

Translate this page into:
Comprehensive in silico discovery of c-Src tyrosine kinase inhibitors in cancer treatment: A unified approach combining pharmacophore modeling, 3D QSAR, DFT, and molecular dynamics simulation
⁎Corresponding authors. s.khamouli@univ-biskra.dz (Saida Khamouli), mrehman@ksu.edu.sa (Md. Tabish Rehman)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Objective
To investigate c-Src, a non-receptor tyrosine kinase dysregulated in various cancer types including colon, breast, and pancreatic cancers, as a potential drug target for cancer therapy.
Methods
Ligand-based pharmacophore modeling and 3D-QSAR analysis on a dataset of 34c-Src tyrosine kinase inhibitors were employed. The established pharmacophore model (DDRRR_1) features two hydrogen bond donor (D) and three aromatic ring (R) features, exhibiting favorable parameters (R2 = 0.926; Q2 = 0.895; F value = 47.9). Hypothesis validation, enrichment analysis, and contour plot analysis were conducted, followed by virtual screening of a PubChem database using the optimized pharmacophore model and filtering based on the Lipinski rule of five.
Results
The most promising inhibitors underwent multistep molecular docking, density Functional Theory (DFT) analysis, ADMET assessments, molecular dynamics simulation, and PCA. CID_70144047 emerged as the most promising hit with all the above favorable properties.
Conclusion
The study provides a comprehensive approach for identifying novel c-Src tyrosine kinase inhibitors, highlighting CID_70144047 as a promising leads with potential therapeutic applications in cancer treatment.
Keywords
c-Src tyrosine kinase
Pharmacophore modeling
Virtual screening
Drug discovery
DFT analysis
Molecular dynamics simulation

1 Introduction
Cancer, characterized by uncontrolled cell growth, presents a global health crisis resulting in significant global mortality (Rashid et al., 2019). The World Health Organization's 2020 cancer report highlighted alarming statistics of 19.3 million new cases and 10 million deaths, with projections suggesting a surge to 28.4 million new cases by 2040 (Sung et al., 2021). This underscores the urgent requirement for innovative anticancer treatments that enhance efficacy, minimize side effects, and address drug resistance (Wang et al., 2019). Consequently, there is a critical need for the development of novel anticancer agents with improved efficacy and reduced toxicities (Carugo and Draetta, 2019).
In modern medicinal chemistry, there is a growing emphasis on nitrogen-containing cyclic structures like pyrimidines (Romasanta et al., 2018). Notably, FDA-approved anticancer drugs (Ibrutinib, Cabozantinib, Gefitinib, Erlotinib, and Sorafenib) incorporate pyrimidine scaffolds to inhibit critical protein kinases. The pyrimidine fragment, a key component of the purine core, holds significance in fragment-based drug discovery for anticancer research (Zhao and Dietrich, 2015). Purine structures, recognized as privileged scaffolds, establish hydrogen bonds with various molecular targets, enhancing pharmacokinetics, pharmacology, and toxicological properties (Khamouli et al., 2022b). Consequently, researchers have explored numerous purine derivatives for their antitumor potential (Yoon et al., 2018).
Protein kinases play a crucial role in cancer biology, regulating cellular functions like proliferation, cell cycle regulation, apoptosis, motility, growth, and differentiation (Cicenas et al., 2018). Among these, SFKs (Src Family Tyrosine Kinases), including c-Src, are particularly pivotal, showing deregulation in various cancers. Tyrosine kinases represent the most targeted protein class in anticancer therapy (Angelucci, 2019). The vertebrate Src kinase family consists of 11 members (Blk, Brk, Fgr, Frk, Fyn, Hck, Lck, Lyn, c-Src, Srm, c-Yes), and their overexpression, deregulation, or mutation has been observed in various human malignancies, including colon, breast, and pancreatic cancers (Amata et al., 2014).
CADD (Computer-Aided Drug Design) methods hold potential for discovering potent anticancer drugs (Yang, 2010). Approaches like pharmacophore modeling, virtual screening, molecular docking, and molecular dynamics simulations expedite the discovery and evaluation of compounds (Samad et al., 2022). Pharmacophore modeling rapidly identifies potential lead compounds (Mohan et al., 2020), while 3D QSAR (3D Quantitative Structure-Activity Relationship) analysis reveals the relationship between biological activity and molecular properties (Lokwani et al., 2012).
In this study, CADD methods were employed to identify potential cancer therapeutic agents. We utilized purine derivatives as c-Src tyrosine kinase inhibitors to create a pharmacophore model, and conducted 3D-QSAR analysis. Virtual screening, and molecular docking were performed to understand interactions with c-Src tyrosine kinase. Validation through chemical reactivity assessments, in silico ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxocity) predictions, and molecular dynamics (MD) simulations highlighted CID_70144047 as a promising c-Src tyrosine kinase inhibitor, with implications for future cancer research and therapy.
2 Materials and methods
2.1 Computational studies
Computational studies were performed using HP Intel(R) i5 CPU with 6.0 GB RAM. Schrodinger Suite 2018–4 was employed for pharmacophore modeling and virtual screening. MD simulations was performed using Gromacs 2020.4 on Intel Xenon E3-1245 processor 32 GB RAM, and NVIDIA Quadro P5000 GPU card (Iqbal et al., 2021).
2.2 Dataset generation and alignment
The dataset comprised 34 purine derivatives, recognized as c-Src tyrosine kinase inhibitors, sourced from literature (Sharma, 2016). ChemSketch was employed to generate 2D molecular structures (Table S1 in supplementary material), which were later converted to 3D using Schrodinger's Ligprep module. Energy minimization for low-energy conformers was carried out by OPLS_2005 forcefield (Dixon et al., 2006). Each ligand generated up to 32 stereoisomers, considering all ionization states at pH 7.0 with Epik. Ligands underwent energy minimization until reaching an RMSD of 0.01 Å, forming the basis for modeling studies.
2.3 Pharmacophore model generation
The pharmacophore model was constructed using the Phase software's “Develop Pharmacophore Hypothesis” protocol, utilizing aligned conformations of purine derivatives. (Mohan et al., 2020). The dataset comprised c-Src tyrosine kinase inhibitors categorized into active (pIC50 > 6.40) and inactive (pIC50 > 5.80) sets based on pIC50 values. The pharmacophore model, featuring 4 to 5 sites, was developed with a maximum of 5 sites and a minimum of 4 sites. Phase (v4.0) performed flexible ligand superposition, with the most active compound as the template, considering default settings of 10 conformations per rotatable bond and up to 100 conformers. Conformations of the compounds were varied, and only one alignment per ligand was retained.
Pharmacophore feature like hydrogen-bond acceptor (A), hydrogen-bond donor (D), hydrophobic group (H), negatively charged group (N), positively charged group (P), and aromatic ring (R) were assigned to the molecules using Phase's predefined features. Multiple common pharmacophore hypotheses were generated, scored, and ranked based on active and inactive survival scores.
2.4 Validation of pharmacophore model
The highly-ranked pharmacophore hypothesis underwent validation via PLS (Partial Least Square) analysis. The Phase module was employed to develop an atom-based 3D-QSAR model for predicting the potential c-Src tyrosine kinase inhibitory activity (Rücker et al., 2007). Atom-based QSAR models consider occupancy properties such as hydrogen bond donor, hydrogen bond acceptor, hydrophobic, negative ionic, and positive ionic features. Molecule alignment for the 3D QSAR model utilized Phase (v4.0) shape screening, aligning c-Src tyrosine kinase inhibitors with DDRRR_1 pharmacophore model. The dataset was split into a 70 % training set and a 30 % test set with default parameter settings. The generated QSAR models were ranked based on statistical parameters including R2 (correlation coefficient of the training set), Q2 (correlation coefficient of the test set), SD (Standard Deviation), Pearson-r values, and Y-randomization (Khamouli et al., 2022a; Khamouli et al., 2018). Additional external validation tests included Tropsha and Golbraikh criteria (Golbraikh and Tropsha, 2002; Tropsha, 2010), rm2 metric analysis (Roy et al., 2013), and PLS factor 5 to establish QSAR model robustness and predictiveness. Contour maps facilitated model visualization.
The pharmacophore model's accuracy and specificity in virtual screening were validated using 1000 decoy molecules enriched with 10 active molecules from the Schrödinger database. The Phase module's “Hypothesis Validation Tool” utilized the hypothesis file, decoy, and active datasets to calculate performance parameters. Statistical parameters included RF (Enrichment Factors), RIE (Robust Initial Improvement), BEDROC (Boltzmann Enhanced Discrimination of Receiver Operating Characteristic), AUC (Area Under Curve), and ROC (Receiver Operating Characteristics) were computed to assess and validate the accuracy of the pharmacophome model in virtual screening (Rizzi and Fioni, 2008).
2.5 Database creation and screening
The 3D coordinates of 111,415 compounds were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/) and processed using Ligprep. Tautomers, ionization states at pH 7.0, and partial atomic charges were computed using OPLS-2005 forcefield. Virtual screening, employing DDRRR_1 pharmacophore and “PhaseLigand Screening” efficiently pinpointed potential c-Src tyrosine kinase binders. Maestro's “Virtual Screening Workflow” was applied with prefiltering based on Lipinski's Rule of 5 (such as Molecular mass < 500 Da, Number of H-bond donor < 5, Number of H-bond acceptor < 10, Octanol-Water partition coefficient (log P) < 5) to eliminate unfit ligands (Friesner et al., 2006). The filtered database underwent a three-step docking process: Glide HTVS (High Throughput Virtual Screening), Glide SP (Standard Precision) of top 10 % hits in HTVS, and Glide XP (Extra Precision) of top 10 % hits in SP. Only compounds with docking scores below −10.5 kcal mol−1 were retained. ADMET properties of the top selected compounds were predicted using Maestro's QikProp and the ProTox-II web server.
2.6 Protein preparation and receptor grid generation
The c-Src tyrosine kinase crystal structure (PDB ID: 1Y57) (Cowan-Jacob et al., 2005) was downloaded from Protein Data Bank (http://www.rcsb.org) having 1.91 Å resolution. “Protein Preparation Wizard” of Schrodinger removed water molecules, added hydrogen atoms, assigned atom charges, and energy-minimized using OPLS-2005 force field keeping RMSD (Root Mean Square Deviation) ≤ 0.3 Å. The glide grid file was generated using Schrödinger, with coordinates extracted from the crystallized ligand MPZ at the active site (X: 18.69 Å, Y: 13.24 Å, Z: −19.81 Å). Docking with the co-crystal ligand in XP mode was performed using Glide, and RMSD was validated (Supplementary Figure S1).
2.7 Molecular docking
Maestro's “Virtual Screening Workflow” was employed to dock and score compounds based on best fitness scores. The workflow offers different levels of docking precision, including HTVS, SP, and XP (Samad et al., 2022). Initially, HTVS calculations were performed, followed by SP and finally XP mode to refine the ligand poses. The ligand-receptor interactions were visualized using Discovery Studio 2020 (Selvaraj et al., 2012).
2.8 DFT calculation
Top screened compounds underwent DFT (Density Functional Theory) calculations using Gaussian 09 software to determine LUMO (Lowest Unoccupied Molecular Orbital), and the HOMO (Highest Occupied Molecular Orbital) quantum properties. B3LYP functional theory with a 6-31G* basis set and hybrid DFT with Becke's 3-parameter exchange potential were applied for HOMO and LUMO calculations.
2.9 In silico ADMET prediction
The top five compounds from virtual screening and docking underwent ADME (Absorption, Distribution, Metabolism, and Excretion) analysis with Maestro's QikProp module. This analysis predicted various ADME properties, including solubility (LogS), partition coefficient (LogPo/w), permeability (PCaco), skin permeability (LogKp), blood–brain partition coefficient (LogBB), human serum albumin binding (LogKhsa), hERG inhibition (loghERG), oral absorption, and Rule of Five and Rule of Three violations. Additionally, toxicity assessment was performed using the ProTox-II web server (https://tox-new.charite.de/protox_II/), predicting mutagenicity, carcinogenicity, immunotoxicity, and hepatic toxicity.
2.10 Molecular dynamics simulation (MDS)
MDS was performed to assess protein–ligand complex stability (Khan et al., 2022). Gromacs 2020.4 was employed to conduct MDS using CHARMM36-all atom force field and TIP3P water. CGenFF (CHARMM General Force Field) was used to generate ligand topology. The complex was placed in a dodecahedron box with 10 Å minimum distance from box boundaries. Neutralization included 0.15 M NaCl and counterions (Na+/Cl-). The system's energy underwent steepest descent minimization (max. 50,000 steps). NVT (Isothermal-Isochoric) and NPT (Isothermal-Isobaric) ensembles were run at 300 K and 1.0 bar, using Berendsen thermostat (Berendsen et al., 1984) and Parrinello-Rahman barostat (Parrinello and Rahman, 1981). A 100 ns production run with 2 fs time-step used the Leap-frog integrator and applied LINCS constraints. RMSD, RMSF (Root Mean Square Fluctuation), and number of hydrogen bonds were analyzed during MDS for system dynamics and stability.
2.11 Principal component analysis (PCA)
Conformational flexibility of c-Src, with and without CID_70144047 was analyzed using Bio3D's PCA (Grant et al., 2021). This involved removing translational and rotational motions, superimposing atomic coordinates onto a reference structure, and calculating a positional covariance matrix to represent atomic coordinate relationships and fluctuations. The diagonalization of this matrix provided eigenvalues and eigenvectors, indicating mean-square fluctuation along each eigenvector. The covariance matrix (C) was calculated to evaluate c-Src's conformational flexibility and its interactions with ligands as follows: where, N, xi/j and < xi/j > represent the number of Cα-atom, the Cartesian coordinate of the ith/jth Cα-atom, and time average of all the conformations respectively.
3 Results and discussion
3.1 Pharmacophore modelling
A dataset of 34 compounds, categorized into active and inactive sets based on experimental data, was used to generate 22 pharmacophore variants. The top five hypotheses, detailed in Table 1, were scored, and pharmacophore hypothesis DDRRR_1 emerged as the best with favorable scores in survival activity (5.723), survival-inactive (2.551), vector (1), volume (0.882), selectivity (2.647), and site matches (10). The alignment of active and inactive compounds with their pharmacophoric features is depicted in Fig. 1A,B.
ID
Survival
Survival-inactive
Site Score
Site Vector
Volume
Matches
Selectivity
DDRRR_1
5.723
2.551
1
1
0.882
10
1.841
ADDRR_2
5.479
2.518
1
1
0.882
10
1.596
ADDRR_3
5.435
2.467
1
1
0.882
10
1.552
AADDR_2
5.163
2.482
1
1
0.882
10
1.280
AADDR_3
5.155
2.531
1
1
0.882
10
1.273
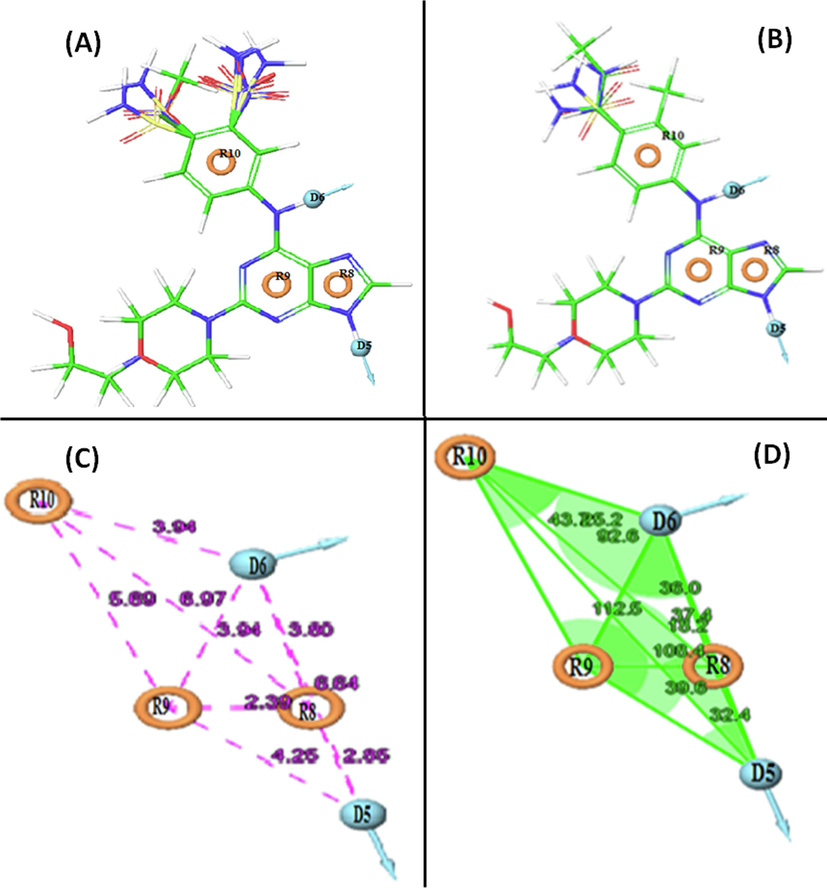
(A) Mapping of the active compounds onto the pharmacophore, and (B) Mapping of inactive compounds onto the pharmacphore. Represents pharmacophore model DDRRR_1 with respect to (C) intersite distances in Å, and (D) angles between the pharmacophoric points.
The identified five-point pharmacophore (DDRRR_1) includes two hydrogen bond donor (D) features related to the amine group at the 9′ position of the purine ring and the amine group between the purine and phenyl rings. Additionally, three aromatic ring (R) features are associated with the purine rings and the phenyl ring on both adjacent sides of the compounds. These chemical features are deemed essential for the biological activity within the dataset, emphasizing their significance for screening and designing potential compounds.
The model's discriminatory ability was evaluated using a test against 1000 decoy molecules from the Schrodinger database. It successfully identified all active compounds in the hit list, showcasing strong performance. The pharmacophore model DDRRR_1 exhibited an RIE value of 18.14, and an EF 1 % value of 101, indicating its superior ranking compared to random distribution. The AUC of the ROC curve, a reliable metric for model evaluation, yielded a favorable value of 1 for DDRRR_1. Additional details on angles and distances among various pharmacophoric features of the selected model are provided in Fig. 1C,D and Table S2 in the supplementary material.
3.2 Atom based 3D- QSAR and validation
The top-scoring hypothesis, DDRRR_1, was subjected to a QSAR study using PLS regression, aligning pharmacophoric features with optimized bioactive ligands. PLS analysis, a chemometric technique, correlated activity (dependent variable) with binary values (independent variable) to develop the QSAR model. The dataset, divided into training (24 molecules) and test (9 molecules) sets, yielded significant statistical outcomes with five factors. The resulting 3D QSAR model was validated on both sets to assess its reliability.
The selection of the best QSAR model was guided by multiple criteria such as R2 and Q2 values, which represent how well the model fits the training data and the predictive power of the generated model respectively. It displayed a robust cross-validated R2 value (>0.6) and a high Q2 value (>0.5). Moreover, the SD values were below 0.3, the root mean square error (RMSE) was minimal, and the F-test value (indicating the overall statistical significance of the model) was substantial. These factors collectively informed the choice of the best QSAR model. The hypothesis exhibited a strong correlation with an R2 value of 0.926 for the training set, accompanied by a high F-test value of 47.9. The Pearson-R value of 0.950 indicated a significant correlation, further reinforced by the corresponding low P-value (4.03 e-10). The SD was 0.085, and the RMSE value was 0.120, as detailed in Table 2. Visual representation of the predicted versus actual pIC50 values for both the training and test sets is presented in Figure S3 (supplementary material), while Table S1 (supplementary material) provides the actual pIC50 and residual pIC50 values for both sets.
ID
PLS
Factors
SD
R2
F
P
RMSE
Q2
Pearson R
DDRRR_1
1
0.494
0.175
4.9
0.037
0.270
0.441
0.686
2
0.415
0.443
8.8
0.001
0.330
0.158
0.528
3
0.347
0.628
11.9
9.28e-005
0.160
0.795
0.897
4
0.213
0.866
32.5
1.17e-008
0.100
0.918
0.963
5
0.162
0.926
47.9
4.03e-010
0.120
0.895
0.950
According to Galbraith and Tropsha criteria along with r2m, matrix the proposed QSAR model exhibits strong predictive abilities (Table 3) (Golbraikh and Tropsha, 2002; Tropsha, 2010). With a robust R2 and Q2 values of 0.903 and 0.895 for the test set, the model surpasses the established thresholds of 0.5. The R2pred exceeds 0.5, and the Degree of Lack of Relationship falls within the satisfactory range of 0.85 to 1.15 for both K and K', indicating a robust relationship between variables. The r2m value, surpassing 0.5, further enhances the model's credibility. External validation results affirm the high agreement for the atom-based 3D-QSAR models, highlighting their potential in predicting the activities of new derivatives.
Parameters
Threshold score
Model score
Q2 LOO
Q2Loo < 0.5
0.895
0.856
0.903
< 0.1
0.069
< 0.1
0.004
K
0.85 ≤ K ≤ 1.15
1.005
K'
0.85 ≤ K' ≤1.15
0.994
< 0.3
0.065
r2m > 0.5
0.666
r'2m > 0.5
0.842
> 0.5
0.754
Furthermore, the Y-randomization test was conducted through 10 random shuffles of the dependent variable i.e. biological activity (supplementary Table S3). These shuffles, preserving the original independent variable matrix, produced new QSAR models with notably lower Q2 and R2 values, highlighting the importance of the initial dependent variable arrangement for meaningful correlations.
3.3 Contour plot analysis
Contour plot analysis revealed key pharmacophoric requirements in the molecular structure, where blue cubes indicated positive contributions, and red cubes represented negative contributions. For highly active compound 20 (Fig. 2A), favorable hydrogen bond donor groups were indicated by blue cubes at specific positions, such as the NH groups of the purine ring (R8 feature) and the NH between the phenyl ring (R9 feature) and pyrimidine (R10 feature) rings, while red cubes around the bridged sulfone group at the 4″ position suggested potential decreased activity. The withdrawing features (Fig. 2B) highlighted by blue cubes were mainly located around the purine structure due to the amine (NH) group at the 6″ position of the purine ring. Numerous blue cubes around the purine and phenyl rings (Fig. 2C) indicated the dominance of hydrophobic moieties in activity. Enhanced activity was associated with positive ionizable groups at the 3″ and 5″ positions of the benzene structure, while the 4″ position of the benzene ring, indicated by red cubes, hinted at potential decreased activity (Fig. 2D).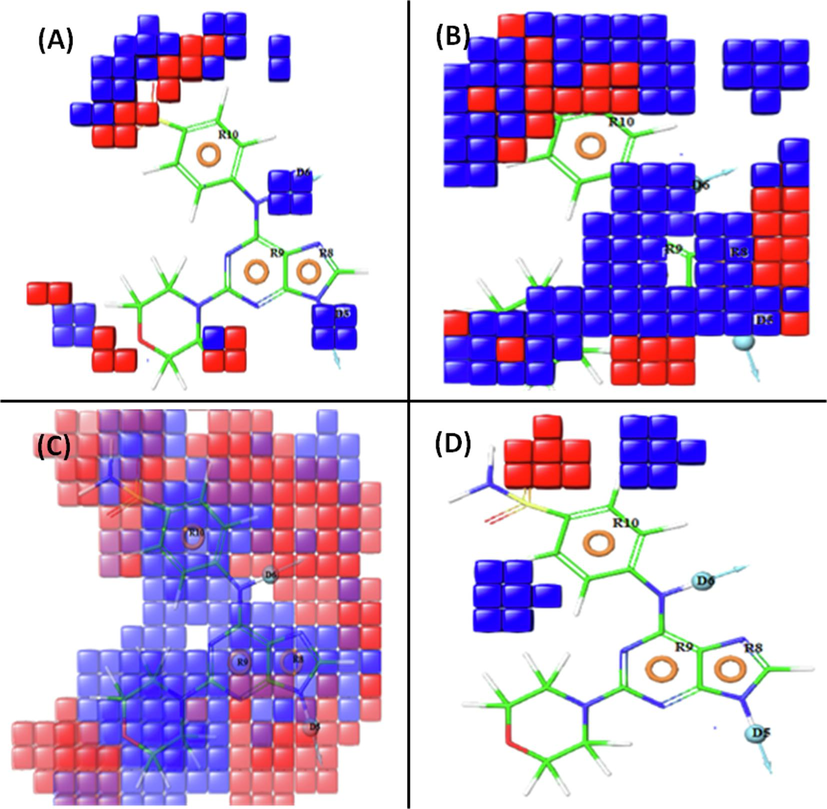
Pictorial representation of the contour plots generated of the active compound (20) in the context of (A) hydrogen bond donor effects, (B) electron withdrawing groups, (C) hydrophobic interactions, and (D) negative ionizable groups.
3.4 Virtual screening and molecular docking
The pharmacophoric model DDRRR_1 guided a 3D database search for compounds meeting its criteria. Initially, compounds with pharmacophore fitness scores ranging between 2 and 1.3 were selected. Further refinement through Glide’s “Virtual Screening Workflow”, including Lipinski's rule of five and molecular docking, resulted in the identification of five hits (CID_70144047, CID_11291395, CID_22844919, CID_134165918, and CID_60151031) characterized by favorable interactions with the active site of Src tyrosine kinase.
Molecular docking explored the binding modes of the top five hits with Src tyrosine kinase. The binding interaction poses were carefully examined and visualized using the Maestro interface and Discovery Studio Visualizer software (Fig. 3). Analysis of docking scores, and binding free energy highlighted substantial contributions from hydrogen bonding, hydrophobic forces, and π-π stacking interactions in the binding process with nearby residues of Src tyrosine kinase (Table 4).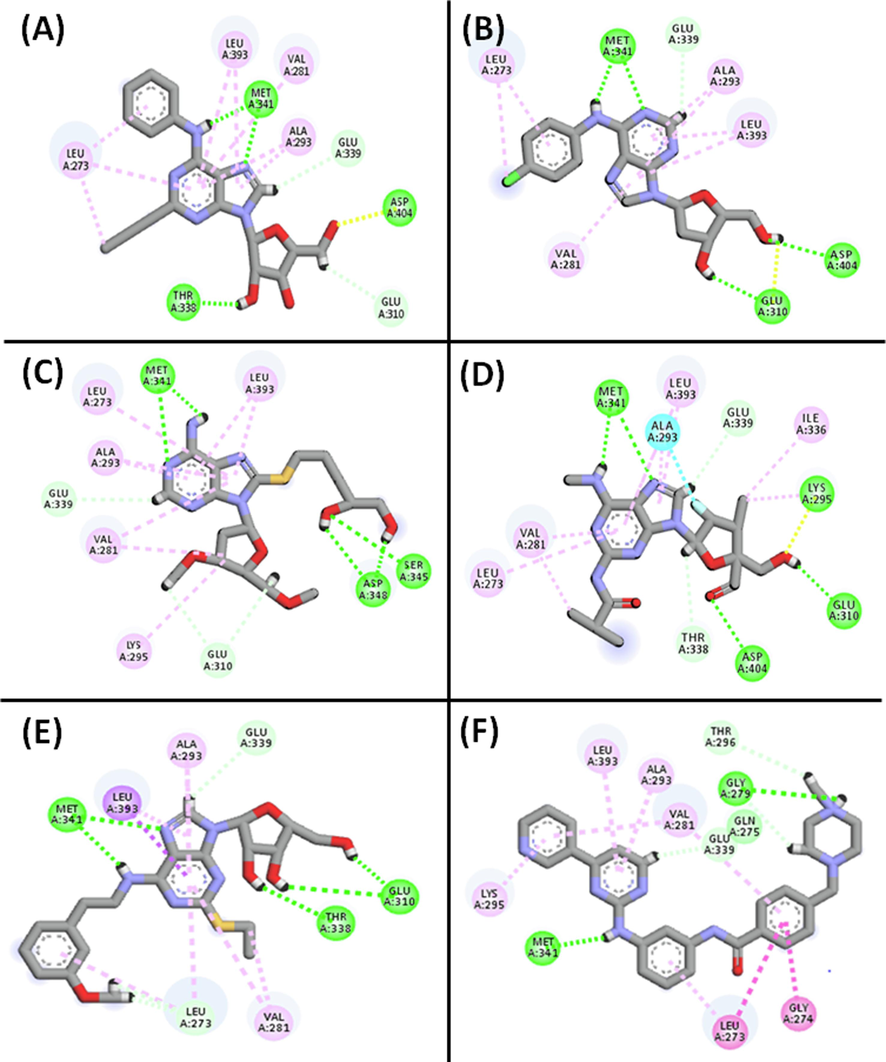
2D and 3D interactions of the top hits with the active sites of c-Src tyrosine kinase. (A) CID_70144047, (B) CID_11291395, and (C) CID_22844919, (D) CID_134165918, (E) CID_60151031, and (F) Co-crystalized ligand respectively.
Pubchem ID
Docking score (kcal/mol)
Glide
Energy
H-bond interaction (Å)
Hydrophobic
interactions
70,144,047
−11.401
−46.306
THR338….HO:Lig (2.36)
MET341….HN: Lig(2.35)
MET341…..N: Lig (2.36)
ASP404….O: Lig (1.97)VAL281, LEU393, VAL173, ALA293, LEU273
11,291,395
−11.134
−52.849
MET341…..N: Lig (1.98)
MET341….HN: Lig(1.80)
ASP404….HO: Lig(2.23)
GLU310….HO: Lig(1.97)
GLU310….HO: Lig(1.82)VAL281, LEU393, ALA293, LEU273
22,844,919
−10.762
−47.721
MET341…..N: Lig (2.32)
MET341….HN: Lig(2.34)
THR338….HO:Lig (1.84)
GLU310….HO: Lig (1.72)VAL281, LEU393, LYS295, ALA 293, LEU273
134,165,918
−10.539
−47.566
MET341…..N: Lig (2.44)
MET341….HN: Lig(2.18)
ASP404…..O: Lig (1.93)
GLU310….HO: Lig (2.00)
LYS295….O: Lig (2.17)VAL281, LEU393, ALA393, LEU273, ILE336
60,151,031
−10.502
−53.731
MET341…..N: Lig(2.31)
MET341….HN: Lig(2.16)
THR338….HO:Lig (2.12)
GLU310….HO: Lig (1.70)
GLU310….HO: Lig (2.85)VAL281, LEU393, ALA293, LEU273
Co-Crystal ligand
−9.488
−45.152
MET341…..N: Lig (2.31)
GLU273….HO: Lig(2.56)VAL281, LYS295, LEU393, ALA293, LEU273, GLY274
The top five hits exhibited high scores toward the target, ranging from −10.502 to −11.401 kcal/mol, surpassing the reference ligand (MPZ) with a docking score of −9.488 kcal/mol, indicating stronger binding affinity. Additionally, these hits demonstrated better binding energies (-46.306 to −53.731 kcal/mol) compared to the reference compound, reflecting robust binding affinity towards c-Src tyrosine kinase. For instance, CID_70144047 formed four conventional hydrogen bonds with ASP404, THR338, and MET341 (two bonds), and engaged in hydrophobic interactions with VAL281, LEU393, VAL173, ALA393, and LEU273. Interactions with key residues like SER345, ASP348, ASP404, THR338, MET341, GLU310, and LYS295 confirmed the placement of hits within c-Src tyrosine kinase's binding pocket (Table 4). All the hits, like reference ligand, formed hydrogen bonds with Met341, indicating their potential as competitive inhibitors (Grant et al., 2006).
3.5 Frontier molecular orbitals (FMO)
FMOs (Frontier Molecular Orbitals) encompass a molecule’s HOMO and the LUMO, representing its electron-donating capacity and electron-accepting capacities respectively. The energy gap (ΔE) between HOMO and LUMO, crucial for understanding reactivity, kinetic stability, and chemical properties, was calculated in this study (Karabacak et al., 2012). The corresponding energy values and contour diagrams of these molecular orbitals can be found in Table S4 (supplementary materials) and Fig. 4, respectively. The DFT analysis revealed HOMO and LUMO values from −4.136 to −7.319 and −1.225 to −2.530, respectively, with an energy gap (ΔE) ranging from 1.877 to 4.979 (Table S4), providing insights into electron-binding properties and indicating kinetic stability and chemical reactivity, with smaller gaps suggesting higher reactivity.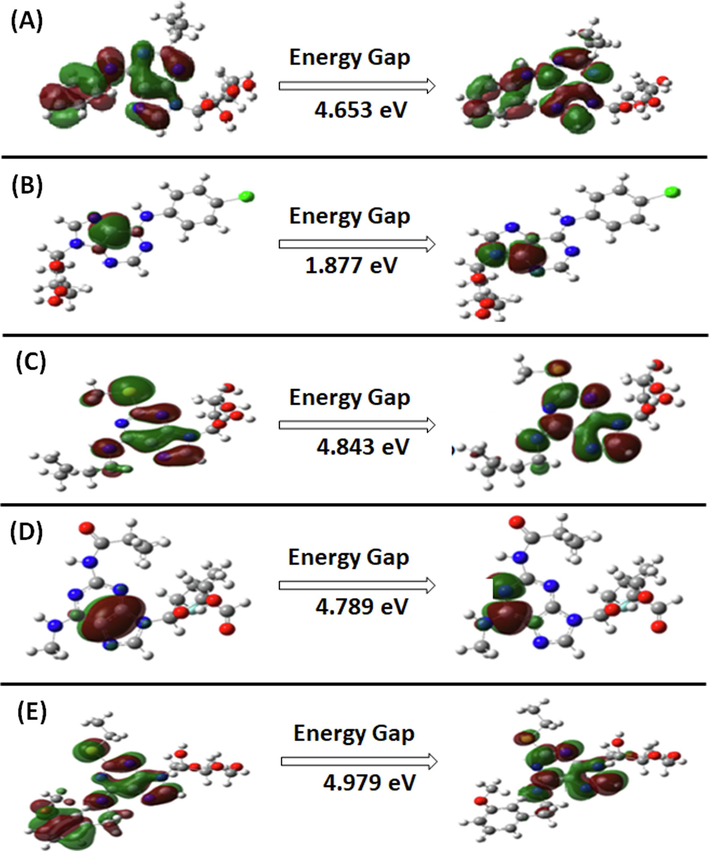
The predicted DFT plots of the HOMO, and LUMO. (A) CID_70144047, (B) CID_11291395, (C) CID_22844919, (D) CID_134165918, and (E) CID_60151031.
3.6 In silico predicted ADMET
We employed in silico tools, including ADME prediction and QikProp (Ioakimidis et al., 2008), and ProTox-II, to access the ADMET features of the top five compounds. All five compounds adhered to Lipinski's rule of five and the rule of three, indicating favorable drug-like properties. QikProp analysis revealed promising pharmacokinetic parameters, with partition coefficient (QPlogPo/w; in 1.085 to 2.039 range), aqueous solubility (QPlogS; in −4.439 to −3.580 range), human serum albumin (QPksha) binding (in −0.541 to 0.238 range), brain/blood barrier values (in −1.890 to −0.997 range), percentage of human oral absorption (in 74.568 to 82.345 range), and permeability across the gut-blood barrier (QPPCaco; in 179.402 to 410.245 range) were within acceptable limits Table S5 (supplementary materials) (Artursson and Karlsson, 1991). Additionally, compound permeability through Madin-Darby Canine Kidney Cells (QPlogMDCK), indicating transport across distal renal epithelia, was within the desired range. Furthermore, predicted toxicity evaluations suggested non-mutagenic, non-carcinogenic, non-immunotoxic, and non-cytotoxic properties for compounds CID_70144047 and CID_22844919, while the reference compound MPZ showed positive immunotoxicity and hepatoxicity (Table S6 supplementary materials).
3.7 Analysis of molecular dynamic simulation
Based-on the findings of multistep molecular docking, DFT analysis, and ADMET assessment, the compound CID_70144047 was found to be the most promising inhibitor of c-Src. Hence, it was selected for further analysis by molecular dynamics simulation.
3.7.1 Root mean square deviation (RMSD)
The stability of a protein–ligand complex can be assessed through RMSD, which measures the deviation of the complex's structure from its initial conformation (AlAjmi et al., 2018; Jairajpuri et al., 2021). For c-Src, Cα-atoms RMSD remained relatively stable during the 20–100 ns simulation, averaging 4.2 Å (Fig. 5A). Similarly, the RMSD of c-Src-CID_70144047 complex fluctuated initially but reached a stable state as favorable interactions were formed. The average RMSD for the c-Src-CID_70144047 complex was 4.8 Å, which was comparable to c-Src alone, confirming the formation of a stable complex within the acceptable threshold of 2.5 Å.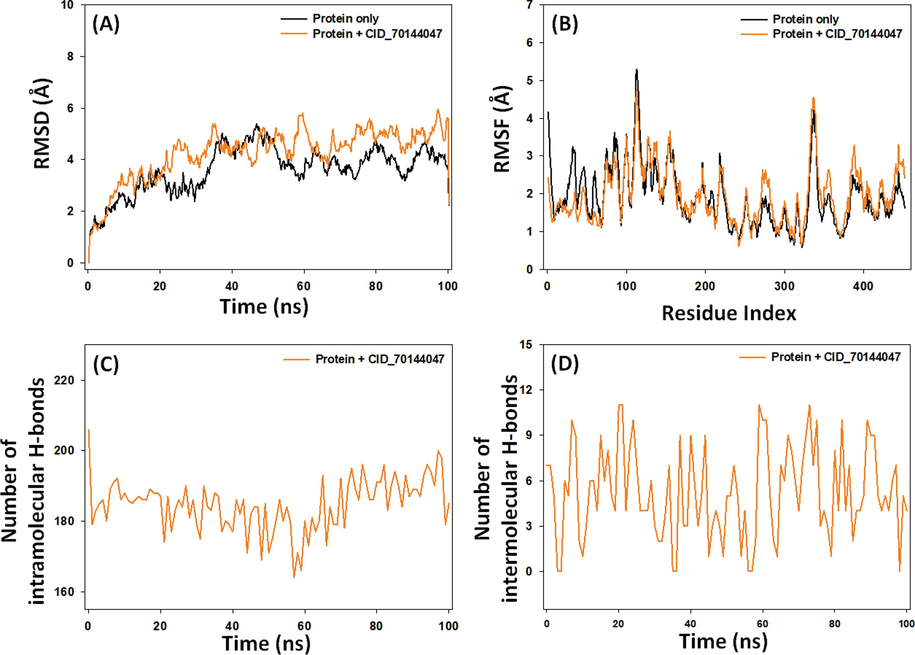
Molecular dynamics simulation of c-Src tyrosine kinase in the absence and presence of CID_70144047. (A) Root mean square deviation (RMSD), (B) Root mean square fluctuation (RMSF), (C) Intramolecular hydrogen bonds, and (D) Intermolecular hydrogen bonds.
3.7.2 Root mean square fluctuation (RMSF)
RMSF, reflecting side chain conformational changes induced by ligand binding (AlAjmi et al., 2021), was measured for c-Src in the absence or presence of CID_70144047 (Fig. 5B). The overlapping RMSF plots for c-Src alone and the c-Src-CID_70144047 complex suggest no significant conformational changes upon ligand binding. The minor fluctuations might be arising due to ligand insertion into the protein’s binding cavity.
3.7.3 Intramolecular and intermolecular hydrogen bonds
Interaction between protein and ligand is crucial a stable protein–ligand complex (Muteeb et al., 2020). Here, we assessed both intramolecular (within the protein) and intermolecular (between the protein and ligand) hydrogen bonding during MD simulation. The intramolecular hydrogen bonds within c-Src in the presence of CID_70144047 ranged from 168 to 198, averaging 188 hydrogen bonds (Fig. 5C). Additionally, hydrogen bonds formed between the c-Src protein and CID_70144047 fluctuated between 0 and 11, with an average value of 5.2 (Fig. 5D).
Overall, the c-Src-CID70144047 complex was stable in nature owing to its lower RMSD and RMSF values, comparable to that of c-Src alone. Further, CID_70144047 form considerable numbers of intra– and inter–molecular hydrogen bonds throughout the simulation time.
3.7.4 Analysis of principal component analysis (PCA)
PCA, a dimensionality reduction method, was employed to analyze the overall motion of c-Src with and without CID_70144047 during simulations. Conformational sampling of c-Src along PC1 (Principal Component 1) and PC2 (Principal Component 2) axes was accessed, red and blue dots representing different conformational states and clusters indicating distinct and energetically favorable regions (Fig. 6). In the absence of any ligand, c-Src occupied a conformational space ranging from −70 to + 50 along PC1 (47.2 %) and −60 to + 40 along PC2 (19.75 %) (Fig. 6A). Conversely, in the presence of CID_70144047, the conformational space covered −70 to + 50 along PC1 (47.84 %) and −50 to + 50 along PC2 (21.75 %) (Fig. 6B). Notably, the first three eigenvalues for c-Src alone and c-Src with CID_70144047 encompassed 80.4 % of the conformational variances (Fig. 6C,D), indicating that the flexibility of c-Src in the presence of CID_70144047 resembles that observed when c-Src is unbound.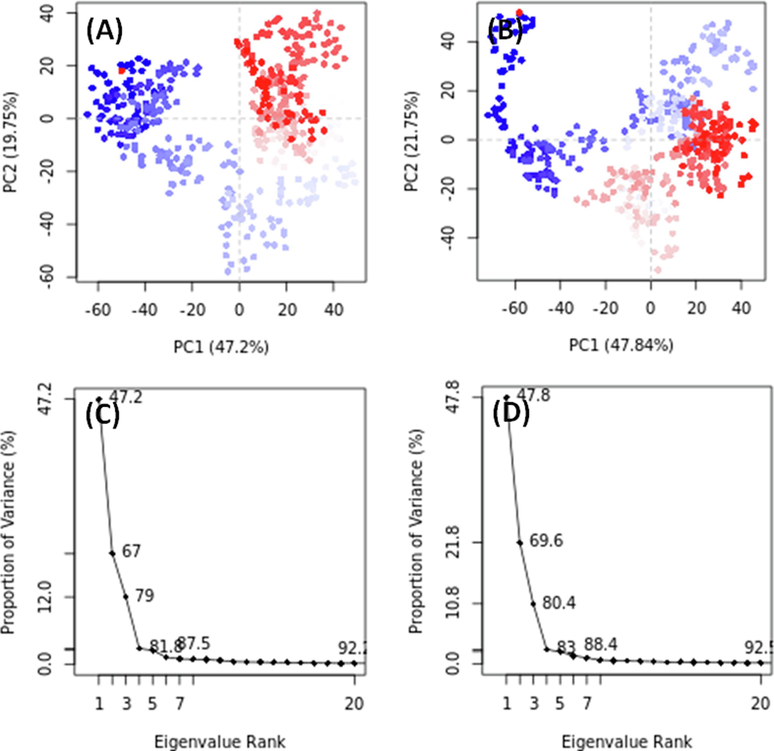
Principal component analysis (PCA) of c-Src tyrosine kinase in the absence and presence of CID_70144047. (A,C) c-Src tyrosine kinase alone, and (B,D) c-Src tyrosine kinase and CID_70144047 complex.
4 Conclusion
The study utilized computational methods to identify potent inhibitors of c-Src tyrosine kinase, integrating diverse techniques such as pharmacophore analysis, 3D QSAR modeling, virtual screening, molecular docking, DFT calculations, and molecular dynamics simulations. The generated atom-based 3D-QSAR model, DDRRR_1, exhibited strong correlation with experimental data. CID_70144047 emerged as a promising inhibitor, showing enhanced bioactivity and favorable pharmacokinetic properties. Despite study limitations, including the lack of experimental validation, the findings hold promise for cancer therapy. Future directions involve experimental validation, expanded pharmacophore analysis, hit optimization, and collaborative efforts for accelerated drug development.
Acknowledgement
“Authors acknowledge the generous support from the Researchers Supporting Project number (RSPD2023R980), King Saud University, Riyadh, Saudi Arabia.”
Disclosure of Funding.
“This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.”
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Pharmacoinformatics approach for the identification of Polo-like kinase-1 inhibitors from natural sources as anti-cancer agents. Int. J. Biol. Macromol.. 2018;116:173-181.
- [CrossRef] [Google Scholar]
- Antiviral potential of some novel structural analogs of standard drugs repurposed for the treatment of COVID-19. J. Biomol. Struct. Dyn.. 2021;39:6676-6688.
- [CrossRef] [Google Scholar]
- Phosphorylation of unique domains of Src family kinases. Front. Genet.. 2014;5
- [CrossRef] [Google Scholar]
- Targeting Tyrosine Kinases in Cancer: Lessons for an Effective Targeted Therapy in the Clinic. Cancers (basel).. 2019;11:490.
- [CrossRef] [Google Scholar]
- Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun.. 1991;175:880-885.
- [CrossRef] [Google Scholar]
- Molecular dynamics with coupling to an external bath. J. Chem. Phys.. 1984;81:3684-3690.
- [CrossRef] [Google Scholar]
- Academic Discovery of Anticancer Drugs: Historic and Future Perspectives. Annu. Rev. Cancer Biol.. 2019;3:385-408.
- [CrossRef] [Google Scholar]
- The Crystal Structure of a c-Src Complex in an Active Conformation Suggests Possible Steps in c-Src Activation. Structure. 2005;13:861-871.
- [CrossRef] [Google Scholar]
- PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided. Mol. Des.. 2006;20:647-671.
- [CrossRef] [Google Scholar]
- Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem.. 2006;49:6177-6196.
- [CrossRef] [Google Scholar]
- Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695-2696.
- [CrossRef] [Google Scholar]
- The Bio3D packages for structural bioinformatics. Protein Sci.. 2021;30:20-30.
- [CrossRef] [Google Scholar]
- Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci.. 2008;27:445-456.
- [CrossRef] [Google Scholar]
- Iqbal, D., Rehman, M.T., Bin Dukhyil, A., Rizvi, S.M.D., Al Ajmi, M.F., Alshehri, B.M., Banawas, S., Khan, M.S., Alturaiki, W., Alsaweed, M., 2021. High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach. Pharm. 2021, Vol. 14, Page 937 14, 937. https://doi.org/10.3390/PH14090937.
- Identification of natural compounds as potent inhibitors of SARS-CoV-2 main protease using combined docking and molecular dynamics simulations. Saudi J. Biol. Sci.. 2021;28:2423-2431.
- [CrossRef] [Google Scholar]
- The spectroscopic (FT-Raman, FT-IR, UV and NMR), molecular electrostatic potential, polarizability and hyperpolarizability, NBO and HOMO–LUMO analysis of monomeric and dimeric structures of 4-chloro-3,5-dinitrobenzoic acid. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2012;93:33-46.
- [CrossRef] [Google Scholar]
- QSAR Studies of amino-pyrimidine derivatives as Mycobacterium tuberculosis Protein Kinase B inhibitors. Turkish Comput. Theor. Chem.. 2018;2:16-27.
- [Google Scholar]
- QSAR modeling, molecular docking, ADMET prediction and molecular dynamics simulations of some 6-arylquinazolin-4-amine derivatives as DYRK1A inhibitors. J. Mol. Struct.. 2022;1258:132659
- [CrossRef] [Google Scholar]
- Multi-combined 3D-QSAR, docking molecular and ADMET prediction of 5-azaindazole derivatives as LRRK2 tyrosine kinase inhibitors. J. Biomol. Struct. Dyn.. 2022;40:1285-1298.
- [CrossRef] [Google Scholar]
- Investigating the binding mechanism of topiramate with bovine serum albumin using spectroscopic and computational methods. J. Mol. Recognit.. 2022;35:e2958.
- [Google Scholar]
- Development of energetic pharmacophore for the designing of 1,2,3,4-tetrahydropyrimidine derivatives as selective cyclooxygenase-2 inhibitors. J. Comput. Aided. Mol. Des.. 2012;26:267-277.
- [CrossRef] [Google Scholar]
- Ligand-based pharmacophore filtering, atom based 3D-QSAR, virtual screening and ADME studies for the discovery of potential ck2 inhibitors. J. Mol. Struct.. 2020;1205:127670
- [CrossRef] [Google Scholar]
- Screening marine algae metabolites as high-affinity inhibitors of SARS-CoV-2 main protease (3CLpro): an in silico analysis to identify novel drug candidates to combat COVID-19 pandemic. Appl. Biol. Chem.. 2020;63
- [CrossRef] [Google Scholar]
- Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys.. 1981;52:7182-7190.
- [CrossRef] [Google Scholar]
- Rashid, H. ur, Xu, Y., Muhammad, Y., Wang, L., Jiang, J., 2019. Research advances on anticancer activities of matrine and its derivatives: An updated overview. Eur. J. Med. Chem. 161, 205–238. https://doi.org/10.1016/j.ejmech.2018.10.037.
- Virtual Screening Using PLS Discriminant Analysis and ROC Curve Approach: An Application Study on PDE4 Inhibitors. J. Chem. Inf. Model.. 2008;48:1686-1692.
- [CrossRef] [Google Scholar]
- When fragments link: a bibliometric perspective on the development of fragment-based drug discovery. Drug Discov. Today. 2018;23:1596-1609.
- [CrossRef] [Google Scholar]
- Some case studies on application of “ r m 2 ” metrics for judging quality of quantitative structure-activity relationship predictions: Emphasis on scaling of response data. J. Comput. Chem.. 2013;34:1071-1082.
- [CrossRef] [Google Scholar]
- y-Randomization and Its Variants in QSPR/QSAR. J. Chem. Inf. Model.. 2007;47:2345-2357.
- [CrossRef] [Google Scholar]
- Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach. J. Biomol. Struct. Dyn.. 2022;40:14-30.
- [CrossRef] [Google Scholar]
- In silico screening of indinavir-based compounds targeting proteolytic activity in HIV PR: binding pocket fit approach. Med. Chem. Res.. 2012;21:4060-4068.
- [CrossRef] [Google Scholar]
- 2D QSAR studies of the inhibitory activity of a series of substituted purine derivatives against c-Src tyrosine kinase. J. Taibah Univ. Sci.. 2016;10:563-570.
- [CrossRef] [Google Scholar]
- Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin.. 2021;71:209-249.
- [CrossRef] [Google Scholar]
- Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform.. 2010;29:476-488.
- [CrossRef] [Google Scholar]
- Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. Https:// 2019
- [CrossRef] [Google Scholar]
- Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov. Today. 2010;15:444-450.
- [CrossRef] [Google Scholar]
- Design, synthesis and anticancer activity of fluorocyclopentenyl-purines and – pyrimidines. Eur. J. Med. Chem.. 2018;155:406-417.
- [CrossRef] [Google Scholar]
- Privileged scaffolds in lead generation. Expert Opin. Drug Discov.. 2015;10:781-790.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2023.103076.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




