

Translate this page into:
Synthesis, characterization and identification of inhibitory activity on the main protease of COVID-19 by molecular docking strategy of (4-oxo-piperidinium ethylene acetal) trioxonitrate
⁎Corresponding author. issaoui_noureddine@yahoo.fr (Noureddine Issaoui)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
In this investigation a single crystal of (4-oxo-piperidinium ethylene acetal) trioxonitrate (4-OPEAN) was synthesized by modifying the mechanism of gradual evaporation at ambient temperature. The operational groupings are found in the complex material in the elaborate substance, according to the infrared spectrum. Single crystal X-ray diffraction suggests, (4-OPEAN) with the chemical formula (C7H12NO2)NO3 belongs to the orthorhombic space group Pnma and is centrosymmetric in three dimensions with the aforementioned network configurations, a = 11.7185(8) Å, b = 7.2729(6) Å, c = 11.0163(8) Å, Z = 4, V = 938.89(12) Å3, R = 0.0725 and wR = 0.1762. Many N—H…O and C—H…O hydrogen bridges, both bifurcated and non-bifurcated, link the 4-oxo-piperidinium ethylene acetal cations to the trigonal (NO3−) anions. Molecular geometry and optimal parameters of (4-OPEAN) have been determined via DFT computations at the theory-level B3LYP/6-311 ++ G(d, p), these have been contrasted with the X-ray data already available. Hirshfeld surface analysis has made it possible for the visualization and quantification of relationships between molecules in the crystal composition. Quantum theory atoms in molecules, electron location function, decreased density gradient, and localized orbital locator research have all been used to explore non-covalent interactions in crystal structure. In order to pinpoint both the nucleophilic and electrophilic locations that support hydrogen bond formation, the molecule electrostatic potential was determined. The greatest and lowest energies of occupied and unfilled molecular orbitals, together with additional derived atomic characteristics, show the material to be extremely stable and hard. According to a molecular docking study, 4-OPEAN may exhibit inhibiting effects on the 6Y84 and 7EJY virus proteins from corona (COVID-19).
Keywords
Synthesis
X-ray diffraction
DFT
Chemical docking
COVID-19
Hirshfeld surface

1 Introduction
Currently, the development of new synthesis techniques combining the principles of solid state chemistry, molecular or supramolecular chemistry, or biochemistry is prompted by the need to find new materials for a variety of applications and new devices. This is especially evident in the realm of hybrid organic–inorganic materials. The relationships between the structural and physical features caused by the molecular entities grafted onto the inorganic layers are frequently well understood using these materials as model systems. We can cite, for instance, the utilization of these resources in fields like opto-electronics (Trindade et al., 2001), catalytic systems (Shipway et al., 2000) and realm of medicine or pharmaceuticals field (Daniel and Astrue, 2004). A fantastic synergy between the properties of these different materials, combining, for instance, the optical, thermal, and/or electrical characteristics of mineral materials in conjunction with the physico-chemical properties of cationic part, while minimizing dispersion and compatibility issues. In this regard, we conducted research on an innovative organic nitrate with the (C7H12NO2)NO3 formula. First and foremost, evaluating the pharmacokinetics and workings of NO3− anion is quite fascinating, due to the fact that they are frequently used in pharmacy, such as glyceryl trinitrate (Novena et al., 2016). Drug design has utilized numerous docking experiments, and having said that suggests, 1EPBN (Gatfaoui et al., 2020) may have inhibitory effects on corona virus (COVID-19) proteins. On the other hand, 1-phenylpiperazin-1,4-diium nitrate monohydrate (Noureddine et al., 2021) penetrates both enzymes' active sites (Kalirin-7 and MOB), therefore we can think of it as a strong inhibitor of Parkinson's, Alzheimer's and schizophrenia disease. The 1,3-Benzenedimethanaminium bis(trioxonitrate) (Gatfaoui et al., 2022) molecule has biological action in silico, indicating that it is regarded as a potent inhibitor of bacteria and fungi. Moreover, some nitrate has interesting antioxidant effects, which are shown by the scavenging of DPPH and ABTS radicals, as well as by its decreasing and somewhat stronger hydroxyl radical scavenging than ascorbic acid (Gatfaoui et al., 2017a, 2017b). On the physical level, recently published results reveal organic nitrates that can have industrial applications in nonlinear optics (NLO) for example the non-centrosymmetric material 3-methylbenzylammonium trioxonitrate (Gatfaoui et al., 2020b). For the creation and synthesis of several types of medications, hexagonal heterocyclic piperidine and its primary derivatives are also utilized (morphine, risperidone, nicotine, strychnine and vitamin B6…). So to investigate these kinds of novel materials, we describe in this paper, the elaboration of the centrosymmetric (4-oxo-piperidinium ethylene acetal) trioxonitrate compound, together with its description using several practical and methodological approach like X-ray diffracting which is the primary method used for determining the configuration of atoms in crystal lattices and their atomic properties, as well as for initially learning about crystallographic features, Hirshfeld surface analysis was employed to determine the number of NCI: non-covalent interactions and to support the significance networks of bonds inside the crystalline composition, spectroscopic measurements (infrared absorption spectroscopy) allowed us to ascertain the material vibrational characteristics, applying the theory of groups, specifically the list of nitrate ion vibrational modes and the assignment of the frequency measurements of the numerous kinds of vibrational alterations in the NO3−group. Additionally, topological theoretical analyses have been conducted throughout this work, greatly enhancing the information already known about the subject matter, specifically AIM, RDG, ELF, LOL, and MEPS. The frontier molecular orbital’s (FMO) enables the comprehension of the molecule's local reactivity and stability. Research used molecular docking to look at our molecule's inhibition mechanism with the newly discovered corona virus illness.
2 Experimental and theoretical data
2.1 Synthesis of (4-OPEAN) and its crystallization
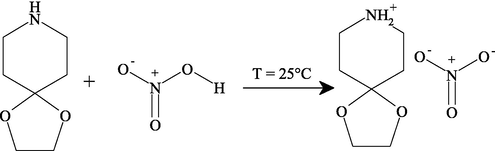
The title chemical is created by adding drops gradually. an aqueous solution of HNO3 acid (1 mmol) to an alcoholic solution (ethanol as solvent) containing 1 mmol of 1,4-dioxa-8-azaspiro [4. 5] decane under magnetic agitation. At room temperature, a gradual evaporation process is applied to the resulting solution. 4 days later forming clear, stable monocrystals of an appropriate size for structural analysis. The reaction can be represented schematically as follows:
2.2 Materials and physical data
The infrared (IR) spectrum in the 4000-400 cm−1 region was recorded at room temperature using a Nicolet IR 200 FTIR spectrophotometer. 150 K was used as the temperature for the X-ray tests. By means of a diffractometer Bruker-AXS APEXII operating with a wavelength of 0.71107 Å for molibdene, collections intensities were measured on solitary crystals. The SADABS software was used to perform absorption corrections using the multi-scan method (Bruker, 2006). Using the SHELXT program (Sheldrick, 2015), the dual-space technique was used to solve the crystal structure, and then improved using F2-based full-matrix least-squares techniques (SHELXL software) (Sheldrick, 2015a)). Anisotropic atomic displacement parameters were used to refine all non-hydrogen atoms. Except Hydrogen atoms linked to Nitrogen atom that were introduced in the structural model through Fourier difference maps analysis, H atoms were finally included in their calculated positions and treated as riding on their parent atom with constrained thermal parameters. A final refinement on F2 with 1159 unique intensities and 84 parameters converged at wR(F2) = 0.1762 (RF = 0.0725) for 1113 observed reflections with I > 2σ. The X-ray data parameters, the method for determining the crystal structure, and the outcomes are all listed in Table 1. ORTEP (Farrugia, 2012) and the Diamond program (Brandenburg, 1998) are used to create the structure graphics.
CCDC
2258214
Chemical formulation
C7H12N2O5
Temperature
150 K
Formula weight (g mol−1)
204.19
Crystal system
Orthorhombic
Space group
Pnma
a, b, c (Å)
11.7185(8), 7.2729(6), 11.0163(8)
Z
4
V(Å3)
938.89(12)
F(000)
432
Radiation type
Mo Kα
μ(mm−1)
0.123
Crystal size (mm)
0.32 × 0.20 × 0.14
Index ranges
-15≤ h ≤ 14, -9 ≤ k ≤ 9, -14 ≤ l ≤ 14
Reflections collected
10163
Independent reflections
1159
Reflections with I > 2σ(I)
1113
Rint
0.0424
Diffractometer
D8 Venture (Bruker-AXS)
Absorption correction
Multi-scan
Tmin, Tmax
0.900, 0.983
Refined parameters
84
R[F2 > 2σ(F2)]
0.0725
wR(F2)
0.1762
Goodness of fit
1.254
Δρmax, Δρmin (eÅ−3)
0.410, −0.293
2.3 Theoretical research
The theoretical computations were performed using the hybrid B3LYP/6-311++G (d,p) approach built within the Gaussian 09 program (Frisch, 2009). A 2D graph that summarizes the intricate information contained in a structure and a 3D graph that shows the area of space where the molecules are in touch form the basis of a Hirshfeld surface analysis. This allows for the identification of each type of interaction. The Crystal Explorer program was used to conduct this study (Wolff et al., 2013). The topological properties in this research were computed via means of the Multiwfn software (Lu and Chen, 2012) in accordance with Bader's theory (Kumar et al., 2016). A multipurpose wave function analyzer called Multiwfn was used to perform the RDG-NCI analysis, and the outcomes were visualized using the VMD molecular visualization tool. To investigate the molecular docking computation, iGEMDOCK software was employed (Yang and Chen, 2004), this application calculates the total energy of the basic ligand–protein interactions. Protein Data Bank (https://www.rcsb.org/pdb/) is where the enzyme blueprints can be found. The visual depictions of the docked ligands are created using molecular operating environment (Molecular, 2015).
3 Findings and commentary
3.1 Structure evaluation and geometry optimization
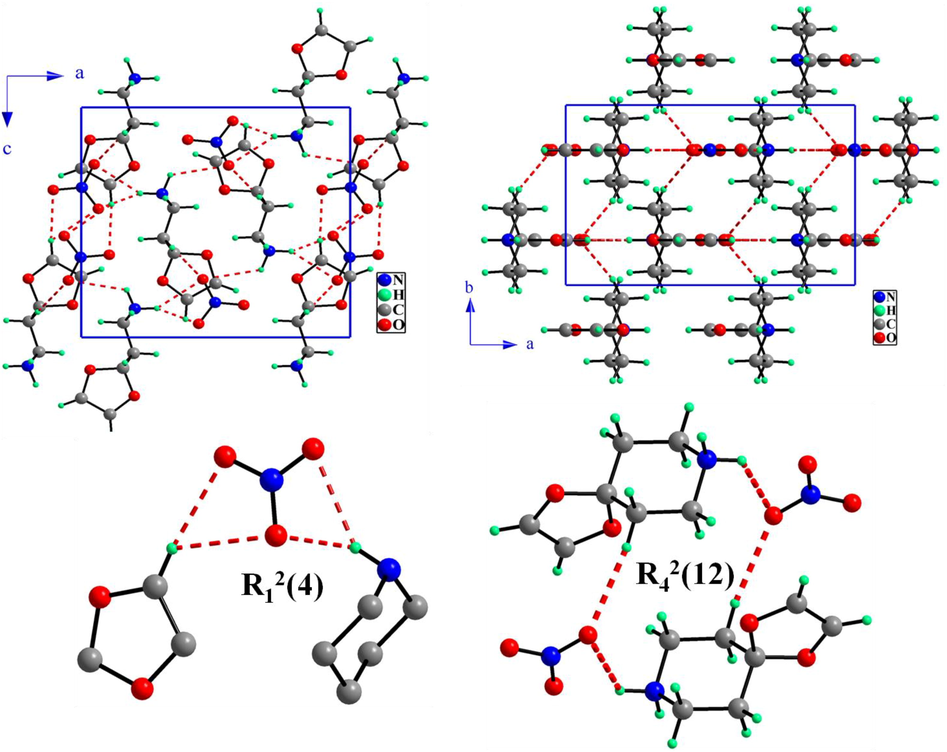
An anionic entity, NO3− and a monoprotonated cationic entity, 4-oxo-piperidinium ethylene acetal, make up the investigated structure's asymmetric unit (Fig. 1a). Medium and weak hydrogen bonds between these entities ensure their connection. The crystal structure of (4-OPEAN) is projected sequentially in the (ac) and (ab) planes in Fig. 2. In contrast to the organic groups, which are organized into dimers along the [0 0 1] direction, the NO3−, nitrate groups form rows that develop in a position equal to 0 and 0.5 along the direction of the
axis. A three-dimensional of a series of hydrogen bandings, highlighting the O…H—N and O…H—C kinds of linkages, which ensures the cohesion and stability of our structure, provides the sequence for the two cationic and anionic halves. Analyzing the nitrate anion (Table S1) reveals that the nitrogen atom (N11) occupying general positions and forming a somewhat planar ionic structure with the oxygen atoms (O12, O3 and O14). In fact, the interatomic N—O bond lengths and O—N—O angle ranges are 1.231(4) – 1.254(4) Å and 119.4(3) – 121.1(3)°, respectively. The anions' geometric shape is similar to that which has been documented in the literature for homologous compounds (Gatfaoui et al., 2014a, b; Jmai et al., 2023). About the 4-oxo-piperidinium ethylene acetal's geometric properties, it is noted that this cation is made up of the combination of the two groups «1,3-dioxolane» and «piperidinium» intermingled at the carbon atom. With a mean plane deviation of zero, the first ring's planar portion contains two oxygen atoms, whereas the second ring displays a chair conformation. The organic cation then exhibits a predictable spatial arrangement, with the normal distances C—C, C—N, C—O, and angles C—C—C, C—C—N, C—C—O, O—C—O, C—O—C changing respectively on the scales [1.374(5)–1.523(4) Å] and [106.1(3)–111.8(3)°]. These distances and angles are equivalent to those in similar constructions such as Tetrakis(4-oxo-piperidinium ethylene acetal) bis sulfate (Marouani et al., 2011) and (4-oxo-piperidinium ethylene acetal) dihydrogen monophosphate monohydrate (Dhaouadi et al., 2010). The crystalline structure of the compound (4-oxo-piperidinium ethylene acetal) trioxonitrate is mostly built on the basis of interconnected system of moderately strong, average and weak hydrogen bonds (Table 2) including two to three centers of the O…H—N design involving the H1A and H1B hydrogen atoms, one with three centers of C—H…O type (C6—H6…O12, C6—H6…O13) and one with two centers involving the H3B atom (dC3…O14 = 3.536(3) Å). Indeed the N1—H1A…O13 hydrogen bond is characterized as being moderately strong with a N1…O13 distance of 2.818(4) and a D—H…A angle that is extremely close to 180 degrees, around 177 (5)°. The crystalline edifices of (4-OPEAN) material are composed of mixed layers that are created by successive R12(4) and R42(12) cycles that are brought about by the interaction of hydrogen bonds between the organic cations and the nitrate anions. Symmetry codes: (ii) x + 1/2, y,−z + 3/2; (iii) − x + 1, −y + 1, −z + 1; (iv) x, y, z − 1.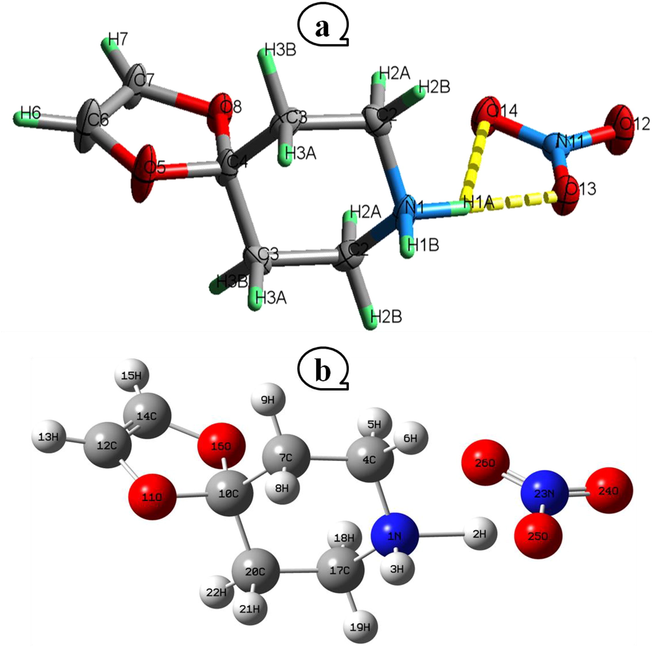
Drawing of the 4-OPEAN using the atom-labeling method in ORTEP. At the 30% probability threshold, displacement ellipsoids are drawn. H atoms are shown as tiny spheres with variable radii (a) and the idealized molecular structure (b).
Projection of the atomic arrangement along the
and
axes and hydrogen bonding motifs of 4-OPEAN.
D—H···A
D—H (Å)
H···A (Å)
D···A (Å)
D—H···A(°)
N1—H1A···O13
0.96(6)
1.86(6)
2.818(4)
177(5)
N1—H1A···O14
0.96(6)
2.59(6)
3.198(4)
122(4)
N1—H1B···O12ii
0.96(6)
1.96(6)
2.874(4)
157(5)
N1— H1B ···O14ii
0.96(6)
2.44(6)
3.282(4)
146(4)
C3—H3B ···O14iii
0.99
2.57
3.536(3)
164.7
C6—H6···O12iv
0.95
2.56
3.173(5)
122.4
C6—H6···O13iv
0.95
2.42
3.369(5)
173.7
Focusing now on the theoretical part, table S1 lists the geometric parameters of the 4-OPEAN that were investigated theoretically by DFT (B3LYP/6-311 ++ G(d, p)) and experimentally by X-ray diffraction, such as the bond lengths and angles. Thus Fig. 1b displays the geometrical optimization. The results show that these parameters nearly match the values obtained through experimentation. The C—C, C—O and C—N bonds in the moiety of the organic cation were calculated to be between 1.3267 and 1.5347 Å, and the C—C—N, C—N—C, O—C—O, C—C—O and C—C—C bond angles were determined to range between 105.7174 and 112.8089 degrees. With regards to the nitrate anion the N1—O24 and N1—O26 calculating distances nearly identical to those found by X-ray diffraction, as long as N1—O25 is elongated by 0.11 relative to the value in experimentation. This is comprehensible by the oxygen atom O25 being a part of the O25…H2—N1 hydrogen molecule link, which is fairly strong. Recapitulating, it can be seen that some theoretical errors exist because calculations are made in the gaseous state as opposed to experimental design is observed during the crystalline stage.
3.2 3D map projections and graphs of 2D fingerprints
Statistical evaluation and visualization of the supramolecular interactions in the title structure were done using Crystal Explorer. Fig. 3 shows the Hirshfeld surface projected over dnorm (Fig. 3a) with values ranging from −0.531 to 1.174 a.u and de (Fig. 3b) from 0.786 to 2.289 a.u. The relative positioning of the nearby atoms in the nitrate and 4-oxo-piperidinium ethylene acetal molecules that interact with one another can be graphically represented using the normalized contact distance. Using a color-coded surface (red, blue, white), this form of study shows the shortest intermolecular interactions detected by the red spots, which are linked to the N—H…O and C—H… O hydrogen bonds. Orange areas on the de map indicate this hydrogen bonding. In relation to the blue hue shown on the two graphs, she relates to the furthest intermolecular contacts in the structure in this area due to the too big of a distance between the nearby atoms and the existence of far-off connections as H… H, C… H, N… H, C… O and O… O (Gatfaoui et al., 2019, 2021; Kurbanova et al., 2023; Kansız et al., 2021 and 2021b). Fig. S1 shows the numerical results of the HS evaluation and includes the percentages of contributions from various types of connections and atoms in the crystal structure. We note that the H…O/O…H intermolecular interactions take up a significant portion of the Hirshfeld surface (55.8%) and appear in 2D fingerprint plots (Fig. 3c) shaped like wings with two equal sides with a significant long tip that is sum (de + di ̴ 1.9 Å) lower than the total of the van der Waals radii of the hydrogen (1.09 Å) and oxygen (1.52 Å) atoms. Such connections are responsible for the generation of N—H…O and C—H…O hydrogen ties. The two-dimensional fingerprint maps for H…H contacts show them as scattered points in the center, making up 35.8% of Hirshfeld's surface. A prolonged interatomic H…H contact is what causes the single round peak at de + di = 2.4 Å in Fig. 3d. By contrasting between the total van der Waals radii of the participating atoms and the sum of the components of the pairs of various contacts (di, de) existing in the structure just H…O/O…H considered to be close and involved in the production of hydrogen bonds which ensure the cohesion and maintenance of the crystalline structure. The enrichment ratio (ER), a novel term to describe a pair of chemical substances that is described as being the ratio between the ratio of actual crystal connections and the assumed ratio of random contacts, has been used to evaluate the various interactions present in our crystal. These enrichment ratios (ER) are computed and provided in Table S2. An inventory of enrichment ratios draws attention to the H…O/O…H contacts (EROH = 1.44), which appear to be preferred in the packing of crystal with the creation of the N—H…O and C—H…O hydrogen links. The enrichment ratio for the H…H interactions is about unity (0.84), which is in good agreement with Jlech's prediction (Jelsch et al., 2014). The EROO ratio is very low, close to zero, this is explained by the O … O contacts which are generally depleted, and thus the contact surface is poor in oxygen (SO < 20%). Although the additional contributions to the Hirshfeld surface (C…O/O…C, N…C/C…N, C…H/H…C and H…N/N…H) are very modest, the ER values are not especially useful for some interactions.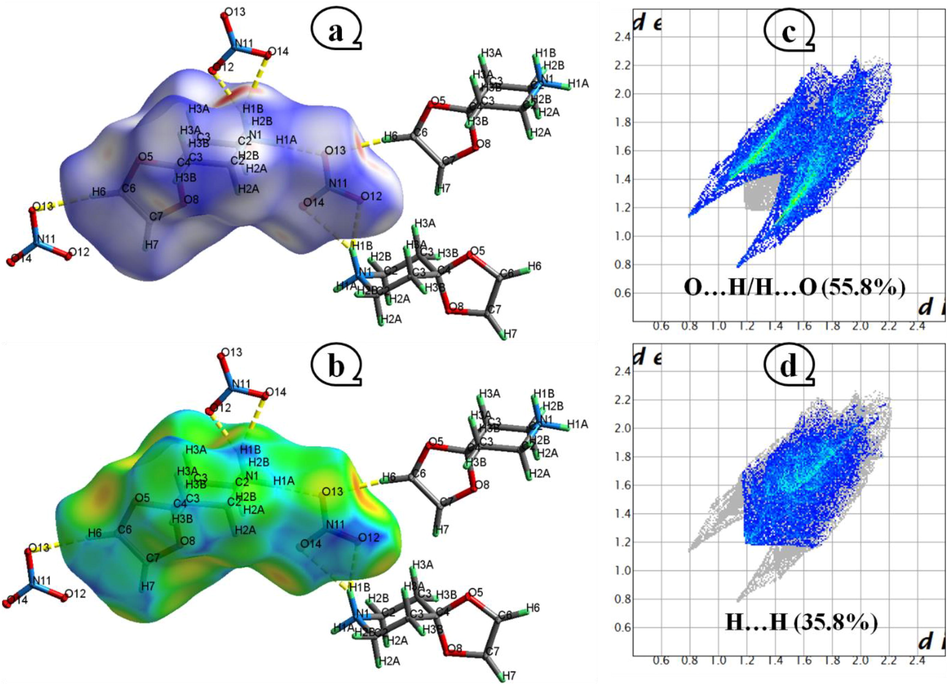
dnorm (a), and de (b) cartography of 4-OPEAN Compound. Fingerprint plot of O…H/H…O (c) and H…H (d) connections.
3.3 Quantum methods of topological analysis
3.3.1 Quantum analysis of atoms in molecules
Based on the investigation of the topology of electron density, Bader's AIM (Atoms-in-Molecules) quantum theory (Bader, 1990) provides a description of molecules and, in particular, of their chemical bonds. By using a variety of topological and energetic properties (such as the electron density ρ(r), the Laplacian of the electron density ∇2ρr), the kinetic energy density G(r), the density total energy H(r) = G(r) + V(r), potential energy density V(r)), it enables us to more clearly understand the nature of non-covalent interactions and precisely the forces that drive hydrogen ties. Hence, Rozas et al. (Rozas et al., 2000) presented the following requirements to understand the nature of hydrogen bonds:
-
o
If ∇2ρ(r) < 0 and H(r) < 0 → Strong hydrogen bonds are considered to exist.
-
o
If ∇2ρ(r) > 0 and H(r) < 0 → moderate hydrogen bonds are considered to exist.
-
o
If ∇2ρ(r) > 0 and H (r) > 0 → weak hydrogen bonds are considered to exist.
Fig. S2 uses the Multiwfn application to display the AIM molecular graph of the (4-OPEAN) molecule. In Table S3, the topological properties of the non-covalent interactions are categorized. According to the findings of the AIM study, the crystalline structure's cohesiveness is supported by three crucial BCPs bonds: two hydrogen bonds of the O…H—C type and one of the O…H—N type. The critical link points are located at the O25…H2—N1, O26…H5—C4 and O26…H18—C17 contacts where the electronic densities are equal to 0.034, 0.006 and 0.006 a.u with Laplacian values about 0.103, 0.022 and 0.022 a.u, respectively. We also note that at these critical points the total energy density (H) is less than zero for the O25…H2—N1 hydrogen bond, on the other hand it is greater than zero for the two other interactions of the O …H—C type. So according to Rozas et al these last two can be included within the category of weak hydrogen bonding. as long as the O25…H2—N1 bond is considered medium.
3.3.2 Gradient of reduced density examination
A unique non-covalent interaction (NCI) descriptor based on a reduced density gradient (RDG) was constructed by Johnson et al (Johnson et al., 2010). It has been proven that this method can tell tiny molecules, molecular complexes, and solids apart when it comes to hydrogen bonds, Van der Waals interactions, and steric repulsion. The following expression is used to compute the RDG quantity:
As a function of the electron density multiplied by the sign of the second eigenvalue of the Hessian matrix, the reduced density gradient of (4-OPEAN) compound is depicted in Fig. 4a. It is easy to distinguish between the three zones thanks to the evolution of the RDG (a.u) as a function of sign λ2*ρ according to a clear color scheme. While the areas that match to a hydrogen bond (ρ > 0, λ2 < 0) or a steric effect (ρ > 0, λ2 > 0) are related to high density, the van der Waals interactions usually display very modest values of the electron density (ρ ≈ 0, λ2 ≈ 0). A closer look at Fig. 4b reveals the existence of light blue dots that indicate strong N—H…O attractive interactions between the atoms of hydrogen and oxygen. The round red spots situated in the center of the two piperidinium and 1,3-dioxolane rings are attributed to describe the destabilizing steric interaction. While the van der Waals bonds are visible as green specks that are situated between the hydrogen atoms.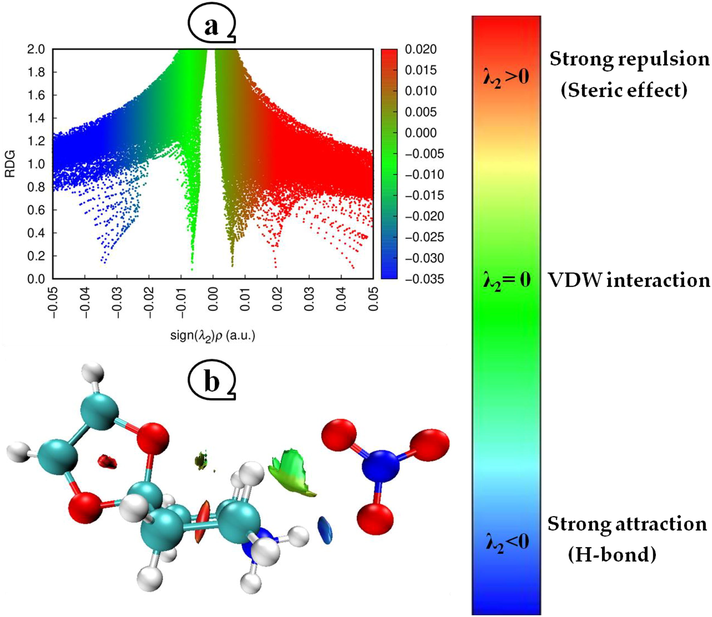
Scale bar with colored data delineating the interaction boundaries for the RSDPN compound is plotted along with the decreased density gradient (a) and the density of an isosurface (b).
3.3.3 Local orbital locator and electron localized function evaluation
The localized orbital localizer (LOL) and the electron localization function are two similar electron localization descriptors because they are reliant on the cinétique energy density, the last two are frequently employed to determine the atomic shell's structure and categorize chemical bonds on molecular surfaces. In Fig. S3, the ELF and LOL pictures for the tested molecule are shown as color shade maps and contour maps. The ELF image is represented by a blue-to-red color scale with values ranging from 0 to 1, while the LOL scale has values from 0 to 0.8. According to the findings of the ELF study, the delocalized electron cloud is shown by the blue regions surrounding a few atoms of carbon, nitrogen, and oxygen. As opposed to that, as shown by the color red, maximum Pauli repulsion is seen to localize strongly electronically around the hydrogen atoms. When the localized orbital locator LOL fingerprint is analyzed, white spots around hydrogen atoms are found, this indicates that the electron density is more than the maximum allowed by the color scale. (0.80). Small LOL values (0.12 to 0.24 a.u) in the blue regions between the organic and inorganic portions emphasize the presence of N—H…O and C—H…O hydrogen bonding.
3.4 Molecular electrostatic potential
Based on electronic density and the locations of a molecule's chemical reactivity, molecular electrostatic potential (MEP) provides details about the total charge distribution of the molecule. This instrument enables the identification of electrophilic (regions of positive electrostatic potential) and nucleophilic (regions of negative electrostatic potential) sites, and as a result, the determination of the likelihood that intra or intermolecular hydrogen bonds will form. Fig. 5a shows the molecular electrostatic potential (MEP) maps of the (4-OPEAN) molecule using the following color-code range: −6.100 10−2 (red denotes the strongest repulsion) to 6.100 10−2 (blue indicates strongest attraction), the green color indicates a neutral zone (zero potential). According to the MEP study, the oxygen atoms in the nitrate unit are where the most negative potential zone is concentrated (red and yellow hue). Contrarily, the most positive sites (in blue) are concentrated mostly around the ammonium NH2+ grouping of the cationic portion and point to the sites that are electron-deficient and the most vulnerable to nucleophilic attack. Then again,the development of hydrogen bonds between the nitrate groups and the 4-oxo-piperidinium ethylene acetal cation and their significance in the stability of (4-OPEAN) are also explained by the electrophilic and nucleophilic sites.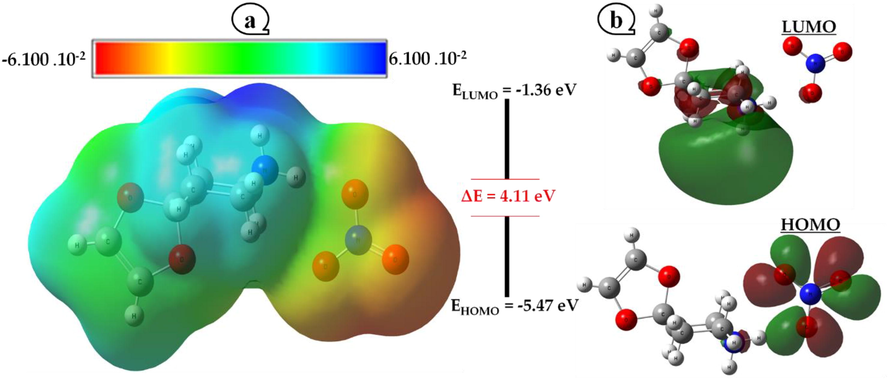
Molecular electrostatic potential (MEP) (a) and Frontier molecular orbital (b) of 4-OPEAN crystal.
3.5 HOMO–LUMO energy
Frontier molecular orbital theory (FMO theory) is a result of Kenichi Fukui's (Fukui, 1975) study of frontier orbitals, namely the impact of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) on reaction processes. These orbitals contribute significantly to the chemical stability of the molecule and are crucial for the electrical characteristics and UV–visible spectra The HOMO-LUMO gap, which measures the energy difference between HOMO and LUMO, is used to forecast the strength and stability of materials, therefore a large molecule's stability is indicated by a high gap, contrariwise small gap is a high-reactivity criterion. Fig. 5b depicts the (4-OPEAN) material's border orbitals (HOMO, LUMO), as well as the gap energy determined in the gas phase. The MO plot's positive and negative phases are portrayed by the colors red and green, respectively. The predicted energies of HOMO and LUMO are approximately −5.47 eV and −1.36 eV respectively, favoring a Gap in the vicinity of 4.11 eV. This high number suggests, in some ways, a low chemical reactivity toward chemical reactions and a high kinetic stability. Chemical hardness and softness are reliable predictors of a molecule's chemical stability in addition to the energy gap. Another crucial element of a material electrical structure is its dipole moment. The results of the calculations are then displayed in Table S4 along with additional quantum characteristics (electronegativity and electrophilic index). Indeed, I = −EHOMO and A = −ELUMO are the relationships that link the energies of the frontier orbitals HOMO and LUMO to their respective ionization potentials I and electronic affinities A. Mulliken (Mulliken, 1934) defines the electronegativity χ as follows:
The correlations shown below (Pearson, 1997) determine chemical hardness and softness:
Parr et al. (Parr et al., 1999) defined the electrophile index ψ as:
The chemical potential represented by in the final statement is given by:
The composite object of our investigation has a chemical softness of 0.49 eV and a hardness of 2.05 eV in the gaseous phase. Low is a sign of a good nucleophile, and vice versa, our chemical can be regarded as an excellent electrophile because the electrophilic index in our case is high at 2.84 eV. Furthermore, the chemical potential is negative (-3.41 eV), which indicates that our crystal is stable and does not spontaneously break down into its constituent parts, a well-known characteristic of biologically active compounds. It's important to note that these outcomes mirror those obtained with other organic nitrates (Gatfaoui et al., 2020, 2017a, b, 2020).
3.6 Infrared spectrum
The investigation of produced nitrate using IR absorption spectroscopy enabled us to identify the vibrational properties of the atomic groups in the (4-OPEAN) molecule. The experimental IR spectra and their theoretical counterpart are shown in Fig. 6. Table S5 provides a list of the estimated and experimental vibrational frequencies as well as descriptions of normal modes. The assignment efforts are based on prior findings in the literature as well as the hypotheses generated via group theoretical research (Marouani et al., 2011; Dhaouadi et al., 2010; Gatfaoui et al, 2017a and 2017b).
-
o
Vibrational modes of the 4-oxo-piperidinium ethylene acetal cation
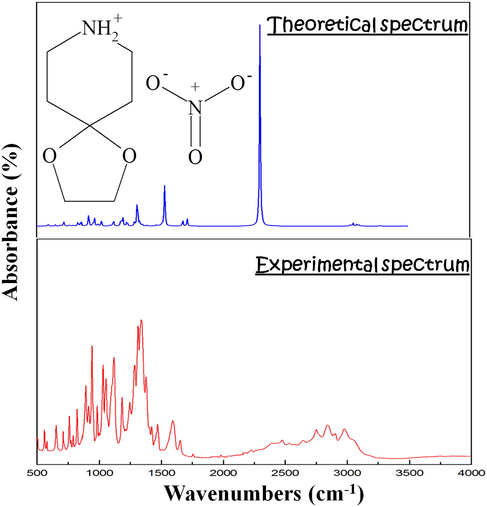
- FT-IR spectra of the 4-OPEAN compound, both theoretical and practical.
In its protonated state, the organic group exhibits valency and bending vibrations between 3400 and 2500 and 1650 and 1300 cm−1, respectively. The theoretical values that correspond to these vibration range between 3100 and 2200 and 1750 and 1300 cm−1. The interaction of the intermolecular hydrogen bond N—H…O, whose mode is calculated to be approximately 3050 cm−1, explains the broad band's mode of stretching, which was experimentally detected at 2842 cm−1. The —CH2 groups' stretching modes can be found in the spectral region [3100–3000 cm−1]. The symmetric stretching of the —CH2 group is responsible for the band at 3077 cm−1; the matching experimental mode can be seen at 2842 cm−1. At 2975 cm−1, the —CH2 asymmetric stretching peaks are visible. Calculations for these modes are done at 3100 cm−1. (—CH2) bending vibrations are responsible for frequencies in the 1700–1300 range. Theoretically, they are worth 1532 and 1309 cm−1. Regarding the C—C, C—N, and C—O groupings that make up the piperidinium and dioxolane nuclei, vibration modes have been experimentally identified in a frequency range between 1200 and 1000 cm−1. These modes are anticipated to range between 1200 and 1080 cm−1 in theoretical terms. δ(CC), δ(CN), and δ(CO) were ultimately demonstrated by experimental peaks at the [915—500 cm−1] domain. Theoretically, these are offered in the spectral space from 915 to 720 cm−1.
-
o
Vibrational modes of the NO3− anion
The nitrate group has D3h symmetry in the free state and has the following normal vibrational modes: A1′(ν1), A2′’(ν2), and E’(ν3 and ν4); ν1 is Raman active, ν2 is IR active, ν3 and ν4 are both IR and Raman active. All of these vibrations are predicted to occur at respective frequencies of 1049, 830, 1355, and 690 cm−1. In the IR spectrum experiment, the strong peak for the NO3− asymmetric vibration that stretches is located at 1338 cm−1. At 1532 cm−1, the DFT technique predicts the same stretching vibration. Experimentally and theoretically, extra stretching vibrations vs(NO3−) were observed at 1181 and 1302 cm−1, respectively. The experimental band that was measured at 765 cm−1 and computed at 825 cm−1 is a result of the plane's distortion. Regarding the out of plane bending vibrations of NO3−, the experimental peaks are located at 832 cm−1 (calculated at 916 cm−1).
3.7 Chemical docking research
The discipline of molecular modeling, and more specifically molecular docking, often known as rapidly brought into the field of biological study with the advent of electronic devices during the previous twenty years. This method is employed to investigate the interactions between two molecules, often a protein and a ligand. These interactions are crucial to the organization of biological systems because they permit the control of some biological processes, the signaling of events, or the catalysis of various biochemical reactions. In our study, a molecular docking analysis of the ligand (4-oxo-piperidinium ethylene acetal) trioxonitrate (4-OPEAN) with COVID-19/7EJY and COVID-19/6Y84 receptors was performed. These proteins are intriguing targets for the development of novel drugs against disease, according to recent research. The outcomes of the calculations allow for the possibility of ten ligand positions within each protein's active site, with the optimal position having the lowest energy and being associated with the most stable complex. These ideal locations are shown in Fig. 7 and Fig. S4. Table 3 compiles information about the computation of conformational interaction energies, including total energy scores, hydrogen bond energies, and VDW interactions. The following findings are obtained from a tabular examination: The overall energy scores for the inhibitor (4-OPEAN) interacting with the 6Y84 and 7EJY proteins, respectively, is −80.0643 and −73.1571 kcal/mol. Their VDW interactions are about −51.133; −54.1381 kcal/mol. High hydrogen bond energy of −20.0932 kcal/mol for (4-OPEAN/7EJY) and −29.6289 kcal/mol for (4-OPEAN/6Y84) is also produced by the molecular interaction of the ligand in the active sites of the two enzymes. Furthermore, Table S6 analysis shows that the root mean square deviation (RMSD) of this compound in each system in the order of 1.0299 (6Y84) and 1. 7153 (7EJY), so since its values are less than three, they help to stabilize the system and support the validity of our docking results. 6Y84 and 7EJY have corresponding binding score values of −3.1968 and −3.0197 kcal/mol. Visual examination of the 3D and 2D representations of the 4-OPEAN ligand in the active sites of 6Y84 and 7EJY (Fig. 7, Fig. S4 and Table S7) demonstrates that the atoms C2, O13 and O5 are connected to the binding residues A: CYS 145, A: ASN 142, TYR 140, LYS 125 and A: SER 126 via conventional hydrogen bonding interactions, with distance ranges varying between 2.74 and 3.81 Å and energy values ranging from −0.7 to −3.5 kcal/mol for the two complexes. As a result, considering the energy scores, all the findings mentioned above, and previous research, we can consider our ligand to be a potential inhibitor in the treatment of COVID-19 diseases because it penetrates well into the active regions of the receptors.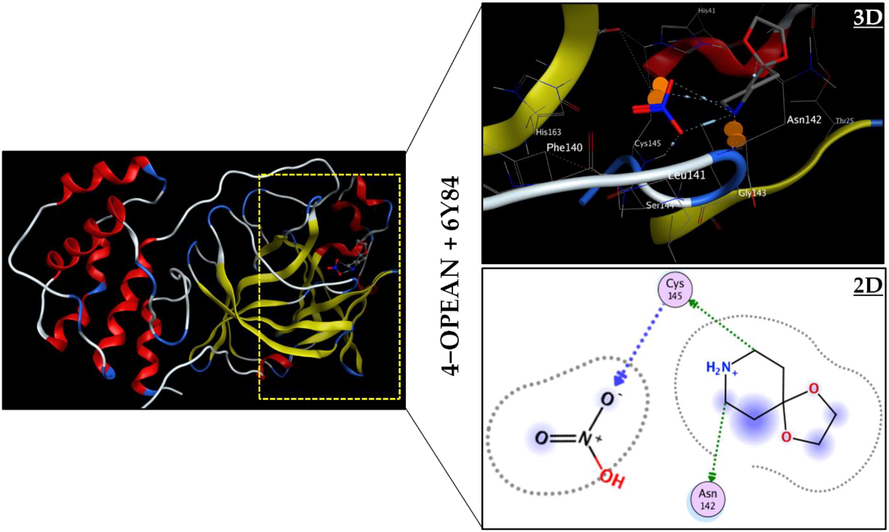
3D best docked poses and 2D interactions of 4-OPEAN compound with the 6Y84 protein.
Ligand
Proteins Code
Total Energie (kcal/mol)
VDW (kcal/mol)
H-Bond (kcal/mol)
Electronic (kcal/mol)
4-OPEAN
6Y84
−80.0643
−51.133
−29.6289
0.697553
7EJY
−73.1571
−54.1381
−20.0932
1.07421
4 Conclusion
Thus, to recap, we created, produced, and studied (4-oxo-piperidinium ethylene acetal) trioxonitrate crystal. This crystal structure reveals a 3D supramolecular network sustained by electrostatic, N—H…O and C—H…O H-bonds as well as van der Waals interactions. The Hirshfeld surface research used in the study of intermolecular interactions reveals that the O…H/H…O and H…H contacts occupy the majority of the Hirshfeld surface. Using the DFT/B3LYP/6-311++G (d,p) basis set, theoretical calculations for the 4-OPEAN compound were performed, and their optimized bond parameters were determined. Good agreement was established between theoretical and experimental results. The vibrational frequencies being tested of basic modes our material has undergone accurate analysis, assigned, and contrasted to the findings from theory. More specifically, the network of N—H… O and C—H…O hydrogen bond was examined using the AIM, RDG and ELF analyses; they make clear the monomeric structure exceptional stability. As a donor-recipient exchange, the MEP surface serves as evidence for the hydrogen bond's creation. It is clear that the hydrogen and nitrogen atoms of the ammonium group are where the possible positive sites are placed, while the oxygen atoms of the nitrate anion are where the potential negative sites are situated. The border orbitals demonstrate the exceptional dynamical stability, inadequate chemical responsiveness (ΔE = 4.11 eV), and considerable biological activity (μ < 0) of our novel material. We investigated the molecular docking of our structure within the same molecular modeling framework. In addition to showing the inhibiting effect of the promoter in the management of the new coronavirus, this molecular model indicates that the 4-OPEAN molecule interacts favorably with COVID-19.
Acknowledgments
Researchers Supporting Project number RSP2023R61, King Saud University, Riyadh, Saudi Arabia. This study was carried out also within the state assignment no. 0287-2021-0012 for the Institute of Chemistry and Chemical Technology, Siberian Branch of the Russian Academy of Sciences.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Atoms in Molecules: A Quantum Theory. Oxford: Oxford University Press; 1990.
- Brandenburg. K, Diamond Version 2.0 Impact, GbR, Bonn, 1998.
- Bruker, APEX2, SAINT and SADABS, Bruker AXS Inc, Madison, Wisconsin, USA, 2006.
- Gold nanoparticles: assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem. Rev.. 2004;104:293-346.
- [Google Scholar]
- Crystal structure and spectroscopic investigations of a new organic monophosphate monohydrate. Phos. Sulf. Sili. 2010;185:609-619.
- [Google Scholar]
- GAUSSIAN 09, Revision A.02. Wallingford, CT: Gaussian, Inc.; 2009.
- Theory of Orientation and Stereoselection. Berlin Heidelberg, New York: Springer-Verlag; 1975.
- Synthesis, characterization, Hirshfeld surface analysis and antioxidant activity of a novel organic-inorganic hybrid material 1-methylpiperazine-1,4-diium bis(nitrate) J. Mol. Struc.. 2017;1139:52-59.
- [Google Scholar]
- Synthesis, structural and spectroscopic features, and investigation of bioactive nature of a novel organic-inorganic hybrid material 1H–1,2,4-triazole-4-ium trioxonitrate. J. Mol. Struct.. 2017;1150:242-257.
- [Google Scholar]
- A proton transfer compound template phenylethylamine: Synthesis, a collective experimental and theoretical investigations. J. Mol. Struc.. 2019;1191:183-196.
- [Google Scholar]
- Synthesis, experimental, theoretical study and molecular docking of 1-ethylpiperazine-1,4-diium bis(nitrate) Solid State Sci.. 2020;106:106326
- [Google Scholar]
- Synthesis, experimental and computational study of a non-centrosymmetric material 3-methylbenzylammonium trioxonitrate. J. Mol. Struc. 2020;1225:129132
- [Google Scholar]
- Self assembly of a novel Cu(II) complex, (C6H9N2)2[CuCl4]: experimental, computational, and molecular docking survey. J. Ira. Chem. Soci. 2021;18:2331-2343.
- [Google Scholar]
- Deciphering non-covalent interactions of 1,3-Benzenedimethanaminium bis(trioxonitrate): Synthesis, empirical and computational study. J. Mol. Struc.. 2022;1250:131720
- [Google Scholar]
- The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ. 2014;1:119-128.
- [Google Scholar]
- Synthesis, empirical and theoretical investigations on new histaminium bis(trioxonitrate) compound. Molec. 2023;28(4):1931.
- [Google Scholar]
- J. Am. Chem. Soc.. 2010;132:6498-6506.
- Crystal structure and Hirshfeld surface analysis of 2-methyl-3-nitro-N-[(E)-(5-nitrothiophen-2-yl) methylidene] aniline. Acta Cryst. Section E. 2021;77(2):138-141.
- [Google Scholar]
- “Two New Copper (II) Carboxylate Complexes Based on n, n, n’, n’-Tetramethylethyleneamine: Synthesis, Crystal Structures, Spectral Properties, Dft Studies and Hirshfeld Surface Analysis. J. Mol. Struc.. 2021;1230:129916.
- [Google Scholar]
- Bader’s theory of atoms in molecules (AIM) and its applications to chemical bonding. J. Chem. Sci.. 2016;128:1527-1536.
- [Google Scholar]
- Synthesis, crystal structure, supramolecular assembly inspection by Hirshfeld surface analysis and computational exploration of 4-phenyl-6-(p-tolyl)pyrimidin-2(1H)-one (PPTP) J. Struc. Chem.. 2023;64(3):437-449.
- [Google Scholar]
- Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem.. 2012;33:580-592.
- [Google Scholar]
- Synthesis and characterization of Tetrakis(4-oxo-piperidinium ethylene acetal) Bis Sulfate Hexahydrate, J. Chem.. 2011;8(4):1930-1936.
- [Google Scholar]
- Molecular Operating Environment (MOE), 2015.10; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2015.
- A new electroaffinity scale; together with data on valence states and on valence ionization potentials and electron affinities. J. Chem. Phys.. 1934;2:782-794.
- [Google Scholar]
- Quantum chemical calculations, spectroscopic properties and molecular docking studies of a novel piperazine derivative. J. King Saud Unive.r – Sci.. 2021;33:101283
- [Google Scholar]
- Improved solubility and bioactivity of Theophylline (a Bronchodilator drug) through its new nitrate salt analysed by experimental and theoretical approaches. J. Mol. Struc.. 2016;1116:45-55.
- [Google Scholar]
- Chemical Hardness. Weinheim: John Wiley-VCH; 1997.
- Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J. Am. Chem. Soc.. 2000;122:11154-11161.
- [Google Scholar]
- SHELXT – Integrated space-group and crystal structure determination. Acta Cryst. A. 2015;71:3-8.
- [Google Scholar]
- Nanoparticle arrays on surfaces for electronic, optical, and sensor applications. Chemplys. Chem.. 2000;1:18-52.
- [Google Scholar]
- Nanocrystalline semiconductors: synthesis, properties, and perspectives. Chem. Mater.. 2001;13:3843-63858.
- [Google Scholar]
- Wolff, S.K., Grimwood, D.J., McKinnon, J.J., Jayatilaka, D., Spackamn, M.A., 2013. Crystal Explorer 3.1, University of Westren Australia, Perth.
- GEMDOCK: a generic evolutionary method for molecular docking proteins. Struct. Funct. Bioinforma.. 2004;55:288-304.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2023.102758.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




