

Translate this page into:
Comprehensive in silico characterization of Arabidopsis thaliana RecQl helicases through structure prediction and molecular dynamics simulations
⁎Corresponding author. amit_geb@ru.ac.bd (Amit Kumar Dutta),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Helicases are ubiquitous enzymes with specific functions that contribute to almost all nucleic acid metabolic processes. The RecQ helicase family is essential for integrity in all organisms through DNA replication, repair, and recombination. This study investigated five RecQ-like helicases in Arabidopsis thaliana (AtRecQl) that exhibit diverse structural and physiochemical attributes and functions. Cis-regulatory element analysis identified stress, hormone, cell cycle, and development-responsive modules involved in various events in plant growth and development. Gene ontology analysis revealed that the five AtRecQl were associated with various cellular components, molecular functions, and biological processes. Protein-protein interaction analysis also implicated some in various abiotic stress processes. Structural analysis and molecular dynamics (MD) simulations were performed to examine conformational stability through root means square deviation and radius of gyration, showing stable AtRecQl protein structures. Free energy landscape analysis validated thermodynamically stable structures throughout the MD simulation. Principle component analysis and probability density functions from MD simulations provided satisfactory structural variational data for the complexes and limited coordinate movements. These insights might greatly benefit future studies.
Keywords
Arabidopsis thaliana
AtRecQl
in silico
cis-elements
molecular dynamics simulation

- GO
-
gene ontology
- HR
-
homologous recombination
- RMSD
-
root means square deviation
- RG
-
radius of gyration
- MD
-
molecular dynamics
- PCA
-
principal component analysis
- FEL
-
free energy landscape
- PPI
-
protein-protein interaction
- GFE
-
Gibbs free energy
Abbreviations
1 Introduction
Helicases are responsible for unfolding the double-strand nucleotide chain and shuffling the secondary structure of RNAs (Tuteja and Tuteja, 2004). They exist in all living organisms, ranging from viruses to humans (Mu et al., 2024; Tapescu et al., 2023). The RecQ helicase family, part of the SF2 helicase superfamily, is one of the most conserved groups of 3′-5′ DNA helicases (Bokhari et al., 2019). They are critical to preserving the genetic integrity of numerous species and the metabolic processes of DNA replication, repair, and recombination (Wiedemann et al., 2018).
RecQ helicase numbers vary across organisms. Among unicellular model organisms, Escherichia coli and Saccharomyces cerevisiae have a single RecQ DNA helicase, while the fission yeast Schizosaccharomyces pombe has two RecQ homologs (Hartung and Puchta, 2006; Mandell et al., 2005). Drosophila melanogaster and Homo sapiens have five RecQ genes, with RecQ5 having three alternatively spliced forms in both species (Hartung and Puchta, 2006; Sekelsky et al., 1999). The Arabidopsis thaliana genome encodes seven RecQ-like genes with homologs in rice and wheat: AtRecQl1, AtRecQl2, AtRecQl3, AtRecQl4A, AtRecQl4B, AtRecQl5, and AtRecQsim. While the AtRecQl4A and AtRecQl4B arose due to a contemporary duplication of a chromosomal region (Bagherieh-Najjar et al., 2003; Hartung et al., 2007; Saotome et al., 2006), the functions of AtRecQl4A and AtRecQl4B are distinct. The loss of AtRecQl4A function enhanced homologous recombination (HR) and genotoxic sensitivity, whereas the loss of ATRecQl4B function showed a lower HR rate, with AtRecQl4B required for crossovers (Bagherieh-Najjar et al., 2005; Hartung et al., 2007). Single-molecule in vivo experiments demonstrated that AtRecQl2 and AtRecQl3 have the same biological activity in strand switching: AtRecQl2 unwinds, AtRecQl3 rewinds, and both exhibit 3′-5′ helicase activity (Klaue et al., 2013; Kobbe et al., 2009). An early report showed that four RecQ homologs had particular functions in the rice DNA repair pathways (Saotome et al., 2006). Single mutations in the RecQ4 gene increased crossovers about threefold in tomatoes, rice, and peas (de Maagd et al., 2020; Mieulet et al., 2018).
While RecQ homologs are common and play vital roles in plant growth and development, their exact roles remain largely unknown. The in silico characterization of RecQ homologs may provide extensive information about these proteins and genes. Therefore, this study comprehensively examined the five AtRecQl genes in A. thaliana. Their potential secondary structures and 3D models were predicted, and the stability of the docked structure was examined using molecular dynamics (MD) simulations. It lays the groundwork for the functional characterization and possible functional significance of RecQ pathways.
2 Experimental methods
2.1 Structure and phylogenetic analyses of the AtRecQl genes
The genomic DNA, coding, and amino acid sequences of previously reported (Hartung et al., 2000) AtRecQl genes were retrieved from the Arabidopsis Information Resource (TAIR) (https://www.arabidopsis.org/) database in FASTA format. The corresponding AtRecQl protein sequences were used as queries in BLASTP searches to examine their phylogenetic relationships with RecQls in other crop plants. The retrieved protein sequences were also used for multiple sequence alignments using ClustalW with default parameters. A phylogenetic tree was constructed using the MEGA X software and the unrooted neighbor-joining method.
2.2 Analysis of cis-elements and gene annotation
The in silico promoter analysis considered one kilobase (kb) upstream of the translation start site of each AtRecQl gene, with sequences obtained from the TAIR database. The cis-elements were identified using three online cis-elements tools—Arabidopsis Gene Regulatory Information Server (AGRIS; http://arabidopsis.med.ohio-state.edu/AtcisDB/), PlantCare (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/), and Plant Cis-Acting Regulatory DNA Elements (PLACE; https://www.dna.affrc.go.jp/PLACE/)—and compared among tools. The AtRecQl genes were annotated with gene ontology (GO) terms using the TAIR search tool (https://www.arabidopsis.org/tools/bulk/go/index.jsp).
2.3 Protein-protein interaction predictions
An interaction network was constructed for the AtRecQl proteins using the STRING website (http://www.string-db.org). The protein-protein interaction (PPI) map of the AtRecQls was predicted with the confidence parameter set to 0.15 using the threshold/molecular action mode with K means clustering.
2.4 Transmembrane, solubility, and thermodynamic properties
Transmembrane topology was predicted using DeepTMHMM (https://dtu.biolib.com/DeepTMHMM). Protein solubility was predicted using Protein-Sol (https://protein-sol.manchester.ac.uk). Thermal resilience was assessed using the SCooP server (http://babylone.3bio.ulb.ac.be/SCooP/index.php).
2.5 Molecular modeling and validation
The secondary structural characteristics of the AtRecQl protein sequences were computed using SOPMA (https://npsa-prabi.ibcp.fr/NPSA/npsa_sopma/) with 17 window widths, 8 similarity thresholds, and 4 conformational states. A hypothetical 3D model was predicted for the protein sequence via comparative homology modeling using SWISS-MODEL (https://swissmodel.expasy.org/). The quality of the modeled protein structures was assessed using MolProbity (http://molprobity.biochem.duke.edu/) assessing. Inaccuracies within the predicted 3D structures were detected based on the model’s atomic coordinates using ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php).
2.6 MD simulations
The AtRecQl proteins were assessed with time-dependent biological conditions for 100 ns using the Yasara Structure software. The protein residue ionizations were set at pH 7.4. A cubic box, located 10 Å away from all atoms, was solvated with the TIP3P water model and 0.9% NaCl to simulate a physiological environment. The MD simulations used the AMBER14 force field for the solute, GAFF and AM1-BCC charges for the ligands, and TIP3P for water molecules. The temperature was maintained at 298 K under a pressure of 1 atm (NPT ensemble) using a Berendsen barostat and thermostat, with time-averaged temperature and density coupling. Non-bonded long-range interactions were computed using the Particle Mesh Ewald algorithm.
3 Results
3.1 Evolutionary analysis of RecQls
The domains of AtRecQl proteins and structures of AtRecQl genes with chromosomal locations were analyzed to obtain information about their evolution (Supplementary Fig. S1). An unrooted phylogenetic tree was constructed among the model plant A. thaliana and nine crop plants (Brassica napus, Camelina sativa, Raphanus sativus, Gossypium hirsutum, Citrus sinensis, Momordica charantia, Solanum tuberosum, Glycine max, and Oryza sativa) to understand the evolutionary connection and predict the possible functions of RecQls in different crops (Supplementary Fig. S2 and Supplementary Table S1). The 44 RecQl proteins of ten plants were partitioned into four groups representing RecQl1, RecQl2, RecQl3, and RecQl4. Two copies of RecQl4 (like AtRecQl4A and AtRecQl4B) were presented only in three crops: B. napus, C. sativa, and R. sativus. Phylogenetic analysis revealed that RecQl2 and RecQl4 were closer among the four groups, consistent with the Simple Modular Architecture Research Tool (SMART) domain structure analysis (Supplementary Fig. S1A). As predicted, the O. sativa RecQls were more distant from all other plant RecQls, except the RecQl2 groups.
3.2 Cis-element analysis of the promoters of AtRecQls
Both reverse and forward strands 1 kb upstream of the five AtRecQl genes were scanned for cis-elements using three programs: PlantCare, AGRIS, and PLACE. Among all AtRecQl genes, development and stress-responsive elements were the most common (Supplementary Fig. S3A). Cis-elements associated with different stress responses, such as dehydration-responsive element, low-temperature-responsive element, early responsive to dehydration ACGTATERD1, and heat shock element (HSE) CCAAT BOX1, were significantly evident in the AtRecQl promoter regions . Drought- and abscisic acid-related MYC and different stress and cell-cycle-related MYB cis-elements were found in all AtRecQl genes. Salicylic acid-induced WRKY DNA binding site W-box/WB-box, abscisic acid-responsive elements (ABREs), auxin response element ARFAT were also common in the AtRecQl promoter regions. Furthermore, light-responsive cis-elements GT1, GATA box, cytokinin-regulated transcriptional activator ARR1, and various organ-specific cis-elements were found in the AtRecQl promoter regions. The AtRecQls promoter regions contained stress, hormone, cell cycle, and development-responsive cis-elements, indicating that these genes are involved in various events in plant growth and development.
3.3 GO annotation of AtRecQls
The five AtRecQl genes were annotated with GO terms using the TAIR database. All three categories of GO enrichment terms (cellular component, molecular function, and biological process) were examined (Supplementary Fig. S3B). Four GO terms were predicted under cellular component categories, of which cytoplasm, nucleus, and other intracellular components were identified for all five AtRecQl genes, whereas other cellular components was identified for only one gene. Four GO terms were also predicted under molecular function categories for the five AtRecQl genes, of which catalytic activity and hydrolase activity were identified for all five genes, whereas nucleic acid binding was identified for four and protein binding for only one. The AtRecQls are involved in various biological processes. The GO annotations revealed that the five AtRecQls are dedicated to DNA metabolic, biosynthetic, cellular components organization, and other cellular and metabolic processes. GO terms related to different stress responses, such as responses to stress, abiotic stimuli, chemicals, and endogenous stimuli, were significantly represented. These results indicate that AtRecQls are involved in various abiotic stresses.
3.4 PPI predictions
The PPI prediction results for the AtRecQls are shown in Fig. 1. The network was divided into three groups. According to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, the 25 proteins constituting the network were present in three main KEGG pathways: 11 in HR, 4 in non-homologous end joining, and 2 in mismatch repair (Supplementary Fig. S4). The top ten predicted functional partners were FANCM, MUS81, TOP3α, UVH1, AT5G19950, ATR, XRCC3, MSH5, AT1G29630, and MRE11 (Supplementary Fig. S5). The FANCM had the highest score and interacted with all examined AtRecQl proteins. The AtRecQls exhibited significant co-expression with FANCM, TOP3α, ATR, MSH5, and AT1G29630 (Supplementary Fig. S6). The GO terms of these 25 proteins were also examined (Supplementary Fig. S7, S8, and S9), indicating that most are DNA-related. Some proteins in this network are also involved in cellular responses to various abiotic stresses stimulus.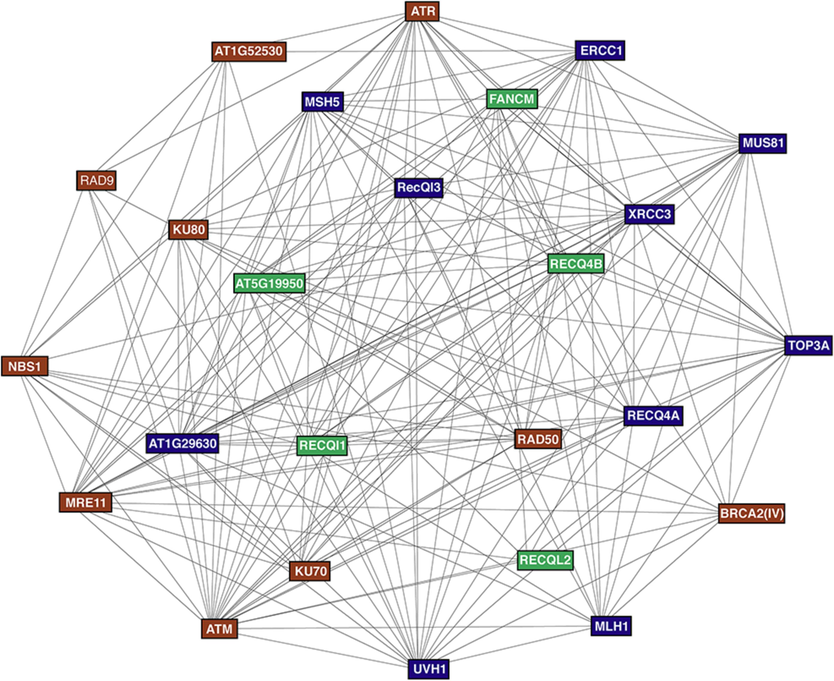
The interaction network of RecQl proteins in A. thaliana. The predicted protein–protein interaction map of AtRecQls was generated with 3 clusters of K-means clustering (clusters-1 is brown, clusters-2 is dark blue, and clusters-3 is green). The “minimum required interaction score” was set to 0.9 (highest confidence); other boxes used default settings.
3.5 Transmembrane, solubility, and thermodynamic properties of the AtRecQls
The transmembrane analysis indicated the absence of transmembrane α-helices and signal regions, while all AtRecQl proteins exhibited an internal localization. The distribution of protein types and regions suggested a predominant protein localization pattern within cellular compartments.
The predicted solubility was 0.521 for AtRecQl1, 0.500 for AtRecQl2, 0.447 for AtRecQl3, 0.447 for AtRecQl4A, and 0.395 for AtRecQl4B (Table 1); proteins with scaled solubility values >0.45 are expected to be soluble, while those with values <0.45 are expected to be less soluble (Niwa et al., 2009). Therefore, AtRecQl1 and AtRecQl2 are expected to be soluble since their scaled solubility values are above this threshold. However, the lower predicted solubilities for AtRecQl3, AtRecQl4A, and AtRecQl4B imply potential structural instability or aggregation tendencies, which could impact their functionality.
AtRecQl1
AtRecQl2
AtRecQl3
AtRecQl4A
AtRecQl4B
predicted solubility
0.5
0.5
0.4
0.4
0.4
Delta Hm (kcal/mol)
-17.7
-26.5
-48.8
-22
-46.5
Delta Cp (kcal/(mol K))
-0.5
-0.7
-1.4
-0.6
-0.8
Tm (°C)
62.5
59.5
58.7
64
64.3
Delta Gr (kcal/mol)
-0.8
-1.5
-2.6
-1
-3.6
Temperature range (°C)
-6.1; 77.5
-22.3; 74.5
-14.7; 73.7
-6.1; 79
-53.7; 79.3
The changes in enthalpy (ΔHm) for the AtRecQls were −17.7 to −48.8 kcal/mol (Table 1). Similarly, their changes in heat capacity (ΔCp) were −0.53 to −1.35 kcal/(mol K). Their melting temperatures (Tm) and changes in Gibbs free energy (ΔGr) were 58.7°C to 64.3°C and −0.8 to −3.6 kcal/mol, respectively. The temperature ranges over which these proteins underwent ΔGr were −6.07–77.52°C for AtRecQl1, −22.29°C–74.49°C for AtRecQl2, −14.72°C–73.67°C for AtRecQl3, −6.12°C–78.99°C for AtRecQl4A, and −53.56°C–79.28°C for AtRecQl4B. AtRecQl3 exhibited the highest ΔHm of −48.8 kcal/mol and ΔGr of −2.6 kcal/mol, suggesting its structure is more stable than the other examined proteins (Pucci and Rooman, 2016). AtRecQl4B also showed a high ΔHm of −46.5 kcal/mol) and ΔGr of −3.6 kcal/mol, indicating high thermodynamic stability. In contrast, AtRecQl1 and AtRecQl2 had lower ΔHm of −17.7 and −26.5 kcal/mol, respectively, and relatively smaller ΔGr of −0.8 and −1.5 kcal/mol, respectively, implying comparatively lower stability. AtRecQl4A fell between these groups, with a moderate ΔHm of −22 kcal/mol and ΔGr of −1.0 kcal/mol. Moreover, AtRecQl4A and AtRecQl4B had higher Tm of 64.0°C and 64.3°C, respectively, compared to 62.5 for AtRecQl1, 59.5 for AtRecQl2, and 58.7 for AtRecQl3, suggesting greater thermal stability (Pucci and Rooman, 2016). The temperature range also varied, with AtRecQl4B exhibiting the widest range of −53.56°C–79.28°C, followed by AtRecQl1, AtRecQl4A, AtRecQl3, and AtRecQl2, in descending order.
3.6 Structural analysis of AtRecQl proteins
The alpha helix content was highest for AtRecQl2 (48.51%) and lowest for AtRecQl4A (37.04%) (Fig. 2 and Table 2). The beta-sheet content was highest for AtRecQl1 (14.85%) and lowest for AtRecQl4A (9.09%). The turn and coil contents also varied among the proteins, with AtRecQl4A having the highest coil content (48.23%) and AtRecQl1 the lowest (35.64%).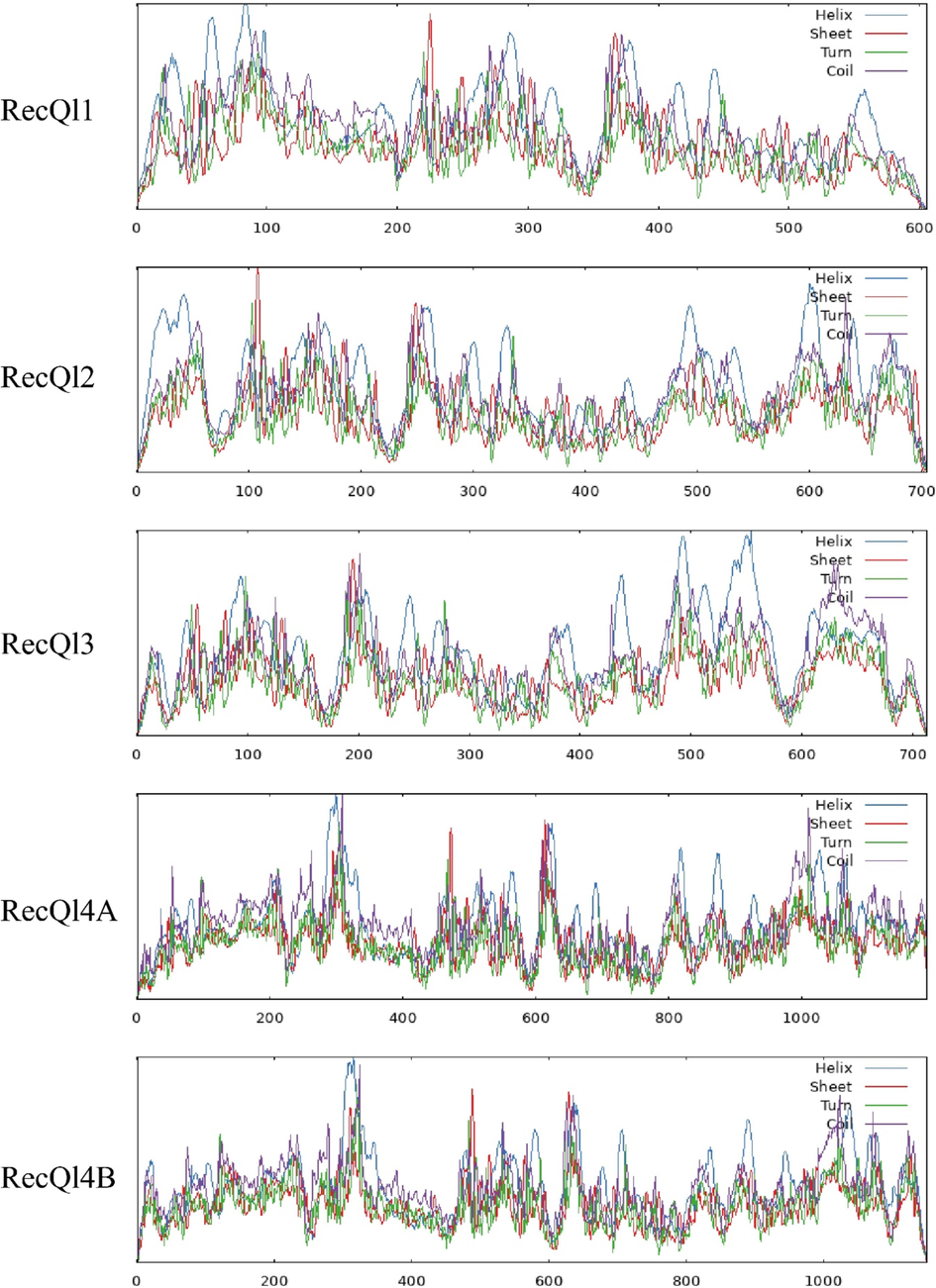
The secondary structure of retrieved AtRecQl amino acid sequences by SOPMA server. The blue, red, green and purple color represents the helix, sheet, turn and coil structure of the proteins.
Alpha helix
Beta sheet
Turn
Coil
AtRecQl1
260 (42.90%)
90 (14.85%)
40 (6.60%)
216 (35.64%)
AtRecQl2
342 (48.51%)
91 (12.91%)
40 (5.67%)
232 (32.91%)
AtRecQl3
326 (45.72%)
81 (11.36%)
44 (6.17%)
262 (36.75%)
AtRecQl4A
440 (37.04%)
108 (9.09%)
67 (5.64%)
573 (48.23%)
AtRecQl4B
465 (40.43%)
125 (10.87%)
63 (5.48%)
497 (43.22%)
Regarding all-atom contacts, the Clashscore values indicated relatively low steric clashes, with AtRecQl2 having the highest and AtRecQl1 the lowest values (Chen et al., 2015) (Supplementary Fig. S10 and Supplementary Table S2). Protein geometry analysis revealed that all proteins had high percentages of favored rotamers, ranging from 92.20% to 96.03%. AtRecQl2 had the lowest percentage of poor rotamers (1.09%) and the highest percentage of Ramachandran favored (94.58%). AtRecQl4A had the highest percentage of Ramachandran outliers (1.09%), while AtRecQl1 had the highest percentage of favored rotamers (96.03%). Ramachandran analysis highlighted minor differences among the proteins, with AtRecQl2 having no outliers, while AtRecQl4A and AtRecQl4B had slightly higher percentages of Ramachandran outliers (Ravikumar et al., 2019). The Rama distribution Z-scores indicated acceptable backbone conformations for all proteins (Supplementary Fig. S11).
3.7 MD simulation and principal component analysis
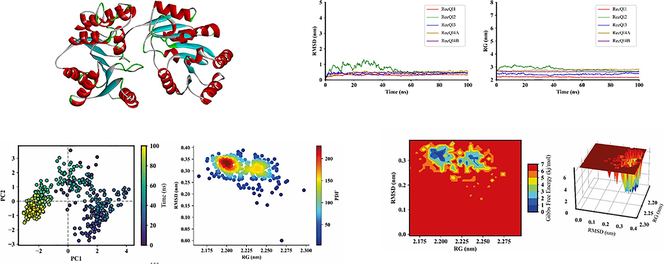
The root means square deviations (RMSD) for the five AtRecQls were calculated to assess their structural variability. The average RMSD varied from 0.3399 to 0.4579 nm, with AtRecQl1 exhibiting the lowest average RMSD of 0.3399 nm and AtRecQl4 showing the highest average RMSD of 0.5422 nm (Fig. 3). The maximum RMSD varied considerably across proteins, with AtRecQl2 having the highest maximum RMSD of 1.3171 nm at 7.5 ns and AtRecQl1 having the lowest maximum RMSD of 0.3884 at 29.75 ns. Similarly, the minimum RMSD values also varied, with AtRecQl2 having the lowest minimum RMSD of 0.1238 nm at 5 ns and AtRecQl1 having the highest minimum RMSD of 0.1528 nm at 7.5 ns. This observation indicated that the AtRecQl proteins had stable carbon backbone conformations throughout the simulation period (Sargsyan et al., 2017). The average radius of gyration (RG) ranged from 2.219 to 2.764 nm across the proteins, with AtRecQl2 having the highest average RG of 2.842 nm and AtRecQl1 having the lowest average RG of 2.219 nm.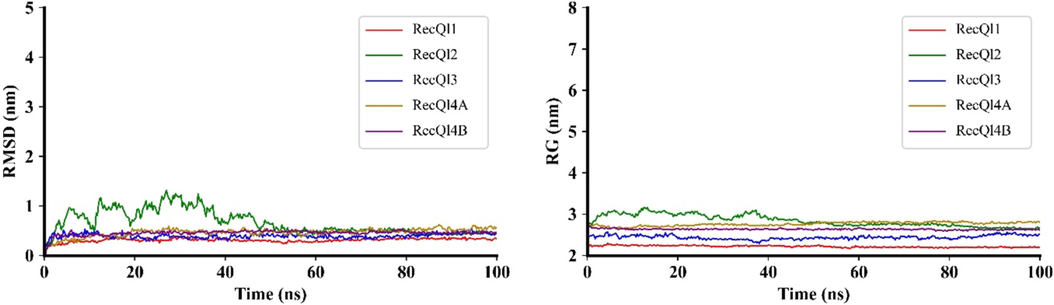
The structural characteristics of RecQ proteins. The MD trajectories were scrutinized utilizing RMSD for assessing structural stability and RG for analyzing rigidity and compactness. Visualization of MD plots was accomplished using Matplotlib v3.7.
A principal component analysis (PCA) score plot was used to measure and identify related motions and dynamic regions within the protein structure to assess fluctuations, changes in atomic locations, and flexibility (Lang et al., 2009). The contributions of eigenvectors to the overall conformational variability of AtRecQl1, AtRecQl2, AtRecQl3, AtRecQl4A, and AtRecQl4B were determined to be cumulative variance ratios of 45.65% (PC1 = 35.37%, PC2 = 11.24%), 79.95% (PC1 = 58.29%, PC2 = 21.66%), 50.8% (PC1 = 36.13%, PC2 = 14.68%), 56.53% (PC1 = 37.8%, PC2 = 18.73%), and 40.24% (PC1 = 22.42%, PC2 = 17.82%), respectively (Fig. 4). These values suggested limited coordinate movements, indicating stability throughout the MD simulation.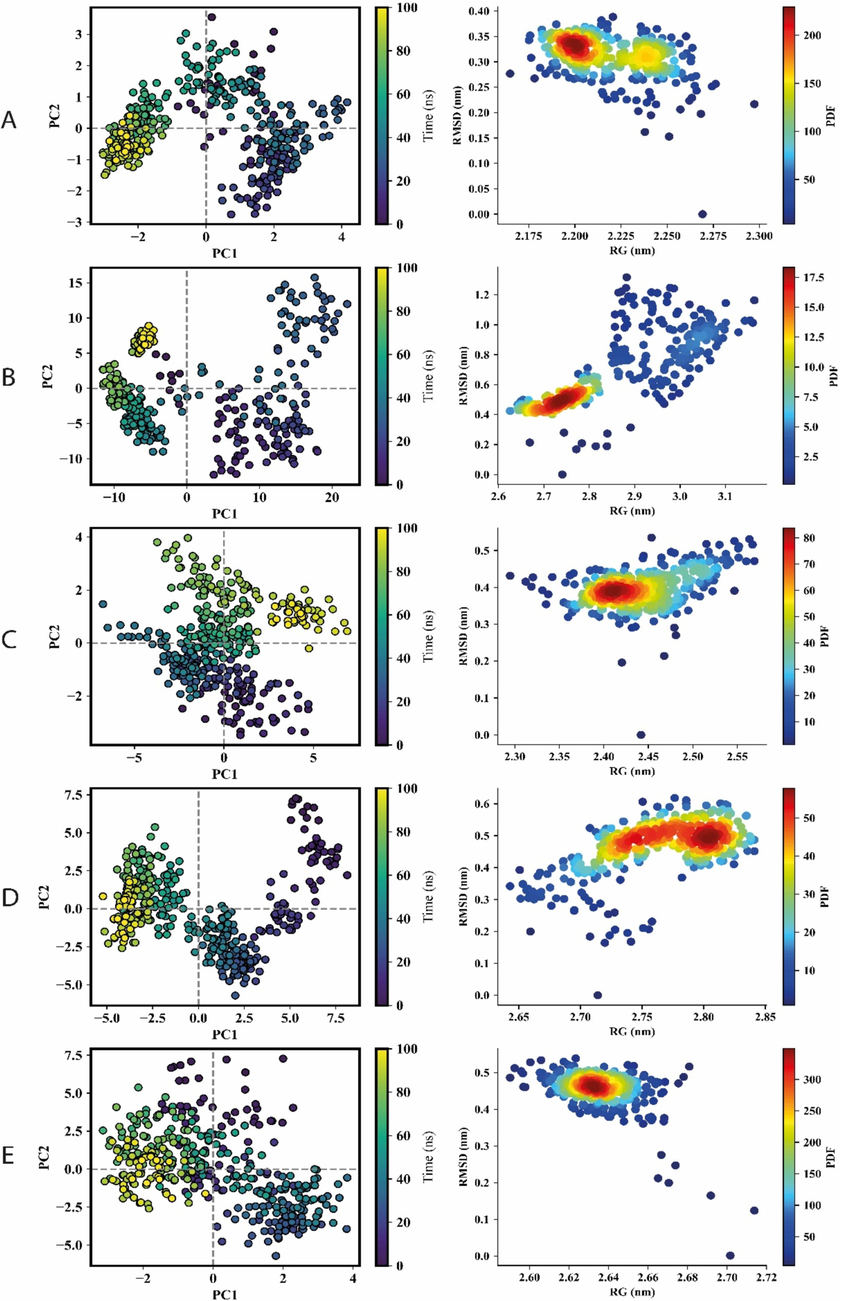
The PCA and the PDF plot of RecQ proteins. A, B, C, D and E represent the RecQl1, RecQl2, RecQl3, RecQl4A and RecQl4B, respectively. Left panel represents PCA where each point on the scatter plot represents the conformation of the complexes along the axes. The color gradient from purple (initial timestep) to teal to yellow (final timestep) corresponds to different time points within the simulation. The right panel shows the PDF plots depicting the distribution of RMSD and RG values for RecQ proteins.
The free energy landscape (FEL) represents the intricate interplay of elements. It reveals the complexes’ most energetically stable states, transition pathways, and widespread dynamic motion (Parra-Cruz et al., 2018). Regions with lower RG and RMSD demonstrated packed and stable structure conformations (Fig. 5); when associated with reduced Gibbs free energy (GFE), these regions signified thermodynamically favorable states. Conversely, higher RMSD suggested structural fluctuations, potentially corresponding to less stable conformations. Increased GFE in such regions indicated higher energy stateand less favorable circumstances. Energy minima, depicted as valleys, represented considerably stable conformations.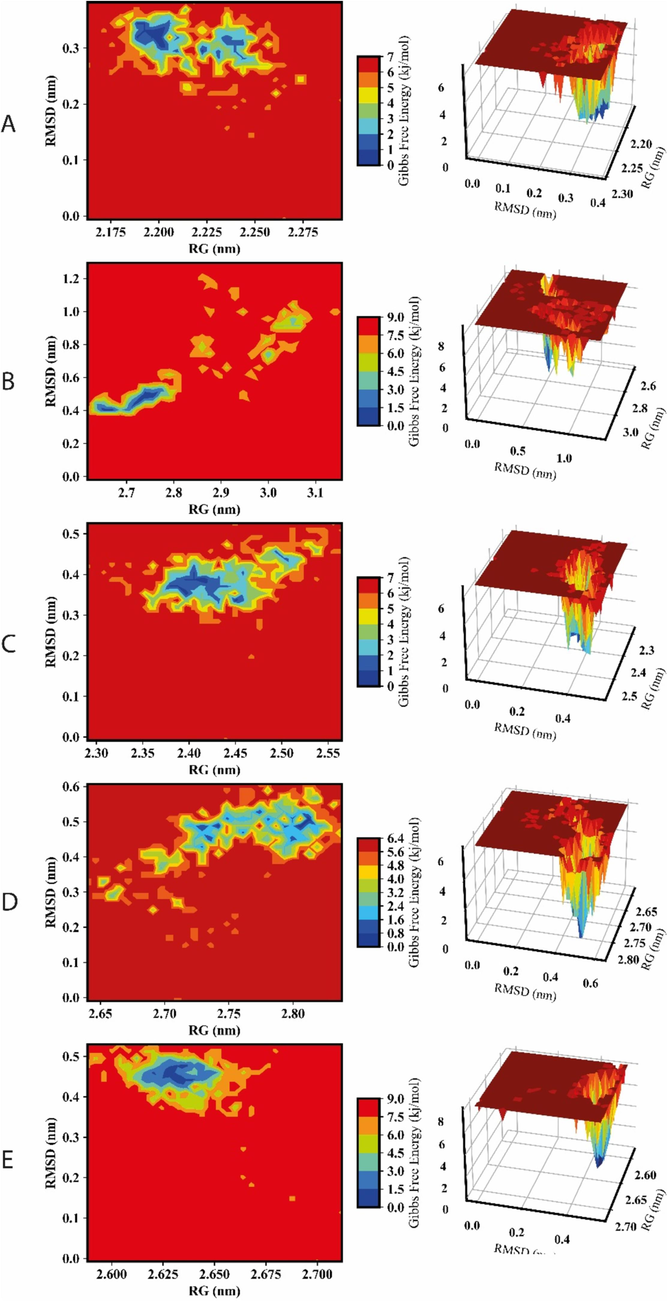
3D and 2D FEL depictions of RecQ, alongside RMSD and RG structural parameters. A, B, C, D and E represent the RecQl1, RecQl2, RecQl3, RecQl4A and RecQl4B, respectively. The left panel shows the two dimensional and the right panel illustrates their corresponding three-dimensional energy surface.
4 Discussion
Helicases are ubiquitous enzymes that perform diverse roles, being engaged in practically every process in nucleic acid metabolism, and the ubiquitous presence of RecQs suggests that they are needed for their vital tasks, which are unknown in plants (Wiedemann et al., 2018). Helicases improve biological processes, stabilize protein synthesis, respond to abiotic stress, and interact with DNA-protein complexes to modulate gene expression (Mohapatra et al., 2023).
Our phylogenetic analysis suggested that RecQls are conserved among related plant species, which may be attributable to their essential functions. Cis-elements function in the transcriptional regulation of several biological processes (Deng et al., 2023). Our results demonstrated that the promoter regions of AtRecQl genes contained significant categories of cis-elements (Supplementary Fig. 3A), such as light-responsive, tissue-specific, cell cycle-related, and abiotic stress-specific cis-elements. While AT-rich sequences, CCAAT boxes, and HSEs moderately affect heat shock gene levels (Nover et al., 2001). Besides heat stress, MYC transcription factors are crucial regulators of plant growth, development, stress adaptation, and secondary metabolite production (Abe et al., 1997). Similarly, W-box elements are important for responses to heat and salinity (Chen et al., 2002), while ABREs are crucial in environmental stress responses during vegetative growth (Suzuki et al., 2005).These diverse cis-elements within the promoter regions of AtrecQl genes demonstrate the remarkable functional complexity of plant gene regulation. These findings were further supported by GO annotation (Supplementary Fig. 3B), which further reinforced the possibility that AtRecQls have roles in several DNA repair and replication processes under environmental stress, including responses to abiotic stress, abiotic stimuli, chemical stress, and endogenous stimuli.
The predicted PPI network revealed that the AtRecQl proteins act as essential elements of the HR pathway (Fig. 1). In A. thaliana, FANCM suppressed spontaneous somatic HR via an AtRecQ4lA independent pathway (Knoll et al., 2012). AtTOP3a was found to be involved in DNA repair in A. thaliana and mammals and played an important role in reducing crossover recombination in somatic cells, along with AtRecQ4l4A and AtRMl1 (Hartung et al., 2008). The BRCA2-RAD51 complex functions in plant immune responses (Wang et al., 2010). Previous reports showed that RecQls are involved in flower and subsequent development in Moses and Arabidopsis (Perianez-Rodriguez et al., 2021; Wiedemann et al., 2018), validating our cis analysis results. The functional annotations, cis-elements, and PPI networks offer a novel perspective for comprehending the biological role of the AtRecQl gene family in plant growth/development and stress responses.
Structural stability reflects a protein’s capacity to maintain its three-dimensional structure, which is essential for its biological function (Beygmoradi et al., 2023). AtRecQl3 and AtRecQl4B showed greater stability and thermal resistance than the other AtRecQl proteins. The MolProbity scores suggested good overall model quality, with values ranging from 1.05 to 1.49. Cβ deviations were minimal across all proteins. Furthermore, the analysis of peptide omegas showed negligible levels of cis prolines and relatively low levels of cis non-prolines, with RecQl4B having the highest percentage of twisted peptides (0.73%). These results suggest that the AtRecQl protein structures exhibit favorable geometry and conformations with minor variations observed among them.
An integrated approach combining molecular modeling and essential MD simulations was employed to elucidate the structural underpinnings of AtRecQl proteins. Understanding the nature of protein stability, notably the free energy of folding, is essential for comprehending proteins’ roles in biological processes since they establish their functional state (Chen et al., 2023). The minimum and maximum RG were observed for AtRecQl1 and AtRecQl2, respectively. A lower RG signifies a more compact and rigid packing of the protein structure (Lobanov et al., 2008). Significant protein shape and compactness disparities may hinder substrate interactions (Ferdous et al., 2023). Our probability density function (PDF) analysis revealed trends, patterns, and potential structural insights into the dynamic behaviors of the AtRecQl proteins. The proximity of contour lines implies a closer relationship or coordinated changes between RG and RMSD, whereas widely spaced contour lines suggest more distinct transitions or less direct correlations between RG and RMSD (Kraml et al., 2021). Transition states, characterized by increases in RMSD and RG, denoted potential structural changes. The slopes of the FEL surface revealed energy barriers to transitions between states (Al-Khafaji and Taskin Tok, 2020). The AtRecQl proteins exhibited thermodynamically stable structures throughout the MD simulation runtime.
Our study examined the distinct functional roles of conserved regions and shed light on the structural mechanisms of the AtRecQ family. Our results provide valuable new insights into the structural and thermodynamic basis of each AtRecQl protein.
5 Conclusions and perspectives
Our study conducted a comprehensive in-silico analysis of five AtRecQl proteins, paving the way for a deeper understanding of RecQ signaling pathways and the potential for enhancing phenotypic properties in A. thaliana and other plants. Cis-regulatory element analysis identified modules associated with various biological processes, suggesting their involvement in plant growth and development. PPI analysis identified some AtRecQl proteins involved in abiotic stress responses. Additionally, structural analysis and MD simulations were conducted to assess conformational stability and dynamics, indicating stable AtRecQl protein structures and limited coordinate movements. Overall, our study provides valuable insights into the diverse roles of AtRecQl proteins in plant biology.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
CRediT authorship contribution statement
Amit Kumar Dutta: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Md Ekhtiar Rahman: Writing – original draft, Visualization, Formal analysis.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Role of Arabidopsis MYC and MYB homologs in drought- and abscisic acid-regulated gene expression. Plant Cell. 1997;9:1859-1868.
- [Google Scholar]
- Molecular dynamics simulation, free energy landscape and binding free energy computations in exploration the anti-invasive activity of amygdalin against metastasis. Comput. Methods Programs Biomed.. 2020;195:105660
- [Google Scholar]
- Arabidopsis RecQsim, a plant-specific member of the RecQ helicase family, can suppress the MMS hypersensitivity of the yeast sgs1 mutant. Plant Mol. Biol.. 2003;52:273-284.
- [Google Scholar]
- Arabidopsis RecQl4A suppresses homologous recombination and modulates DNA damage responses. Plant J.. 2005;43:789-798.
- [Google Scholar]
- Recombinant protein expression: Challenges in production and folding related matters. Int. J. Biol. Macromol.. 2023;233:123407
- [Google Scholar]
- Role of Zinc-Binding Domains of RecQ Helicases. Helicases from All Domains of Life: Elsevier Inc; 2019.
- Protein folds vs. protein folding : Differing questions, different challenges. PNAS. 2023;120:1-4.
- [Google Scholar]
- Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell. 2002;14:559-574.
- [Google Scholar]
- CRISPR/Cas inactivation of RECQ4 increases homeologous crossovers in an interspecific tomato hybrid. Plant Biotechnol. J.. 2020;18:805-813.
- [Google Scholar]
- Genome - wide systematic characterization of the NRT2 gene family and its expression profile in wheat (Triticum aestivum L.) during plant growth and in response to nitrate deficiency. BMC Plant Biol.. 2023;23:1-20.
- [Google Scholar]
- Assessment of the hypoglycemic and anti-hemostasis effects of Paederia foetida (L.) in controlling diabetes and thrombophilia combining in vivo and computational analysis. Comput. Biol. Chem.. 2023;107:107954
- [Google Scholar]
- Molecular characterisation of RecQ homologues in Arabidopsis thaliana. Nucleic Acids Res.. 2000;28:4275-4282.
- [Google Scholar]
- Two closely related RecQ helicases have antagonistic roles in homologous recombination and DNA repair in Arabidopsis thaliana. Proc. Natl. Acad. Sci.. 2007;104:18836-18841.
- [Google Scholar]
- Topoisomerase 3α and RMI1 suppress somatic crossovers and are essential for resolution of meiotic recombination intermediates in Arabidopsis thaliana. PLoS Genet.. 2008;4
- [Google Scholar]
- Fork sensing and strand switching control antagonistic activities of RecQ helicases. Nat. Commun.. 2013;4:1-9.
- [Google Scholar]
- The fanconi anemia ortholog FANCM ensures ordered homologous recombination in both somatic and meiotic Cells in Arabidopsis. Plant Cell. 2012;24:1448-1464.
- [Google Scholar]
- Biochemical characterization of AtRECQ3 reveals significant differences relative to other RecQ helicases. Plant Physiol.. 2009;151:1658-1666.
- [Google Scholar]
- X-Entropy: A Parallelized Kernel Density Estimator with Automated Bandwidth Selection to Calculate Entropy. J. Chem. Inf. Model.. 2021;61:1533-1538.
- [Google Scholar]
- Reduced Order Model Based on Principal Component Analysis for Process Simulation and Optimization. Energy & Fuels. 2009;23:1695-1706.
- [Google Scholar]
- Radius of gyration as an indicator of protein structure compactness. Mol. Biol.. 2008;42:623-628.
- [Google Scholar]
- Expression of a RecQ helicase homolog affects progression through crisis in fission yeast lacking telomerase. J. Biol. Chem.. 2005;280:5249-5257.
- [Google Scholar]
- Helicase: A genetic tool for providing stress tolerance in plants. Plant Stress. 2023;9:100171
- [Google Scholar]
- Genome-wide systematic survey and analysis of the RNA helicase gene family and their response to abiotic stress in sweetpotato. BMC Plant Biol.. 2024;24:1-24.
- [Google Scholar]
- Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. Proc. Natl. Acad. Sci. U. S. A.. 2009;106:4201-4206.
- [Google Scholar]
- Arabidopsis and the heat stress transcription factor world: How many heat stress transcription factors do we need? Cell Stress Chaperones. 2001;6:177-189.
- [Google Scholar]
- Rational Design of Thermostable Carbonic Anhydrase Mutants Using Molecular Dynamics Simulations. J. Phys. Chem. B. 2018;122:8526-8536.
- [Google Scholar]
- An auxin-regulable oscillatory circuit drives the root clock in arabidopsis. Sci. Adv.. 2021;7:1-10.
- [Google Scholar]
- Towards an accurate prediction of the thermal stability of homologous proteins. J. Biomol. Struct. Dyn.. 2016;34:1132-1142.
- [Google Scholar]
- Stereochemical Assessment of (φ, ψ) Outliers in Protein Structures Using Bond Geometry-Specific Ramachandran Steric-Maps. Structure. 2019;27:1875-1884.e2.
- [Google Scholar]
- Characterization of four RecQ homologues from rice (Oryza sativa L. cv. Nipponbare) Biochem. Biophys. Res. Commun.. 2006;345:1283-1291.
- [Google Scholar]
- How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput.. 2017;13:1518-1524.
- [Google Scholar]
- Drosophila and human RecQ5 exist in different isoforms generated by alternative splicing. Nucleic Acids Res.. 1999;27:3762-3769.
- [Google Scholar]
- Quantitative statistical analysis of cis-regulatory sequences in ABA/VP1- and CBF/DREB1-regulated genes of arabidopsis. Plant Physiol.. 2005;139:437-447.
- [Google Scholar]
- The RNA helicase DDX39A binds a conserved structure in chikungunya virus RNA to control infection. Mol. Cell. 2023;83:4174-4189.e7.
- [Google Scholar]
- Prokaryotic and eukaryotic DNA helicases: Essential molecular motor proteins for cellular machinery. Eur. J. Biochem.. 2004;271:1835-1848.
- [Google Scholar]
- Arabidopsis BRCA2 and RAD51 proteins are specifically involved in defense gene transcription during plant immune responses. Proc. Natl. Acad. Sci. U. S. A.. 2010;107:22716-22721.
- [Google Scholar]
- RecQ Helicases Function in Development, DNA Repair, and Gene Targeting in Physcomitrella patens. Plant Cell. 2018;30:717-736.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2024.103479.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




