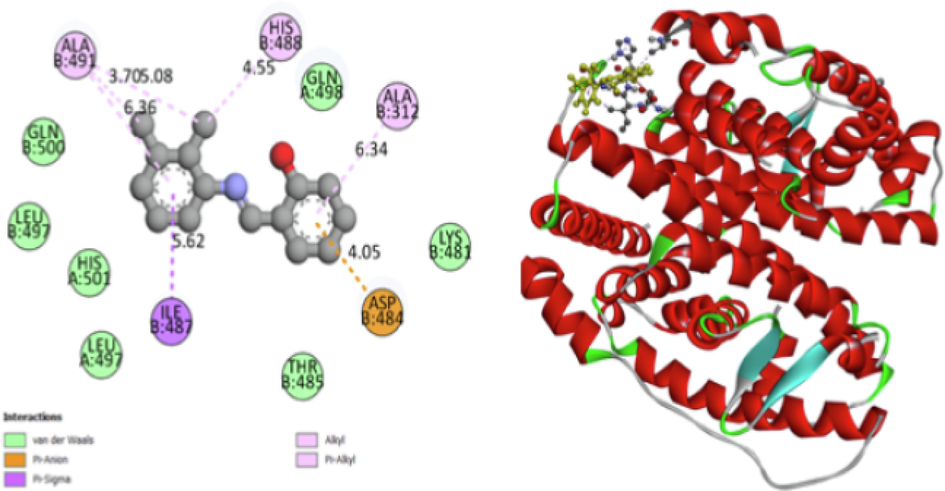
Protein and Ligand in dock bind interaction.
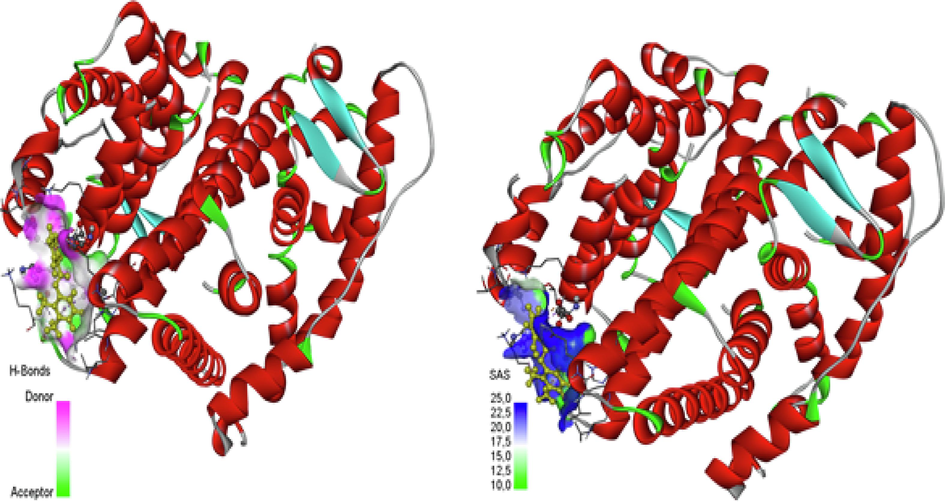
H-Bonds (Donor −Acceptor) and SAS.
4 Conclusion
The stability of the DNHBA molecule, due to the contribution of intermolecular force is confirmed through the data obtained from the molecular geometry, natural bond analysis and hirshfeld surface analysis. The lattice energy due to the pairing of molecules through energy framework is confirmed. The covalent character within the aromatic ring presence is confirmed from electron density ρ(r) and also from the natural bond analysis. On comparison to the RCP, strong repulsive steric effect indication is sustained from the report of AIM function. The substance which interacts with other molecules reactive are taken from the visualization site map of electrostatic potential. The strong kinetic stiffness and the implementation of low organic reactivity are observed in orbital analysis such as Fukui function. The efficacy of the DNHBA molecule is obtained through the process of molecular docking and molecular dynamics. The molecular dynamics study helps to understand the movement of molecules during interaction process between the protein receptor (Homo Sapiens) and ligand (DNHBA). To conclude, these studies make confirmation of the subjected molecule is highly reactive with the protein receptor by giving the best binding affinity at the scale of −7.8 kcal/mol, which can be used for the clinical analysis further through invivo /invitro studies.
CRediT authorship contribution statement
A. Prabakaran: Conceptualization, Methodology, Writing – original draft, Writing – review & editing, Visualization. C. Uma Maheswari: Validation, Methodology, Resources. Noureddine ISSAOUI: Funding acquisition, Project administration. Omar M. Al-Dossary: Formal analysis, Writing – review & editing, Visualization. T. Rajamani: Conceptualization, Software, Resources, Editing Original Draft. T. Gnanasambandan: Software, Methodology, Review, Resources. P. Saravanan: Writing – review & editing, Visualization. M. Vimalan: Writing – review & editing, Writing – original draft. A. Manikandan: Writing – review & editing, Writing – original draft, Visualization.
Acknowledgements
This study was supported by the Researchers Supporting Project no. RSP2024R61 of King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Molecular dynamics simulation on protein using gromacs, international journal of computer science and information. Security. 2011;9(2)
- [CrossRef] [Google Scholar]
- A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys.. 1990;92(9):5397-5403.
- [CrossRef] [Google Scholar]
- Synthesis, quantum computational analysis and molecular docking of 3-(2-hydroxyphenyl)-1-phenyl propanone: a combined experimental and theoretical analysis. Polycycl. Aromat. Compd.. 2023;43(10):8903-8932.
- [CrossRef] [Google Scholar]
- Role of solvents in molecular level interaction, reactivity and spectral characterisation of 2-Amino-3- (((E)-4-(dimethylamino) benzylidene) amino) maleonitrile: anti depressant agent. J. Mol. Liq. 2023;389:122937
- [CrossRef] [Google Scholar]
- Metal chalcogenides: a rich source of non-linear optical materials. Chem. Mater. 2014;26:849-869.
- [CrossRef] [Google Scholar]
- Empirical and computational studies on newly synthesis cyclohexylammonium perchlorate. J. Mol. Struc. 2021;1230:129820.
- [CrossRef] [Google Scholar]
- GaussView, version 5. Shawnee Mission KS: Semichem Inc.; 2009.
- Computational, reactivity, Fukui function, Molecular docking, and spectroscopic studies of a novel (E)-1-Benzyl-3-(2-(Pyrindin-2-yl)Hydrazono)Indolin-2-one. Polycycl. Aromat. Compd.. 2023;1–21
- [CrossRef] [Google Scholar]
- Frisch. M.J., Trucks. G.W., Schlegel. H.B., 2009, Gaussian 09, Revision E.01, Gaussian Inc., Wallingford, CT.
- Synthesis, structural and spectroscopic features, and investigation of bioactive nature of a novel organic-inorganic hybrid material 1H-1,2,4-triazole-4-ium trioxonitrate. J. Mol. Struct.1150 2017:242-257.
- [CrossRef] [Google Scholar]
- Experimental and theoretical study of p-nitroacetanilide. Spectrochim. Acta A Mol. Biomol. Spectrosc.. 2014;117:557-567.
- [CrossRef] [Google Scholar]
- Investigation of hydrogen bonded structure of urea-water mixtures through Infra-red spectroscopy and non-covalent interaction (NCI) theoretical approach. Computational and Theoretical Chemistry. 2021;1199:113218.
- [CrossRef] [Google Scholar]
- Theoretical investigation of the relative stability of Na+He n (n = 2–24) clusters: Manybody versus delocalization effects. The Journal of Chemical Physics. 2014;141:174316.
- [CrossRef] [Google Scholar]
- ́ Anharmonic effects on theoretical IR line shapes of medium strong H(D) bonds. Int. J. Quant. Chem.. 2017;1135(3):483-499.
- [CrossRef] [Google Scholar]
- Effects of the molecular structure on the second-order nonlinear optical properties of stilbazolium derivative single crystals: a review. J. Mater. Chem. C. 2020;8:16668-16690.
- [CrossRef] [Google Scholar]
- Optimization of guar gum galactomannan sulfation process with sulfamic acid. Biomass Conversion and. 2023;Biorefinery,13(11),:10041-10050.
- [CrossRef] [Google Scholar]
- Über die zuordnung von wellenfunktionen und eigenwerten zu den einzelnen elektronen eines atoms. Physica. 1934;1:104-113.
- [CrossRef] [Google Scholar]
- Synthesis, growth, spectral, thermal, mechanical and optical properties of 4-chloro-4′dimethylamino-benzylidene aniline crystal: a third order nonlinear optical material, spectrochim. acta - Part a Mol. Biomol. Spectrosc. 2009;74:78-83.
- [CrossRef] [Google Scholar]
- Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem.. 2012;33(5):580-592.
- [CrossRef] [Google Scholar]
- An introduction of the quantum theory of atom in molecule. Wiley-VCH Verlag Gmbh; 2007. p. :1-33. https://doi.org/10.1002/9783527610709.ch1
- H.J. Michal4 Program Warsaw Vibrational Energy Distribution Analysis VEDA 2004.
- DFT and molecular docking study of the effect of a green solvent (water and DMSO) on the structure, MEP, and FMOs of the 1-ethylpiperazine-1,4-diium bis(hydrogenoxalate) compound. J. Mol. Liq.. 2023;369:120851.
- [CrossRef] [Google Scholar]
- Synthesis, DFT, solvent effect and biological attributes of NLO active 4- bromo-2-((2-(2, 4-dinitrophenyl) hydrazono) methyl) phenol-potent drug anti-brain cancer. J. Mol. Struct. 2023;1289:135839
- [CrossRef] [Google Scholar]
- First principal studies of spectroscopic (IR and raman, UV-visible), molecular structure, linear and nonlinear optical properties of L-arginine p-nitrobenzoate monohydrate (LANB): a new non-centrosymmetric material, spectrochimica acta. Part a, Mol. Biomol. Spectrosc.. 2015;147:84-92.
- [CrossRef] [Google Scholar]
- Quantum chemical design of non-linear optical materials by sp 2-hybridized carbon nanomaterials: issues and opportunities. J. Mater. Chem. C. 2013;1:5439-5449.
- [CrossRef] [Google Scholar]
- Principles and applications of Nonlinear optical materials. Chapman & Hall, London. 1993
- [CrossRef] [Google Scholar]
- Scaled quantum chemical studies of the molecular structure and vibrational spectra of minoxidil. Spectrosc. Lett.. 2015;48(1):63-73.
- [CrossRef] [Google Scholar]
- Hardness, softness, and the fukui function in the electronic theory of metals and catalysis. Proc. Natl. Acad. Sci.. 1985;82:6723-6726.
- [CrossRef] [Google Scholar]
- Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1998;88:899-926.
- [Google Scholar]
- Rekik , N., Ghalla, H., Issaoui, N., Oujia, B., Wojcik, M.J., 2007. Infrared spectral density of hydrogen bonds within the strong anharmonic coupling theory: Quadratic dependence of the angular frequency and the equilibrium position of the fast mode. J. Mol, Struct. THEOCHEM 821 (1–3), 58 – 70, theochem.2007.06.025. https://doi.org/10.1016/j.
- Properties and reactivities of niclosamide in different media, a potential antiviral to treatment of COVID-19 by using DFT calculations and molecular docking. Biointerface Res. Appl. Chem.. 2020;10(6):7295-7328.
- [CrossRef] [Google Scholar]
- Classification of chemical bonds based on topological analysis of electron localization functions. Nature. 1994;371:683-686.
- [CrossRef] [Google Scholar]
- J. Appl. Cryst.. 2021;54(3):1006-1011.
- [CrossRef]
- Synthesis, growth and characterization of 4-bromo-4′chloro benzylidene aniline—A third order non linear optical material, spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011;78:935-941.
- [CrossRef] [Google Scholar]
- 2-[(E)-(2, 3-dimethylphenyl) iminomethyl] phenol. Acta Crystallogr. E. 2010;66:o2439
- [CrossRef] [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem.. 2010;30, 31(2):455-461.
- [CrossRef] [Google Scholar]
- Combined quantum-mechanics/molecular-mechanics, molecular docking studies on hemorrhoid drug. Results in Chemistry. 2023;5:100962
- [CrossRef] [Google Scholar]
- Weinhold. F, Landis.C.R., 2005, Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective, Cambridge University Press, Cambridge. https://doi.org/10.1017/CBO9780511614569.
- The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986;108:5708-5711.
- [CrossRef] [Google Scholar]
- Growth and characterization of l-histidine nitrate single crystal, a promising semiorganic NLO material. Mater. Chem. Phys. 2008;108:192-195.
- [CrossRef] [Google Scholar]
- Correlations between molecular structures and thirdorder non-linear optical functions of heterothiometallic clusters: a comparative study. Coord. Chem. Rev.. 2007;25:111-141.
- [CrossRef] [Google Scholar]
- N. Mhadhbi, N. Issaoui, H. Walid S., A. Jahoor M., E. Abdelmonein S., A. Mohd, N. Houcine, R. Badraoui, Physico-Chemical Properties, Pharmacokinetics, Molecular Docking and In-Vitro Pharmacological Study of a Cobalt (II) Complex Based on 2-Aminopyridine, ChemistrySelect, 7, 321 (2022) e202103592. DOI:10.1002/slct.202103592.
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2024.103196.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




