

Translate this page into:
Computational probing of Nigella sativa bioactive metabolites against chickungunya nsP2 cysteine protease
⁎Corresponding authors. skg1979@gmail.com (Sushil Kumar), mjavedansari@gmail.com (Mohammad Javed Ansari), khalizan@ums.edu.my (Mohd Khalizan Sabullah)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background and Aim
Instances of chikungunya reported throughout the world in the past two decades of the present century. There is a lack of effective medicine or vaccine for chikungunya treatment. Non-structural protein, the nsP2 cysteine protease (nsP2pro) is an attractive target for inhibitors. It is a key enzyme for proteolytic cleavage of polyprotein precursors and produces functional proteins for replication and multiplication of the virus. Bioactive metabolites from Nigella sativa L; a popular spice and well-known medicinal plant, were selected for the current study against nsP2pro to search for potent non-toxic natural inhibitors of nsP2pro.
Experimental procedure
Out of 54 bioactive metabolites from N. sativa 27 qualified drug likeliness properties. Virtual screening of 27 selected molecules was performed using AutoDock Vina. Top four molecules Kaempferol, (-)-Epicatechin, (+)-Catechin, and Apigenin with the least binding energy were taken for molecular docking employing AUTODOCK4. These metabolites were subjected to molecular dynamics simulation and MMPBSA, and the resilience of protein–ligand complexes had been assessed in terms of RMSD, RMSF, Rg, SASA, and hydrogen bonding.
Results and Conclusions
Drug likeliness, molecular docking, molecular dynamics simulation properties, and MMPBSA analyses made clear that Kaempferol, (-)- Epicatechin, (+)- Catechin, and Apigenin all seem to be potential nsP2pro potent inhibitors and strong candidates for chikungunya virus drug development.
Keywords
Chikungunya Virus
nsP2 cysteine protease
Drug likeliness
Molecular docking
Molecular dynamics simulation
Molecular Mechanics Poisson Boltzmann Surface Area

- CHIKV
-
Chikungunya Virus
- nsP2pro
-
Non-structural protein 2 cysteine protease
- MD
-
Molecular Dynamics Simulation
- RMSD
-
Root Mean Square Deviation
- RMSF
-
Root Mean Square Fluctuation
- Rg
-
Radius of Gyration
- SASA
-
Solvent Exposed Surface Area
- MMPBSA
-
Molecular Mechanics Poisson Boltzmann Surface Area
Abbreviations
1 Introduction
Chikungunya was first identified from southern Tanzania in 1952 (https://www.who.int/health-topics/chikungunya#tab = tab_1). It is characterized by abrupt fever mostly accompanied by joint pain. The symptoms may continue for a few days and occasionally stay for weeks, months, or even years. Additional manifestations comprise muscular pain, swelling at joints, nausea, headache, rash, and fatigue (Pastorino et al., 2008). Chikungunya virus (CHIKV), the causal agent of chikungunya, is a member of the family Togaviridae and belongs to the genus Alphavirus. About 50 years of its initial isolation, CHIKV caused occasional outbreaks in Africa and Asia. Chikungunya re-emerged in the first decade of the present century in the Indian subcontinent, Caribbean islands, and French Guiana (Bortel et al., 2014). During 2004–2006,five hundred thousand people were reported infected from Kenya and surrounding locations. Subsequently, it spread to Indonesia, Maldives, Sri Lanka, Myanmar, and Thailand due to viremic travelers. In 2007, one hundred ninety-seven cases were reported from Italy in Europe. In 2014, Europe had the highest chikungunya burden; with approximately 1500 reported cases. In 2015 and 2016, 37,480 and 146,914 confirmed cases were reported from the United States. Pakistan had 8387 instances, whereas India had 62000. Chikungunya episodes have also been confirmed in Sudan, Yemen, Cambodia, and Chad more recently (WHO 2022).
The vectors to carry the virus from viremic to native hosts were identified as mosquitoes Aedes aegypti and A. albopictus, recognized to be the most anthropophilic mosquitoes with a tight linkage to humans. After getting infected, they transmit the virus to humans through their bites, right into the bloodstream. This virus is an envelope, icosahedral entity with a diameter of 65–70 nm. Its genome is composed of positive-sense ssRNA of approximately 11.8 Kb. There are two open reading frames (ORFs), one of them coding for nonstructural proteins starting translation just after the entry into a host cell. This results in the synthesis of nonstructural polyprotein precursor nsP1-4. The ns polyprotein precursor was further processed by proteolytic cleavage into constituent nonstructural proteins nsP1, nsP2, nsP3, and nsP4 by virus-encoded nsP2 cysteine protease (nsP2pro). Replication of viral genome and subgenomic expression depend on these nsPs (nonstructural proteins). The nsP2pro is a multifunctional two-domain protein. The N-terminal domain is responsible for RNA helicase, RNA dependent 5′-triphosphatase, and nucleoside triphosphatase activities, whereas the C-terminal domain is liable for polyprotein cleavage into constituent nsPs (Strauss et al.,1983; Strauss et al., 1984). Cys478-His548 is the catalytic dyad of nsP2pro. Asn476, Cys478, Asn547, and Tyr544 are the main putative binding residues, while Met707, Asp711, and Leu670 as well play a significant role in substrate binding and identification. Trp549 is the most conserved residue in alphaviruses. Since the C-terminal domain of nsP2 is pivotal for the viral replication cycle, it is a tempting target for drugs and inhibitors (Narwal et al., 2018). In addition, nsP2 protease enters into the nucleus of the host cell and stops the expression of antiviral genes (Fros et al., 2010; Fros et al., 2013). To date, there is no vaccine or potent drug against CHIKV.
Several herbs have been identified to possess unique antiviral, antibacterial, antifungal and anti-cancer properties (Padhye, 2008; Ahmad, 2021). Nigella sativa L. (black cumin or black seed), widely used as spice and medicine in various systems of medicines since ancient times (Randhawa, 2011), is also reported as “a cure for every disease except death” (Butt and Sultan, 2010). It is indigenous to North Africa, Southern Europe, south and Southwest Asia (Padhye, 2008) with several bioactive metabolites. The oil extracted from black cumin seeds suppressed H9N2 (influenza virus) pathogenicity and improved immunity in poultry (Umar et al., 2016). N. sativa seeds exhibited antiviral effects against Laryngotrachietis Virus (ILTV) in Chicken Embryo Rough Cells (Zaher et al., 2008). The diabetic patients infected with hepatitis C virus when treated with N. sativa were found with lower viral load and improved glycemic control (Barakat et al., 2013). When used against HIV findings confirmed the efficacy and potency of N. sativa seeds as potent anti-HIV (Onifade et al., 2013) agents. This led to screening the bioactive metabolites against CHIKV using in silico approach. In the present study, we carried out molecular docking, atomistic explicit solvent MD simulations (molecular dynamic simulations), and MMPBSA (Molecular Mechanics Poisson Boltzmann Surface Area) based free energy calculations to investigate the potential inhibiting efficiency of selected bioactive metabolites from N. sativa to the nsP2pro, an important enzyme that participates in processing of nonstructural polyprotein complex. High binding scores, stability of the nsP2pro-chosen bioactive metabolite complexes, and high binding free energy values indicate that bioactive metabolites can be effective inhibitors for the nsP2pro.
2 Materials and methods
2.1 Metabolite library
A metabolite library of 54 structurally diverse phytomolecules was constructed through a literature survey to identify potential CHIKV treatments (Salehi et al., 2021; Avato and Tava, 2022) (Supplementary Table 1). The chemical and structural properties such as molecular weight, chemical formulae, and canonical smiles of these metabolites were retrieved from the PubChem Database (Kim et al., 2016).
2.2 ADME analysis
The ADME (absorption, distribution, metabolism, and excretion) properties of phytomolecules were calculated by the SwissADME tool (Guex and Peitsch, 1997). The pharmacokinetic and pharmacodynamic properties of the molecules are established by Lipinski's rule of five (Lipinski et al., 1997).The molecules that did not violate Lipinski's rule were considered for molecular docking. Based on selected drug likeliness parameters 27 molecules were taken for docking studies.
2.3 Protein and ligand preparation
The 3D coordinates of CHIKV protease was retrieved from the RCSB database [PDB: 4ZTB]. For further analysis, water and other nonspecific molecules were removed by using UCSF Chimera(Pettersen et al., 2004).The structure of the protein was optimized using the clean geometry module of Discovery Studio(San Diego, CA, USA) and saved in pdb format. The hydrogen atoms and Kollman Charges were added using MGL tool 1.5.6and saved as pdbqt format. The 3D structures of selected phytomolecules were converted by using the OpenBabel tool where canonical smiles were inputs. Explicit hydrogen atoms were added, and the pdb structure was generated using the OpenBabel tool(O'Boyle et al., 2011).The steepest descent algorithm of PyRx0.8 tool with inbuilt OpenBabel was used to perform energy minimization and optimization of molecules, and structures were saved in pdbqt format (Dallakyan and Olson, 2015).
2.4 Molecular docking of ligands
Molecular docking for the identification of potential antiviral molecules was performed by using AutoDock Vina (Trott and Olson,2010). The grid centre point was set as X = 20.223 Y = 1.321 Z = -16.419. The box dimensions were 16 Å × 16 Å ×16 Å and exhaustiveness was set at 16. The other parameters were left at their default values. The binding energy was distributed from −4.1 to −6.1 kcal/mol. The molecules having the least binding energies (-6.0 kcal/mol and lower) were analyzed for docking interactions. For molecular dynamics simulation (MD simulation), molecules with binding energies of −6.0 and −6.1 kcal/mol were chosen. The 2D and 3D interactions of selected complexes were plotted using the receptor-ligand interactions module of Discovery Studio (San Diego, CA, USA) and PyMol (Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.).
2.5 Molecular dynamics simulation
MD simulation studies were carried out to evaluate the dynamic properties of the Phytomolecule-protease complexes. GROMACS 5.1.4 suite (Van Der Spoel et al., 2005) was used to run all simulations, which used GROMOS 43a1 force field. PRODRG server was used to generate ligand topology files (Schüttelkopf and Van Aalten, 2004). Solvation of prepared protein complexes was performed in the cubic box and a sufficient number of ions were incorporated to maintain system neutrality. To eliminate any steric collisions among atoms, the system's energy was minimized using the steepest descent algorithm with a convergence criterion of less than 1000 kJ/mol/nm. Long-range electrostatic interactions were computed using PME (Hess, 2008) and for both van der Waals and Coulombic interactions, a cut-off radius of 9 Å was used. Equilibration was carried out in two stages. The solvent and ions were kept unrestrained in the NVT ensemble for 100 ps in the first phase, while the restraint weight from the protein and protein–ligand complexes was gradually reduced in the NPT ensemble for 100 ps in the second phase. The LINCS algorithm was used to keep all hydrogen bonds constrained (Hess et al., 1997; Hess, 2008). Utilizing Berendsen's temperature and Parrinello-Rahman pressure coupling, the temperature and pressure of the system were kept at 300 K and 1 atm, respectively (Berendsen et al., 1984). Following equilibration, a production simulation was run using a LeapFrog dynamics integrator with a step size of 2 fs and periodic boundary conditions for a timescale of 200 ns. MD simulation trajectory was analyzed by the available GROMACS program and in-house Python-3.5 scripts.
2.6 MMPBSA free energy calculation
The binding energy and per residue energy contribution were determined using MMPBSA. The polar fraction of solvation energy (ΔGpsolv) was assessed in MMPBSA by solving the Poisson-Boltzmann equation; meanwhile, the non-polar component (ΔGnpsolv) was calculated by using the linear relation to the solvent accessible surface area (SASA). The g_mmpbsa module of GROMACS was used in this study to estimate various components of complexes' binding free energy (Kumari et al., 2014). For the analysis, the last 10 ns of the trajectory were used.
3 3. Results and discussion
3.1 Drug likeliness
In silico drug-likeliness analyses, consider many physicochemical and functional factors determining a given molecule's probable drug-like behavior. The physicochemical characters include mainly molecule size, molecular flexibility, lipophilicity, electronic distribution, and hydrogen bonding characteristics; whereas transport, absorption, metabolic stability, distribution, affinity to proteins, reactivity, toxicity, etc., the pharmacophoric characters determine the molecular interaction with the living system. Analysis of molecules using this information reduces the effort while screening a large pool of metabolites and reduces the probability of failures in vivo experiments. Twenty-seven molecules out of 54 molecules of the library exhibited zero violation of Lipinski’s rule of five (Supplementary Table 2).
3.2 Molecular docking
The binding energy of 27 phytomolecules was distributed from −4.1 to -0.6.1 kcal/mol (Supplementary Table 3). Four molecules with minimum binding energies Kaempferol (-6.0 kcal/mol), (-)-Epicatechin, (+)-Catechin and Apigenin (-6.1 kcal/mol) respectively. These molecules form H-bonds, van der Waals forces, π-π, π-σ, and π-alkyl interactions with active site residues of nsP2pro. Kaempferol forms five H-bonds with four amino acid residues Asn476, Cys478 (two H-bonds), Tyr512, and Tyr544, and van der Waals interactions with Asn547, Trp549, Trp479, and Met707. Kaempferol forms two π-σ interactions with Ala511 and π-alkyl with Leu670 (two) Cys478 (one).(-)-Epicatechin and (+)-Catechin both form three H-bonds with the amino acid residues Cys478, Tyr512 and Tyr544, vander Waals interactions with Asn476, Asn547, Trp549, Met707, and Leu670; π-σ interaction with Ala511. Alkyl interactions with Cys478 and Ala511. (-)-Epicatechin and (+)-Catechin are diastereoisomers found in plants that have two chiral centers and remain in cis and trans configurations, respectively (Tsao, 2010). Due to structural similarities both displayed similar interactions with active site residues of nsP2pro. Apigenin forms four H-bonds with Ala475, Cys478, Tyr512, and Trp549; van der Waals interactions with Asn476, Trp479, Tyr544, Asn547, Leu670, and Met407; two π-S bonds with Cys478; π -alkyl and π-σ with Ala511 (Fig. 1 and Supplementary Figure 1).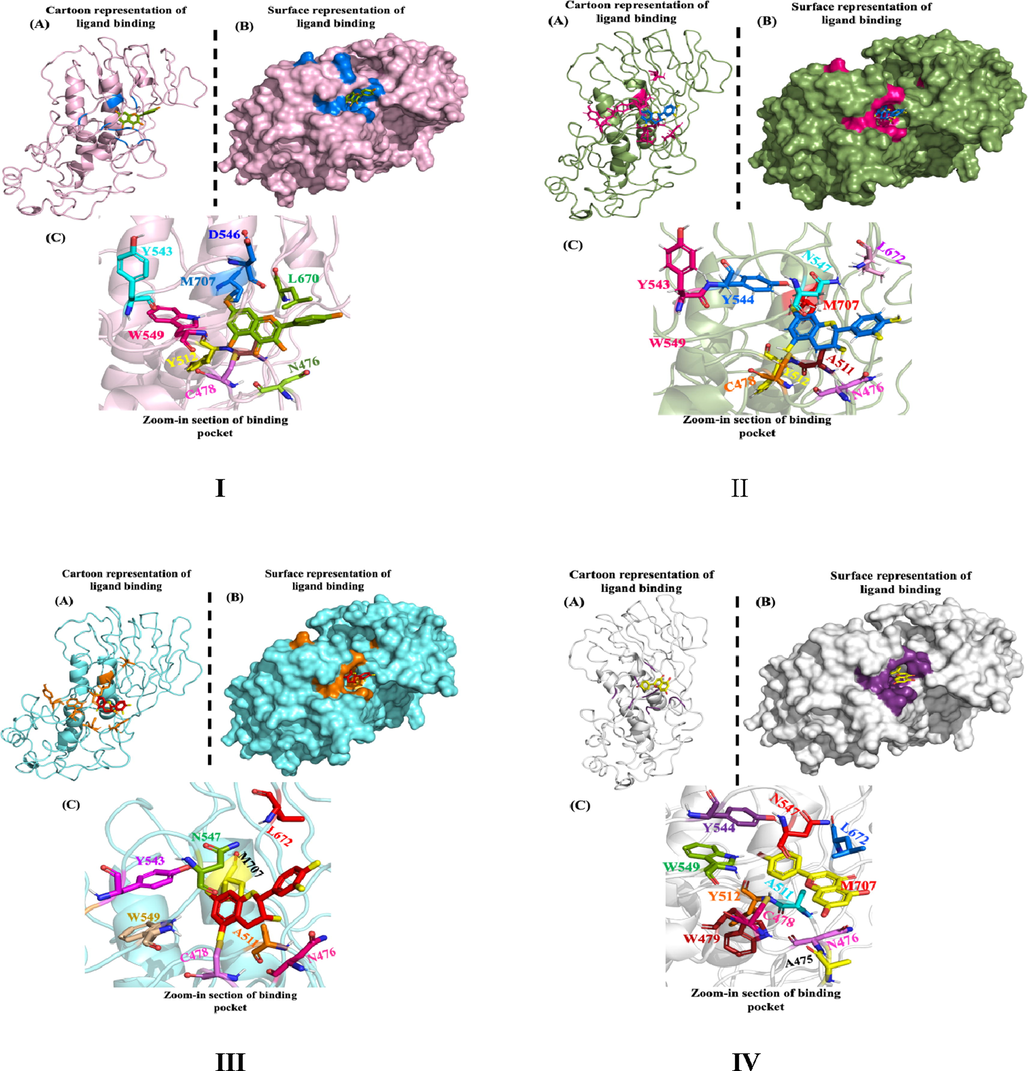
Molecular Docking (I) Kaempferol, (II) (-)- Epicatechin, (III) (+)- Catechin, and (IV) Apigenin with nsP2pro.
The interaction analysis also revealed that these molecules interact, preferably with His548, the catalytic residue, Trp549, and Asn547 residues involved in substrate binding and recognition. It is important to mention that most of the selected compounds Kaempferol, (-)-Epicatechin, (+)-Catechin, and Apigenin are the phytochemical constituents of N. sativa well known to possess antibacterial, antifungal, and antiviral properties (Colomer et al., 2017).
3.3 Molecular dynamics simulations
The docked molecules with minimum binding energy Kaempferol, (-)-Epicatechin, (+)-Catechin, and Apigenin were selected for further validation. The binding of a ligand may introduce a broad range of conformational perturbations in protein. Therefore, to examine the ligand-induced structural changes in protein and stability of protein–ligand complexes, we have calculated various parameters like Root-mean-square deviation (RMS deviation), Root-mean-square fluctuation (RMSF), and Radius of gyration (Rg) for free and all protein–ligand complexes.
3.3.1 Root-mean-square deviation
The RMS deviation calculation of proteins leads to quantifying the degree of conformational changes that may occur during MD simulations. The RMS deviation of the backbone Cα atoms from its starting structures was calculated to elucidate the stability of complexes. The time-dependent mean RMS deviation of backbone atoms of nsP2pro in free and ligand-bound from its starting structure varies in the range of 0.35–0.40 nm, which reaches to plateau in 20 ns. These low values of RMS deviation indicate the stability of nsP2pro in both free and ligand-bound states. The average RMS deviation of nsP2pro backboneand nsP2pro-ligand complexes wereranging ∼ 0.33–0.4 nm, whereas the average RMS deviation of ligands such as compounds Kaempferol (∼0.921 nm), (-)-Epicatechin (∼1.094 nm), (+)-Catechin (∼0.870 nm) and Apigenin (∼0.507 nm Since the RMSD of all systems were less than > 0.4 nm, therefore it can be concluded that during the simulation time the enzyme has not much differed from its starting conformation. Further, the binding of phytomolecules imparts no major conformational changes in the protein backbone (Fig. 2).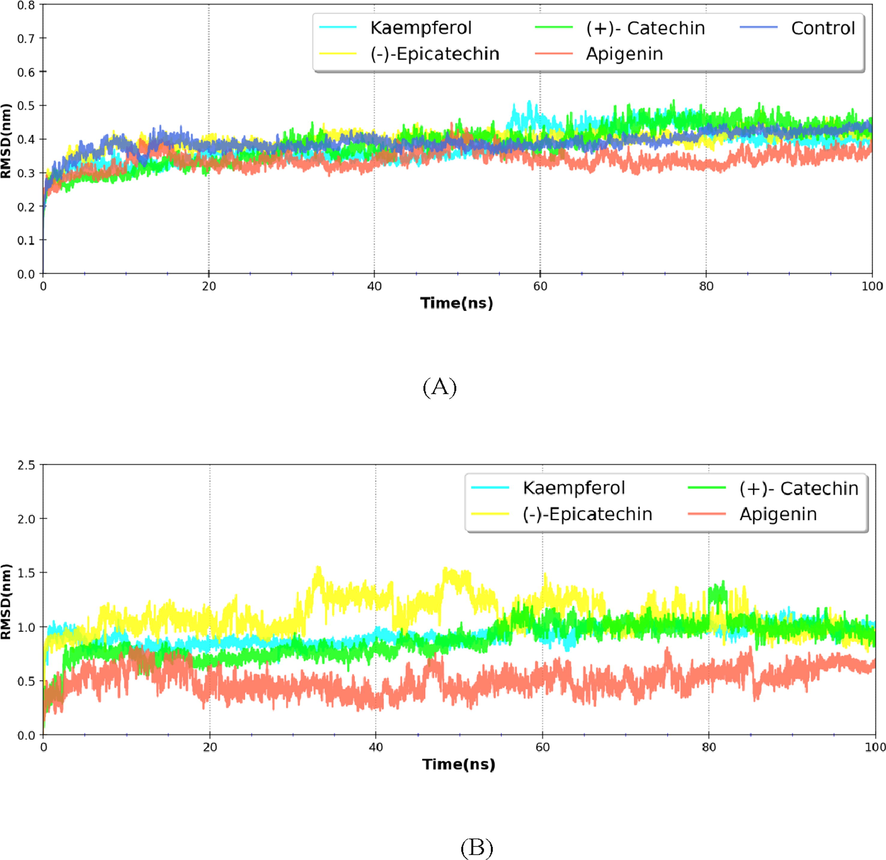
Line Diagram of RMSD profile of nsP2pro backbone atoms (A) in both control and ligand bound states. Panel B depicts the RMSD of ligands during the simulation100ns.
3.3.2 Root-mean-square fluctuation
The RMSF is a measure of residue level fluctuation at its average position, thus indicating the dynamics of a system. The RMSF of most residues in the enzyme fluctuates with an intensity of 0.12 to 0.17 nm (Fig. 3). The overlapping profile of residue fluctuation indicates that the binding of the phytomolecules at the active site does not affect the amino acid residues' position. Thus, it can be inferred that binding of ligand has not influenced the dynamics of the protein.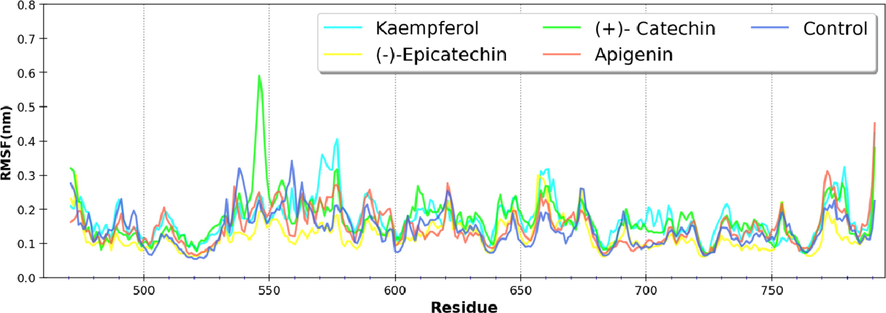
RMSF of nsP2pro in unbound and ligand bound states.
3.3.3 Radius of gyration
The radius of gyration represents the compactness of protein, where low Rg values are associated with its stability. Since binding of ligand might result in the unfolding of a protein, hence Rg variation was determined in all cases during the simulation length. Rg, for the unbound nsP2pro, was centered around ∼ 1.95 nm, and similar values (∼1.98 to 2.03 nm) (Fig. 4) were noted for nsP2pro bound to phytomolecules indicating that the binding of proposed phytomolecules doesn't impart any change in the integrity and compactness of nsP2pro.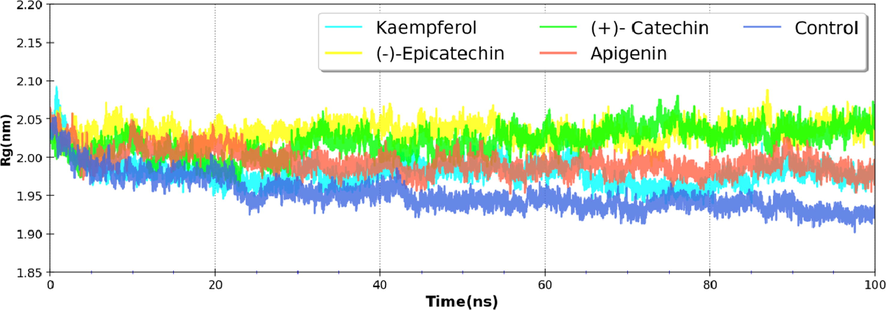
Variation in Radius of Gyration (Rg) of nsP2pro and along with proposed inhibitors.
3.3.4 Solvent accessible surface area
By protecting non-polar amino acids in hydrophobic cores from the aqueous environment, hydrophobic interactions between non-polar amino acids promote the stability of globular proteins in solution. SASA can theoretically be used to determine changes in protein accessibility to solvent. It measures the contribution of free energy of solvation of each atom of the system such as water, polar and non-polar amino acids of the protein. The SASA profile of unbound nsP2pro was found to be peaked at ∼ 140 nm2, which further shifts to ∼ 146–149 nm2 in the presence of ligands (Fig. 5).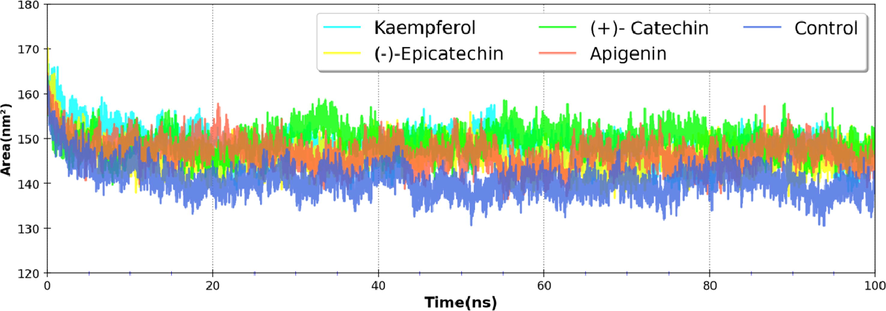
Variation of the SASA of nsP2pro complex with phytomolecules during the course of the simulation.
Since the SASA values are less skewed than in the control, the binding of these compounds does not affect protein folding.The mean values of molecular dynamics simulation parameters are summarized in Table 1.
Control
Kaempferol
(-)-Epicatechin
(+)- Catechin
Apigenin
Backbone RMSD
0.389 ± 0.029
0.378 ± 0.050
0.396 ± 0.030
0.384 ± 0.058
0.338 ± 0.027
Complex RMSD
–
0.386 ± 0.049
0.407 ± 0.030
0.391 ± 0.059
0.339 ± 0.027
Ligand RMSD
–
0.921 ± 0.087
1.094 ± 0.153
0.870 ± 0.166
0.507 ± 0.114
Radius of Gyration
1.952 ± 0.022
1.980 ± 0.017
2.033 ± 0.012
2.020 ± 0.018
1.994 ± 0.016
SASA
140.648 ± 3.316
148.049 ± 3.647
146.031 ± 3.151
149.376 ± 2.765
146.633 ± 3.107
RMSF
0.141 ± 0.052
0.170 ± 0.057
0.123 ± 0.044
0.167 ± 0.065
0.147 ± 0.052
3.4 Hydrogen bond analysis
The hydrogen bond formed between the nsP2pro and bioactive metabolites was calculated by using h_bond module of GROMACS. The hydrogen bonds were distributed along with the 100 ns simulation and maximum hydrogen bonds formed 0.25 to 0.35 nm.Hydrogen bonds distribution graph revealed that hydrogen bond formation initiates at 0.25 nm distance among the hydrogen bond donor and acceptor and maximum distribution was shown at the distance 0.30 to 0.35 nm (Fig. 6).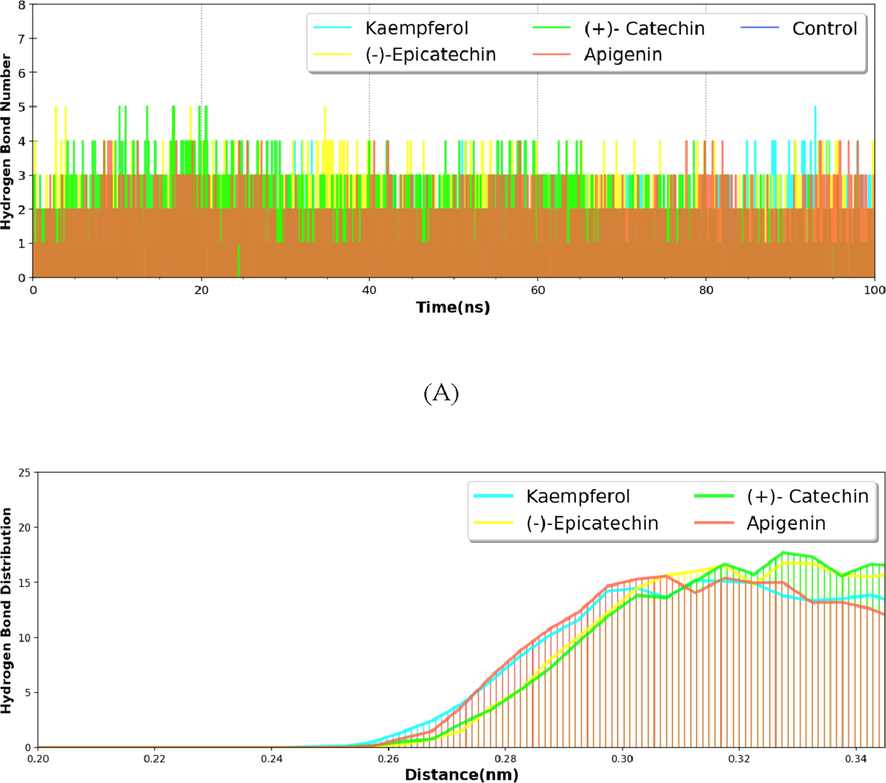
Hydrogen bond number and distribution between selected bioactive metabolites and nsP2pro.
3.5 Binding free energy calculation
The binding free energy (ΔGbind) was calculated using MMPBSA analysis which indicates the binding potential of ligands (Table 2).Results of MMPBSA confirmed that all molecules bind to the active site of nsP2pro to form a stable complex. Among all the four tested molecules Kaempferol expressed the least binding free energy hence maximum affinity with nsP2pro. The difference in ΔGbind majorly comes from the ΔEvdw (energy of van der Waals interactions) interaction component and ΔGpsolv free energy components, playing an essential role in lower binding free energy. Various energy components such as van der Waals energy (ΔEvdw), Electrostatic energy (EEEL), Polar solvation energy (ΔGpsolv), and nonpolar solvation energy (ΔGnpsolv) contributed to the binding free energy of enzyme-ligand complexes. MMPBSA results of ligands were given in Table 2.
Compounds
ΔEvdw
EEEL
ΔGpsolv
ΔGnpsolv
ΔGbind
Kaempferol
−226.417 ± 15.845
−4.798 ± 3.854
121.6
± 14.62
−45.569 ± 0.775
−201.512 ± 15.374
(-)-Epicatechin
−191.998 ± 10.716
−2.077 ± 5.98
46.807 ± 6.649
−15.020 ± 1.095
162.288 ± 11.873
(+)-Catechin
−162.852 ± 12.359
−1.429 ± 1.812
31.403 ± 4.901
−13.169 ± 0.870
−146.04 ± 12.123
Apigenin
−207.827 ± 16.177
−13.0871 ± 7.612
56.006 ± 9.425
−15.203 ± 0.856
−180.896 ± 17.564
4 Conclusions
The phytomolecules Kaempferol, (-)-Epicatechin, (+)-Catechin, and Apigenin from N. sativa provide conviction in the articulation of binding energy from molecular docking and MMPBSA. These molecules also provide stability and interaction with nsP2pro during the course of MD simulation. Drug likeliness properties of these molecules unveiled no violation of the Lipinski rule indicating Kaempferol, (-)-Epicatechin, (+)-Catechin, and Apigenin to be strong candidates for drug development against chikungunya virus. The findings provide a base to evaluate these metabolites for further research to obtain a drug for chikungunya treatment validating through in vitro and in vivo experiments.
Disclosure statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Acknowledgment
This project was supported by Researchers Supporting Project number (RSP2023R230) King Saud University, Riyadh, Saudi Arabia. The authors would like to acknowledge the support provided by Research Management Centre (RMC), University Malaysia Sabah (UMS), Malaysia, through grant Number SKIM DANA NICHE SDN0020-2019.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Molecular docking, simulation and MM-PBSA studies of Nigella sativa compounds: a computational quest to identify potential natural antiviral for COVID-19 treatment. J. Biomol. Struct. Dyn.. 2021;39(12):4225-4233.
- [Google Scholar]
- Rare fatty acids and lipids in plant oilseeds: occurrence and bioactivity. Phytochem. Rev.. 2022;21:401-428.
- [CrossRef] [Google Scholar]
- Effects of Nigella sativa on the outcome of hepatitis C in Egypt. World J. Gastroenterol.. 2013;19(16):2529-2536.
- [Google Scholar]
- Molecular dynamics with coupling to an external bath. J. Chem. Phys.. 1984;81(8):3684-3690.
- [Google Scholar]
- Najioullah, Chikungunya outbreak in the Caribbean region, December 2013 to March 2014, and the significance for Europe. Euro. Surveill.. 2014;19(13):20759.
- [CrossRef] [Google Scholar]
- Nigella sativa: Reduces the risk of various maladies. Crit. Rev. Food Sci.. 2010;50:654-665.
- [Google Scholar]
- Natural polyphenols and their synthetic analogs as emerging anticancer agents. Curr. Drug Targets. 2017;18(2):147-159.
- [CrossRef] [Google Scholar]
- Small-molecule library screening by docking with PyRx. Methods Mol. Biol.. 2015;1263:243-250.
- [Google Scholar]
- Chikungunya virus nonstructural protein2 inhibits type I/II interferon-stimulated JAK-STAT signaling. J. Virol.. 2010;84:10877-10887.
- [Google Scholar]
- The C-terminal domain of chikungunya virus nsP2 independently governs viral RNA replication, cytopathicity, and inhibition of interferon signaling. J. Virol.. 2013;87(15):10394-10400.
- [Google Scholar]
- SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714-2723.
- [Google Scholar]
- P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput.. 2008;4(1):116-122.
- [Google Scholar]
- LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem.. 1997;18(12):1463-1472.
- [Google Scholar]
- https://www.who.int/news-room/fact-sheets/detail/chikungunya.
- PubChem substance and compound databases. Nucleic Acids Res.. 2016;44(D1):D1202-D1213.
- [Google Scholar]
- Open Source Drug Discovery Consortium, Lynn A. g_mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J. Chemical Information Model.. 2014;54(7):1951-1962.
- [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 1997;23(1–3):3-25.
- [Google Scholar]
- Crystal structure of chikungunya virus nsP2 cysteine protease reveals a putative flexible loop blocking its active site. Int. J. Biol. Macromol.. 2018;116:451-462.
- [Google Scholar]
- Nigella sativa concoction induced sustained sero-reversion in HIV patients. Afr. J. Tradit. Complement. Altern. Med.. 2013;10(5):332-335.
- [Google Scholar]
- From here to the eternity-the secret of Pharaohs: therapeutic potential of black cumin seeds and beyond. Cancer Ther.. 2008;6:495-510.
- [Google Scholar]
- Expression and biochemical characterization of nsP2 cysteine protease of chikungunya virus. Virus Res.. 2008;13:293-298.
- [Google Scholar]
- UCSF Chimera visualization system for exploratory research and analysis. J. Comput. Chem.. 2004;25(13):1605-1612.
- [Google Scholar]
- Anticancer activity of Nigella sativa (black seed) - a review. Am. J. Chin. Med.. 2011;39(6):1075-1091.
- [CrossRef] [Google Scholar]
- Nigella Plants-traditional uses, bioactive phytoconstituents. Preclin. Clin. Stud. Front. Pharmacol.. 2021;12:625386
- [CrossRef] [Google Scholar]
- PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. D. Biol. Crystallogr.. 2004;60(8):1355-1363.
- [Google Scholar]
- Sequence coding for the alphavirus nonstructural proteins is interrupted by an opal termination codon. Proc. Nat. Acad. Sci. USA. 1983;80:5271-5275.
- [Google Scholar]
- Complete nucleotide sequence of the genomic RNA of Sindbis virus. Virol.. 1984;133:92-110.
- [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31(2):455-461.
- [Google Scholar]
- Protective and antiviral activities of Nigella sativa against avian influenza (H9N2) in turkeys. Sci: J. Saudi Soc. Agric; 2016. p. :10.
- Observations on the biological effects of black cumin seed (Nigella sativa) and green tea (Camellia sinensis) Global Vet.. 2008;2(4):198-204.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2023.102651.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




