

Translate this page into:
Intersection of genomics and health informatics approaches in identification of diseases’ biomarkers
⁎Address: Department of Public Health, College of Applied Medical Sciences, Majmaah University, PO Box 7921, Majmaah 15341, Saudi Arabia. r.abdullah@mu.edu.sa (Raed Abdullah Alharbi)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Genomes are one of the most essential sensitive molecular biomarkers that are used to be discovered in a very small amount in a sample to identify a specific type of diseases. Since genomics is the science that studies structures, interactions, and functions of all genomes, genomics approach is used to identify genomes as diseases’ biomarkers. However, health informatics approach, especially bioinformatics, has a main rule in data analysis. The purpose of this review is to describe briefly the technologies and methods that are used in both genomics and health informatics approaches to identify these biomarkers. Therefore, this paper is based on a computerized database search. In genomics approach, after collecting samples, first start with extraction and purification of DNA to get a purified DNA that is extracted from the nucleus of cells. Then, DNA amplification is to produce many copies of a specific DNA sequence. Next is sequencing of DNA to determine and read the sequence of nucleic acid sequence in DNA. Once the DNA sequence is determined, the following procedure is the health informatics approach and called bioinformatics pipeline steps, which is DNA data analysis steps. Finally, there are several different techniques and methods of genomics approach to identify a genome as a disease’s biomarker. However, next generation techniques and methods whether extraction, purification, amplification, and sequencing of DNA or bioinformatics pipeline are more accurate, faster, and cheaper from other generation sequencing of genomic approach.
Keywords
Genomics
Health Informatics
Bioinformatics
Biomarkers Identification

1 Introduction
Biomarkers, also known as biological markers, are indicators that can detect the presence of biological activities and processes, allowing them to be used to identify biological situations (BDWG, 2001; Strimbu and Tavel, 2010). There are many uses and advantages of biomarkers in different sciences. These biomarkers, on the other hand, are used in different medical fields such as; disease identification, drug discovery, and patients care. Diagnostic biomarkers, predisposition biomarkers, prognostic biomarkers, and predictive biomarkers are only a few of the numerous types of biomarkers available (Brody, 2016; Huss, 2015). Genomes can be classified as molecular biomarkers depending on their physicochemical characteristics (Huss, 2015; Davis et al., 2013).
Among all these types of biomarkers, genomes are one of the most essential sensitive molecules that are used to be revealed in a very small amount in a sample to identify a specific type of diseases. Therefore, it is very important to understand genomics approach as well as its technologies and methods in order to have the ability of using them to identify a genome as a disease’s biomarker.
Genomics is the science that studies structures, interactions, and functions of all genomes (NCI, 2021; NHGRI, 2020). Genomes are complete set of deoxyribonucleic acids (DNAs) while genes are a part of the DNA. DNA is a molecule containing the instructions required for practically all living organisms to develop and direct their activities (NHGRI, 2020; Yourgenome, 2021). Every DNA molecule is a double helix, which means it is made up of two twisting linked strands. Adenine, Thymine, Guanine, and Cytosine are the four chemical components that make up each DNA strand. A and T are always linked together on opposing strands, while C and G are always linked together on opposite strands (NHGRI, 2020; Yourgenome, 2021).
In fact, there are several purposes of genomics approach technologies including: agriculture science, forensic science, gene manipulation, metagenomic applications, identifying genes, endonuclease maps, and DNA data bank (Drmanac et al., 2010; Bisht and Panda, 2014). However, in applications of medical research, genomics approach technologies are highly used to diagnose and identify different genes that are associated with different diseases as biomarkers (Drmanac et al., 2010; Bisht and Panda, 2014; Mardis, 2017).
According to World Health Organization, “Health Informatics is an umbrella term used to encompass the rapidly evolving discipline of using computing, networking and communications, methodology and technology, to support the health-related fields, such as medicine, nursing, pharmacy and dentistry” (WHO, 2001).
Branches of health informatics include biomedical informatics, clinical informatics, bioinformatics, nursing informatics, medical informatics, pharmacy informatics, public health informatics with different goals of each branch of them (PHD, 2020).
According to Professor Miller from Yale University, the Yale Center Genome Informatics, the researches area of biomedical informatics include genome informatics, neuroinformatics, and clinical informatics (Miller, 2000). Also, he concluded that, there are many scientists in genomics' fields are using bioinformatics to point on informatics applications in their fields (Miller, 2000).
Since that it is very significant to understand genomics and health informatics approaches, technologies, and methods in order to have the ability of using them to identify a genome as a disease’s biomarker, the purpose of this paper is to collect and describe briefly the technologies and methods that are used in both approaches to identify these biomarkers resulting in bioinformatics pipelines in genomics approach.
2 Genomics approach
So, after collecting the samples, which include hair, blood, tissue, fluid, saliva and urine samples (NRC, 1997; NIJ, 2021; OSU, 2021), there are four major stages in the process of genomics and health informatics approaches in order to identify a gene. These stages are (I) extraction and purification of DNA, (II) amplification of DNA, (III) sequencing of DNA, and (IV) data analysis of DNA. (Figs. 1 and 2) (Bisht and Panda, 2014; Lowe and Reddy, 2015).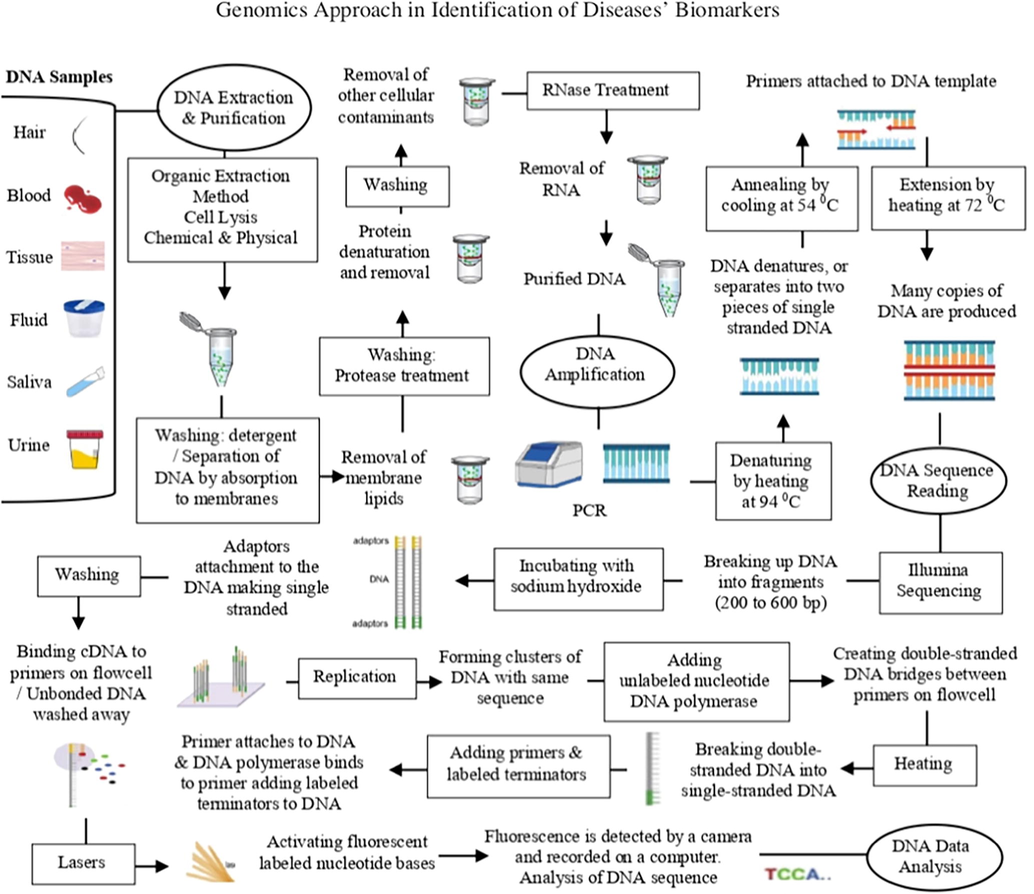
Genomic approach to identify disease’s biomarkers.
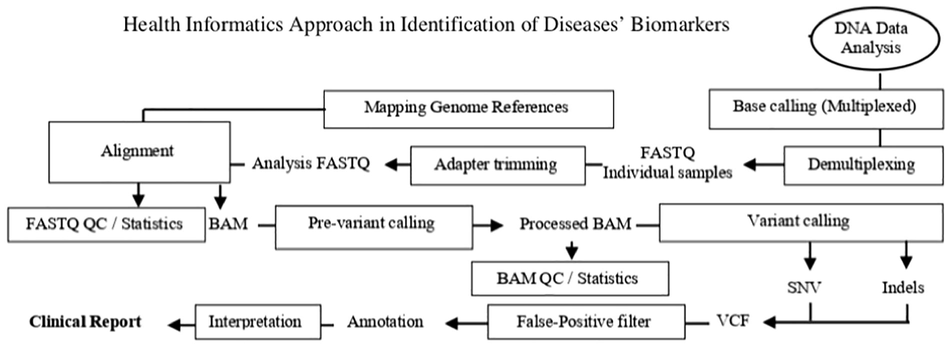
Health informatics approach to identify disease’s biomarkers.
2.1 Extraction and purification of DNA
This step is used to get a purified DNA that is extracted from the nucleus of cells. There are several methods as well as many different kits for DNA extraction and purification. However, methods are used must give effective extraction of DNA, enough amount of DNA, effective elimination of contaminants, high purity and quality of DNA (Dhaliwal, 2013). Since the sensitivity of polymerase chain reaction (PCR) and the following procedures of DNA sequencing, it is very critical to select the appropriate kit for DNA extraction as well as DNA purification depending on the purpose of using, type of method, sample type and quantity, humic content, and simplicity (Dhaliwal, 2013; Thatcher, 2015; Mullegama et al., 2018).
Even if there are different methods and steps of DNA extraction and purification, there is a basic procedure of DNA extraction and purification in genomic approach (Fig. 1) (Dhaliwal, 2013; Thatcher, 2015; Mullegama et al., 2018). Different methods of DNA extraction and purification as the following: (Walsh et al., 1991; Budelier and Schorr, 1998; Tan and Yiap, 2009; Akbarzadeh et al., 2012; Dhaliwal, 2013; Ma et al., 2013).
2.1.1 Organic extraction method
In this method, after cell lysis, centrifuge would be used to remove any cell debris (Fig. 1). Then, proteins would be digested by protease and precipitated by an organic solvent such as phenol before using centrifugation to remove the protein precipitate (Dhaliwal, 2013). Finally, precipitation by ethanol or isopropanol would be used to recover the purified DNA (Fig. 1). This method uses dangerous organic solvents, takes long-time, and these organic solvents may affect other procedures such as PCR (Dhaliwal, 2013).
2.1.2 Silica-Based technology method
In the present, kits based on silica-based technologies are commonly used (Tan and Yiap, 2009; Dhaliwal, 2013). In this method, when a specific chosen salt is added along with typical pH, DNA would adsorb to silica beads. Then, cellular contaminants would be eliminated by washing. Finally, purified DNA is eluted in an elution buffer. Silica-based technology method has simple principle, cheap, takes short-time, and suitable for automation (Tan and Yiap, 2009).
2.1.3 Magnetic separation method
In this method, DNA would bind to a magnetic surface beads covered with either functional materials that interact with DNA or DNA binding antibodies (Akbarzadeh et al., 2012; Dhaliwal, 2013). Then, these magnetic beads would be separated from other contaminating components. Then, after washing, ethanol would be used to elute purified DNA. Magnetic separation method is expensive, rapid, simple, and suitable for automation (Dhaliwal, 2013; Ma et al., 2013).
2.1.4 Anion exchange technology method
In this method, there is an interaction occurs between surface molecules on the substrate (positive charged) and phosphates of the nucleic acid (negative charged) (Budelier and Schorr, 1998; Dhaliwal, 2013). So, when there is a low concentration of salt, DNA would bind to the substrate. Then, by washing and using low salt buffer, contaminants would be removed. Final step is using high salt buffer to elute purified DNA (Budelier and Schorr, 1998).
2.1.5 Salting out method
This method is commonly used for isolation of DNA from the whole blood samples (Miller et al., 1988; Shokrzadeh and Mohammadpour, 2018). After the cell lysis, proteins K and RNase would be added. Then, to precipitate out the proteins the solution, saturated salt is required. After then, to separate the DNA, centrifugation is used before washing by using ethanol. Finally, after washing, purified DNA would be eluted. The salting-out method is simple and non-toxic (Shokrzadeh and Mohammadpour, 2018).
2.1.6 Cesium chloride density gradients method
Cesium chloride density gradients method is simple. So, by using centrifugal force, cesium chloride (CsCl) would be separated. So, the heavy materials would migrate to the end and to the top of the tube causing in a surface density gradient. Then, DNA would move to the level where gradient’s same density (Hernandez, 2017).
2.1.7 Chelex 100 resin method
This method is simple, saves time, and it can be used for many different types of samples. The principle of this method is to prevent destroying DNA from enzymes and other contaminants. So, after cell lysis, next step would be washing by phosphate buffer and then adding Chelex resin solution. After then, using centrifugation and avoiding chelex resin beads before eluting the purified DNA (Walsh et al., 1991).
2.2 Amplification of DNA
DNA amplification is the procedure that is used to produce multiple copies of a specific DNA sequence. Even if there are different methods and steps of DNA amplification, there is a basic procedure of DNA amplification in genomic approach (Fig. 1). Different methods of DNA amplification as the following: (Fakruddin et al., 2013).
2.2.1 Polymerase chain reaction (PCR)
PCR is simple, suitable, and fast for DNA sequencing with Sanger sequencing (Fig. 1). In this method, heating at 94 °C is used to separate the DNA into two pieces of single strand. Then, cooling the temperature to 54 °C for primers to bind. Then, heating at 72 °C Taq polymerase, which is an enzyme that is used synthesize DNA strands, resulting in duplicating the original DNA using these new strands of DNA for other copies of the DNA and so on (Garibyan and Avashia, 2013). ELISA and gel electrophoresis can detect the amplicons of PCR. Although there are different methods of amplification other than PCR, these different methods are not widely used because they have not proved themselves in validation as PCR (Fakruddin et al., 2013).
2.2.2 Nucleic acid sequence-based amplification (NASBA)
The principle of NASBA depends on transcription system. This method is designed for RNA targets detection but it can also amplify DNA. NASBA uses different enzymes such as; RNase enzymes. This leads single-stranded RNA to be amplified (Fahy et al., 1991; Deiman et al., 2002; Guatelli et al., 1990). In the amplification process in this method, temperature is 41 °C (Fakruddin et al., 2013). The process of NASBA depends on a given DNA to multiple transcription of RNA copies. Gel electrophoresis can detect the amplicons of NASBA. This is more effective than other methods, which can increase binary per cycle (Sooknanan and Malek, 1995).
2.2.3 Rolling circle amplification (RCA)
This is an isothermal method. In a single temperature, this method amplifies more than 109 of DNA sequences in solution phase as well as solid phase (Lizardi et al., 1998; Schweitzer et al., 2000; Wiltshire et al., 2000; Fakruddin et al., 2013). In this method, Ø29 DNA polymerase leads to replication of the sequence of the nucleotides again and again (Demidov, 2002). RCA is contamination-resistant, and it needs small assay optimization. It's also ideal for storing morphological data (Fakruddin et al., 2013). This method permits signals’ localization. Therefore, it represents specific genetic traits in single molecules (Lizardi et al., 1998; Zhong et al., 2001) or biochemical features (Schweitzer et al., 2000; Wiltshire et al., 2000). Gel electrophoresis can detect RCA products. RCA is considered to have less amplification errors than other amplification methods (Lievens et al., 2005).
2.2.4 Ramification amplification method (RAM)
This is an isothermal method. Multiple ramification points give power to this method as well as primer extension and strand displacement (Fakruddin et al., 2013). By hybridization to a target, circular probe that is used in RAM is designed that 3′ end would be brought along with 5′ ends. Then, linking those ends by T4 DNA ligase leading to the production of a closed DNA circle. The bonded strand is then extended and the downstream strand is displaced, resulting in a multimeric ssDNA, which serves as a template for many primers resulting in a huge ramified DNA complex. Finally, the ramification process proceeds until all single-stranded DNAs are double-stranded. In this method, significant amplification car be achieved within 1 h at 35 °C because of Ø29 DNA polymerase (Fakruddin et al., 2013).
2.2.5 Ligase chain reaction (LCR)
LCR depends on cyclic DNA template reaction. It is similar to PCR. However, rather than production of amplicon by polymerization process, LCR amplifies the probe molecule. Enzymes like DNA ligase and DNA polymerase enzyme are used in LCR. Also, this method uses oligonucleotides in order to hybridize target fragments (Wu and Wallace, 1989; Fakruddin et al., 2013). Once oligonucleotides hybridize the correct target sequence, the rest of nick is ligated by DNA ligase. So, when probes are ligated, the product of ligation serves as the template for annealing and ligation. LCR is similar to PCR in the need of thermal circler for the reaction leading to doubling of DNA (Fakruddin et al., 2013). However, LCR has greater specificity than PCR (Wu and Wallace, 1989; Khanna et al., 1999). ELISA as well as gel electrophoresis can detect LCR products (Csako, 2006). There are many limitations of this method including: specificity of the ligase reaction, risk of contamination, lack of conformation, lack of sensitivity (Dean et al., 1998).
Other amplification methods of DNA include: strand displacement amplification (SDA) (McHugh et al., 2004), multiple displacement amplification (MDA) (Dean et al., 2001; Hughes et al., 2005), helicase dependent amplification (HDA) (Vincent et al., 2004; An et al., 2005), and loop mediated isothermal amplification (LAMP) (Fakruddin and Chowdhury, 2012).
2.3 Sequencing of DNA
Sequencing of DNA is the used procedure to determine and read the sequence of nucleic acid sequence in DNA. It determines the order of the four bases (Behjati and Tarpey, 2013; NHGRI, 2020). DNA sequence reading methods can be divided into three categories including: traditional basic methods, whole genome (large-scale) methods, and high-throughput methods (Bisht and Panda, 2014; Mardis, 2017; Straiton et al., 2019).
2.3.1 Traditional basic methods
Traditional basic methods are known as first-generation sequencing methods (Thakur et al., 2018; Straiton et al., 2019). Traditional basic methods include: Maxam–Gilbert method and Sanger method (Maxam and Gilbert, 1977; Sanger et al., 1977; Bisht and Panda, 2014).
2.3.1.1 Maxam-Gilbert sequencing method
This method was developed in 1977 by Allan Maxam and Walter Gilbert. The principle of this method depends on chemical reaction as well as radioactive labeling. Sequencing the DNA in this method is based on two steps of catalytic procedure including: piperidine and two selective chemicals attacking pyrimidines and purines (Maxam and Gilbert, 1977; Bisht and Panda, 2014). So, as chemical reaction, pyrimidines and purines react with hydrazine and dimethyl sulfate respectively leading to displacement of the base by breaking the glycoside bond between the ribose sugar and the base. Then, in the place that the base has been displaced, piperidine would catalyze cleavage of phosphodiester bonds (Maxam and Gilbert, 1977; Bisht and Panda, 2014). In the formic acid, piperidine and hydrazine cleave thymine and cytosine nucleotides as well as piperidine and dimethyl sulfate cleave guanine and adenine nucleotides. However, radioactive label is at one 5′ end. Finally, all that would be loaded into polyacrylamide gels to resolve the fragments with electrophoresis. By placing the gel on a light proof X-ray film cassette and place them in a freezer for several days, labeled fragment would be seen and results would be inferred (Maxam and Gilbert, 1977; Bisht and Panda, 2014).
2.3.1.2 Sanger sequencing
This method was developed in 1977 by Frederick Sanger. It is also called chain termination method because of its principle. The principle behind chain termination method was to generate all possible single-stranded DNA molecules complementary to a template that starts at a common 50 base and extends up to 1 kilobase in the 30 direction (Sanger and Coulson, 1975; Sanger et al., 1977; Bisht and Panda, 2014). Sanger sequencing method became common because it uses fewer chemicals, lower radioactivity, easier, and more reliable than the Maxam and Gilbert sequencing method. So, instead of using chemical cleavage reactions, this method is using a third form of the ribose sugar, which dideoxyribose in the hydroxyl group is missing from both the 20 and the 30 carbons (Sanger and Coulson, 1975; Sanger et al., 1977; Bisht and Panda, 2014). So, in this common method, denaturing double stranded DNA (dsDNA) into two single-stranded DNA (ssDNA) before attachment of a primer to one end of the sequence. Then, the four polymerase solutions with four types of dNTPs but only one type of ddNTP are added. Then, synthesis of DNA starts and the chain extends until a termination nucleotide is randomly incorporated. That would result in denaturing DNA fragments into ssDNA. Finally, by using gel electrophoresis, fragments can be separated and the sequence would be determined (Michel, 2008). Automation can be used in Sanger sequencing method that instead of using the gel, ddNTPs or the primer can be labeled by a fluorescent dye that the laser machine read the results (Bisht and Panda, 2014).
2.3.2 Whole genome (large-scale) methods
Whole genome methods are known as large-scale sequencing methods. These methods include: clone-by-clone sequencing and shotgun sequencing (Bisht and Panda, 2014; Nickle and Ng, 2021).
2.3.2.1 Clone-by-clone sequencing method
In clone-by-clone sequencing method, before splitting the DNA into fragments and star sequencing these DNA fragments, a map of all chromosomes of the genome must be created (Bisht and Panda, 2014; Nickle and Ng, 2021). After drawing the map of the genome, the genome would be splitted into small bits with overlapped bits between them in order to reassemble the genome after sequencing (Bisht and Panda, 2014). In order to make a lot of DNA copies, the small bits would be inserted into Bacterial Artificial Chromosomes (BACs) inserted inside bacterial cells to grow. So, each time the bacteria grow and divide, identical copies of DNA become more and more (Nickle and Ng, 2021). Finally, DNA fragments would be sequenced after putting them in a known DNA sequence vector (Bisht and Panda, 2014; Nickle and Ng, 2021). In this method, mapping can take a long time as well as high cost (Bisht and Panda, 2014).
2.3.2.2 Shotgun sequencing method
Shotgun sequencing method is also known as whole genome shotgun (WGS). This method was developed by Fred Sanger in 1982 (Bisht and Panda, 2014). The principle of shotgun sequencing method is very similar to clone-by-clone sequencing method with minor differences (Bisht and Panda, 2014; Nickle and Ng, 2021). In this method, DNA would be splitted into fragments followed by sequencing these fragments to determine the order of the DNA bases. Then, by using certain computer programs and softwares, these sequenced fragments would be assembled together to find overlapping of the fragments (Staden, 1979; Bisht and Panda, 2014; Nickle and Ng, 2021). In shotgun sequencing method, prior mapping of the genome is not required. However, before the end of the project, assembling cannot be produced. Shotgun sequencing method is widely used for bacterial genome projects (Staden, 1979; Bisht and Panda, 2014).
2.3.3 High-throughput methods
High-throughput methods can be divided into two categories including: short-read sequencing methods and long-read sequencing methods. Short-read sequencing methods, which are known as next or second-generation sequencing methods, including: massively parallel signature sequencing (MPSS), Illumina sequencing, SOLiD sequencing, pyrosequencing, ion torrent semiconductor sequencing, polony sequencing, and heliscope sequencing (Thakur et al., 2018; Straiton et al., 2019). Long-read sequencing methods, which are known as third-generation sequencing methods, including: single molecule real time (SMRT) sequencing. Also, long-read sequencing methods but is known as fourth-generation sequencing method including: nanopore DNA sequencing as well as short-read sequencing methods but is known as fourth-generation sequencing method including: nanoball DNA sequencing (Thakur et al., 2018; Straiton et al., 2019).
2.3.3.1 Massively parallel signature sequencing (MPSS)
This method is considered as next or second-generation sequencing method (Thakur et al., 2018; Straiton et al., 2019). Lynx Therapeutics company developed MPSS in 1992 by Sydney Brenner and Sam Eletr. MPSS principle depends on applying adapter ligation followed by adapter decoding resulting in reading the sequence in increasing of four nucleotides (Brenner et al., 2000; Thakur et al., 2018). MPSS counts individual mRNA molecules that are produced by each gene in order to analyze the level of gene expression (Reinartz et al., 2002; Torres et al., 2008). So, complementary DNA produce products that are tagged PCR, which are amplified, to be used in attaching the PCR products to microbeads. Then, ligation-based sequence determination for many rounds would identify a sequence signature bead. After several rounds of ligation-based sequence determination using the type IIs restriction endonuclease BbvI, a sequence signature is identified from each bead performing it in parallel obtaining sequence signatures called MPSS tag (Reinartz et al., 2002; Torres et al., 2008). These MPSS tags would be analyzed in the MPSS dataset as well as compared with all other signatures. MPSS datasets are additive meaning mRNA can be combined from multiple analyses. In MPSS, Lynx Megaclone technology can clone cDNA fragments. So, starting with a number of mRNA, Megaclone would produce the same number of beads cloning copies of cDNA from each mRNA molecule attaching these molecules to the microbeads for the sequencing reactions (Reinartz et al., 2002; Torres et al., 2008).
2.3.3.2 Illumina sequencing method
This method is considered as next or second-generation sequencing method (Thakur et al., 2018; Straiton et al., 2019; Illumina, 2021). In 2006, Illumina company purchased Solexa sequencing technology and developed it into a main sequencing technology on the market. Now, Illumina provides different sequencing systems including: MiSeq, HiSeq 2500, HiSeq 3000, and HiSeq 4000. Illumina sequencing technologies are considered the most successful technologies with >70 % dominance of the market (Thakur et al., 2018; Illumina, 2021). In Illumina sequencing technology, breaking up the DNA into fragments. Then, attachment of adaptors to the DNA fragments leading to single stranded by incubating with sodium hydroxide (Fig. 1). Next, washing DNA fragments that cDNA would bind to primers on the surface of the flowcell. Then, attached DNA should be replicated before creating bridges of double-stranded DNA between the primers on the flowcell surface by adding DNA polymerase and unlabeled nucleotide bases. Then, breaking double-stranded DNA into single-stranded DNA by heating. Next, adding primers and fluorescent labelled terminators for binding DNA polymerase to these primers and adding fluorescent labelled terminator to the new DNA strand. Fluorescent label on the nucleotide base would be activated by lasers before is detected by a camera and recorded on a computer (Fig. 1). Finally, DNA sequence is analyzed (Thakur et al., 2018; Illumina, 2021; Yourgenome, 2021).
2.3.3.3 SOLiD method
This method is considered as next or second-generation sequencing method (Thakur et al., 2018; Straiton et al., 2019). SOLiD method is based on sequencing by ligation. In this method, all oligonucleotides of specific length would be labeled according to the position of sequence. Then, by DNA ligase, oligonucleotides would be ligated and annealed to match the results of sequences. Next would be amplification of the DNA. Finally, each resulting bead would contain a single copy of the same DNA. However, there is a report on SOLiD method that it has an issue in palindromic sequences (Thakur et al., 2018).
2.3.3.4 Pyrosequencing Method
This method is considered as next or second-generation sequencing method (Thakur et al., 2018; Straiton et al., 2019). Pyrosequencing method is similar to Sanger sequencing method. However, Sanger sequencing depends on termination with dideoxynucleotides but Pyrosequencing depends on detecting pyrophosphate release and generating a light on nucleotide incorporation (Simner et al., 2015). In this method, a mix of firefly luciferase, ATP sulfurylase, DNA polymerase, and nucleotides would be added to the sequencing of single stranded DNA. Integration of these nucleotide is regulated by the released light, which its intensity would show the number of complementary nucleotides on a template strand. Then, the mix of nucleotides should be a way, before the next mix of nucleotides is added. This procedure should be performed again with each of the four nucleotides until the determination of the DNA sequence (Thakur et al., 2018).
2.3.3.5 Ion torrent semiconductor sequencing method
This method is considered as next or second-generation sequencing method (Rusk, 2010; Gupta and Gupta, 2014). In this method, polymerization of a DNA template within a microwell releases hydrogen ions. So, the principle of ion torrent semiconductor sequencing is to detect these hydrogen ions (Rusk, 2010; Gupta and Gupta, 2014). In this method, a single type of nucleotides would be added to template DNA strand in a microwell leading to the growing of complementary strand. This process leads to the release of hydrogen ions indicating an activity by the hypersensitive ion sensor (Rusk, 2010; Gupta and Gupta, 2014).
2.3.3.6 Polony sequencing method
This method is considered as next or second-generation sequencing method (Porreca et al., 2006; Thakur et al., 2018; Straiton et al., 2019). This method was developed at the laboratory of George M. Churchlaboratory in Harvard in 2005. It was applied for sequencing full genome E. coli (Thakur et al., 2018). Polony sequencing method depends on polymerases and ligases. In this method, with emulsion PCR, mate-paired in vitro are produced and amplified on microbeads. These are used as templates for sequencing by fluorescent ligation reactions in a microscope. Results of sequencing would be aligned to the reference genome that allows comparing of differences between sequences (Porreca et al., 2006). Polony sequencing method is accurate and cheap method (Porreca et al., 2006; Thakur et al., 2018).
2.3.3.7 Heliscope sequencing
This method is considered as third-generation sequencing method. It is a single-molecule fluorescent sequencing method (Thompson and Steinmann, 2010; Thakur et al., 2018). This method is developed by Helicos Biosciences company. This method uses DNA polymerase in a combination of synthesis sequencing and hybridization sequencing. First, DNA would be splitted and tailed with poly A. Then, hybridization of that splitted DNA to a flow cell surface with oligo-dT to sequence by synthesis. Then, attachment of fragments of DNA fragments that tailed with poly A to the bound of oligo-dT50. Finally, incorporation of terminating nucleotides with fluorescent nucleotides discontinue the cyclical process until one nucleotide is captured from the DNA sequence, and then sequencing of the fragments would be continued until the sequence is completed (Thompson and Steinmann, 2010; Thakur et al., 2018).
2.3.3.8 Single molecule real time (SMRT) sequencing
This method is considered as third-generation sequencing method (Thakur et al., 2018). This method is developed by Pacific Biosciences company. (Ben-Ari and Lavi, 2012). The principle of this method depends on measuring the incorporation of nucleotides in real time. SMRT has the ability to differentiate between adenine and cytosine modification states (Prater and Hamilton, 2019). This method works at single molecule resolution with main steps starting with enabling the observation of individual fluorophores by SMRT Cell. Then, synthesis of DNA through building blocks by phospho-linked nucleotides. Finally, enabling single molecule for real-time detection by a detection platform. SMRT is considered as accurate, fast, and cheap method (Ben-Ari and Lavi, 2012).
2.3.3.9 Nanopore DNA sequencing
This method is considered as fourth-generation sequencing method (Thakur et al., 2018). This method is a single-molecule sequencing with the ability to detect epigenetic modifications (Okoniewski et al., 2016). The principle of this method is DNA passing through the nanopore changes it to ion current depending on the length, size, and shape of the DNA sequence (Thakur et al., 2018).
2.3.3.10 Nanoball DNA sequencing
This method is considered as fourth-generation sequencing method (Thakur et al., 2018). This method can be used for the determination of the complete genomic sequence. In order to get DNA nanoballs in this method, Nanoball DNA sequencing amplifies small fragments of genomic DNA by rolling circle replication. Then, ligation is used to determine the nucleotide sequence (Thakur et al., 2018). Nanoball DNA sequencing method is cheap and give a large number of DNA nanoballs to be sequenced per run (Porreca, 2010).
3 Health informatics approach
So, after sequencing of DNA in the genomics approach, health informatics approach would start in data analysis of DNA that is called (Fig. 2) (Oakeson et al., 2017; Roy et al., 2018; Miller, 2000).
3.1 Data analysis of DNA
Data analysis of the DNA is also known as bioinformatics pipelines (Oakeson et al., 2017; Roy et al., 2018). There are several routes of bioinformatics pipelines in the genomics approach including; next-generation sequencing bioinformatics pipeline (NGS-bioinformatics pipeline). NGS-bioinformatics pipeline can be complicated but it can be more accurate, faster, and cheaper from other generation sequencing of genomic approach (Oakeson et al., 2017; Roy et al., 2018). So, starting form next-generation sequencing methods in the step of the DNA sequencing including: Illumina method and then transferring to NGS-bioinformatics pipeline (Fig. 2). NGS-bioinformatics pipeline consists of six major steps including: sequence generation, sequence alignment, variant calling, variant filtering, variant annotation, and variant prioritization (Roy et al., 2018).
3.1.1 Sequence generation
Sequence generation step include signal processing, base calling, and FASTQ files (Fig. 2). This step transforms data from the sequencing platform as well as identifies the sequence of short DNA fragments in the prepared sample of analysis. In this step, Phred-like quality score, which is a measure of identification quality of nucleobases produced by automated DNA sequencing, is assigned for each short DNA fragment sequence. Then, FASTQ files, which are format for storing nucleotide sequences and their quality score, would store Phred-like quality scores and read nucleotide sequences (Roy et al., 2018).
3.1.2 Sequence alignment
Sequence alignment step includes alignment, reference genome mapping, and binary alignment map (BAM) (Fig. 2). This step determines the alignment of each short fragment DNA sequence with a reference genome assigning a Phred-scale mapping quality score for each short sequence. Moreover, the genomic context for each aligned sequence is provided to be used in calculating the proportion mapped sequence. Then, these data would be stored in BAM file, which is a format the sequence alignment map (Roy et al., 2018).
3.1.3 Variant calling
Variant calling step includes pre-variant calling, single-nucleotide variants (SNVs), small insertions and deletions (indels), and variant calling format (VCF) (Fig. 2). This step identifies variations or differences in sequences. When aligned sequence in BAM is called typical input. VCF is used to represent variants for SNVs and indels. There are different types sequence variants including: SNVs, indels, copy number alterations, and large structural alterations. So, variant calling is known as a heterogeneous collection of algorithmic strategies depends on these types. This step depends on quality of called bases and aligned sequence. Therefore, pre-variant calling is needed to guarantee an effective and accurate variant calling (Roy et al., 2018).
3.1.4 Variant filtering
Variant filtering step filters false-positive variant of the NGS method from the original VCF. It is known as post-variant calling step (Fig. 2). This process is used to allow annotation and review true variants only (Roy et al., 2018).
3.1.5 Variant annotation
Variant annotation step categorizes each variant with a huge set of metadata. This metadata includes variant location, predicted cDNA and amino acid sequence change, and prevalence in different variant databases (Fig. 2). All that would be used in next step for classification and interpretation of variants (Roy et al., 2018).
3.1.6 Variant prioritization
Variant prioritization step includes classification and interpretation of variants. This step uses annotations to identify clinically insignificant variants such as synonymous deep intronic variants (Fig. 2). Therefore, it is important to present the remaining variants for further review and interpretation. Finally, clinical reports would be issued (Roy et al., 2018).
4 Conclusion
Intersection of genomics and health informatics approaches is a great combination in the era of diseases’ biomarkers identification. Genomics is the science that studies structures, interactions, and functions of all genomes. So, genomics is used to identify genomes as diseases' biomarkers. In genomics approach, after collecting samples, first start with extraction and purification of DNA to get a purified DNA that is extracted from the nucleus of cells. Then, amplification of DNA that is used to produce multiple copies of a specific DNA sequence. Next is sequencing of DNA to determine and read the sequence of nucleic acid sequence in DNA. Once the DNA sequence is determined, the following procedure is the health informatics approach, called bioinformatics pipeline steps, which is DNA data analysis steps. Finally, there are several different techniques and methods of integration of genomics and health informatics approaches to identify a genome as a disease’s biomarker.
Acknowledgements
The author would like to thank Deanship of Scientific Research at Majmaah University, Al Majmaah, 11952, Saudi Arabia for supporting this work under the Project Number R-2022-238.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Magnetic nanoparticles: preparation, physical properties, and applications in biomedicine. Nanoscale Res. Lett.. 2012;7(1):144.
- [CrossRef] [Google Scholar]
- Characterization of a thermostable UvrD helicase and its participation in helicase-dependent amplification. J. Biol. Chem.. 2005;280(32):28952-28958.
- [CrossRef] [Google Scholar]
- What is next generation sequencing? Archives of disease in childhood. Education and Practice Edition. 2013;98(6):236-238.
- [CrossRef] [Google Scholar]
- Ben-Ari, G., Lavi, U., 2012. Marker-assisted selection in plant breeding, in: Altman, A., Hasegawa, M.P. (Eds.), Plant Biotechnology and Agriculture. Elsevier, pp. 163–184. doi: 10.1016/B978-0-12-381466-1.00011-0.
- Biomarkers and surrogate endpoints: Preferred definitions and Conceptual Framework. Clinical Pharmacology; Therapeutics. 2001;69(3):89-95.
- [CrossRef] [Google Scholar]
- Bisht S.S., Panda A.K., 2014. DNA Sequencing: Methods and Applications. In: Ravi I., Baunthiyal M., Saxena J. (eds) Advances in Biotechnology. Springer, New Delhi. Doi: 10.1007/978-81-322-1554-7_2.
- Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol.. 2000;18(6):630-634.
- [CrossRef] [Google Scholar]
- Biomarkers. Clinical trials. Elsevier Academic Press; 2016. p. :377-419.
- Purification ofdnaby anion-exchange chromatography. Curr. Protocols Mol. Biol.. 1998;42(1)
- [CrossRef] [Google Scholar]
- Present and future of rapid and/or high-throughput methods for nucleic acid testing. Clin. Chim. Acta. 2006;363(1–2):6-31.
- [CrossRef] [Google Scholar]
- Davis, M. A., Eldridge, S., Louden, C., 2013. Biomarkers. Haschek and Rousseaux's Handbook of Toxicologic Pathology, 317–352. doi: 10.1016/b978-0-12-415759-0.00010-8.
- Comparison of performance and cost-effectiveness of direct fluorescent-antibody, ligase chain reaction, and PCR assays for verification of chlamydial enzyme immunoassay results for populations with a low to moderate prevalence of Chlamydia trachomatis infection. J. Clin. Microbiol.. 1998;36(1):94-99.
- [CrossRef] [Google Scholar]
- Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res.. 2001;11(6):1095-1099.
- [CrossRef] [Google Scholar]
- Characteristics and applications of nucleic acid sequence-based amplification (NASBA) Mol. Biotechnol.. 2002;20(2):163-180.
- [CrossRef] [Google Scholar]
- Rolling-circle amplification in DNA diagnostics: the power of Simplicity. Expert Review of Molecular Diagnostics. 2002;2(6):542-548.
- [CrossRef] [Google Scholar]
- Human genome sequencing using Unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327(5961):78-81.
- [CrossRef] [Google Scholar]
- Self-sustained sequence replication (3SR): an isothermal transcription-based amplification system alternative to PCR. Genome Res.. 1991;1(1):25-33.
- [CrossRef] [Google Scholar]
- Pyrosequencing-an alternative to traditional Sanger sequencing. Am. J. Biochem. Biotechnol.. 2012;8(1):14-20.
- [CrossRef] [Google Scholar]
- Nucleic acid amplification: Alternative methods of polymerase chain reaction. J. Pharmacy Bioallied Sci.. 2013;5(4):245-252.
- [CrossRef] [Google Scholar]
- Isothermal, in vitro amplification of nucleic acids by a multienzyme reaction modeled after retroviral replication. Proc. Natl. Acad. Sci.. 1990;87(19)
- [Google Scholar]
- Next generation sequencing and its applications. Anim. Biotechnol.. 2014;345–367
- [CrossRef] [Google Scholar]
- Hernandez, V., “Equilibrium Density Gradient Centrifugation in Cesium Chloride Solutions Developed by Matthew Meselson and Franklin Stahl”., 2017. Embryo Project Encyclopedia. ISSN: 1940-5030.
- The use of whole genome amplification in the study of human disease. Prog. Biophys. Mol. Biol.. 2005;88(1):173-189.
- [CrossRef] [Google Scholar]
- Illumina. Illumina Sequencing Methods. 2021. Retrieved from www.illumina.com (accessed 17 oct 2021).
- Lievens, B., Grauwet, T. J. M. A., Cammue, B. P. A., Thomma, B. P. H. J., 2005. Recent developments in diagnostics of plant pathogens: A review. In S. G. Pandalai (Ed.), Recent Research Developments in Microbiology (pp. 57-79). (Recent Research Developments in Microbiology; No. 9).
- Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat. Genet.. 1998;19(3):225-232.
- [CrossRef] [Google Scholar]
- Genomic approaches for understanding the genetics of complex disease. Genome Res.. 2015;25(10):1432-1441.
- [CrossRef] [Google Scholar]
- Magnetic nanoparticles-based extraction and verification of nucleic acids from different sources. J. Biomed. Nanotechnol.. 2013;9(4):703-709.
- [CrossRef] [Google Scholar]
- DNA sequencing technologies: 2006–2016. Nat. Protoc.. 2017;12(2):213-218.
- [CrossRef] [Google Scholar]
- Maxam, A. M., Gilbert, W., 1977. A new method for sequencing DNA. Proceedings of the National Academy of Sciences of the United States of America, 74(2), 560–564. doi: 10.1073/pnas.74.2.560.
- Prospective evaluation of BDPROBETEC strand displacement amplification (SDA) system for diagnosis of tuberculosis in non-respiratory and respiratory samples. J. Med. Microbiol.. 2004;53(12):1215-1219.
- [CrossRef] [Google Scholar]
- Michel G., 2008. Simulation of polymer translocation through small channels: A molecular dynamics study and a new Monte Carlo approach. Diss. University of Ottawa, Canada.
- Opportunities at the intersection of bioinformatics and health informatics: a case study. J. Am. Med. Informatics Assoc.: JAMIA. 2000;7(5):431-438.
- [CrossRef] [Google Scholar]
- A simple salting out procedure for extracting DNA from human nucleated cells. Nucl. Acids Res.. 1988;16(3):1215.
- [CrossRef] [Google Scholar]
- Nucleic acid extraction from human biological samples. Methods Mol. Biol.. 2018;359–383
- [CrossRef] [Google Scholar]
- National Cancer Institute (NCI)., 2021. Genomics. Retrieved from https://www.cancer.gov (accessed 16 Sep 2021).
- National Human Genome Research Institute (NHGRI). National Human Genome Research Institute of Health., 2020. DNA sequencing fact sheet. Retrieved from https://www.genome.gov (accessed 12 Oct 2021).
- Nickle T, Ng I B., 2021. Mount Royal University & University of Calgary. 11.3. Whole Genome Sequencing. 2021.Biology. LiberText.
- National Institute of Justice (NIJ)., 2021. DNA evidence basics: Types of samples suitable for DNA testing. Retrieved, from https://nij.ojp.gov (accessed 09 oct 2021).
- National Research Council (US) (NRC) Committee on Human Genome Diversity., 1997. Sample collection and data management. Evaluating Human Genetic Diversity. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK100428/ (accessed 09 Oct 2021).
- Bioinformatic Analyses of Whole-Genome Sequence Data in a Public Health Laboratory. Emerg. Infect. Dis.. 2017;23(9):1441-1445.
- [CrossRef] [Google Scholar]
- Future directions. Clinical Applications for Next-Generation Sequencing. 2016;281–294
- [CrossRef] [Google Scholar]
- Oklahoma State University (2021), 2021. DNA sample collection. DNA Sample Collection | Oklahoma State University. Retrieved from (accessed 15 Oct 2021).
- Public Health Degrees (PHD). 2020. Retrieved April, 2022, from https://www.publichealthdegrees.org/careers/health-informatics-vs-bioinformatics/.
- Epigenetics: Analysis of cytosine modifications at single base resolution. Encyclopedia of Bioinformatics and Computational Biology. 2019;341–353
- [CrossRef] [Google Scholar]
- Massively parallel signature sequencing (MPSS) as a tool for in-depth quantitative gene expression profiling in all organisms. Briefings in Functional Genomics. 2002;1(1):95-104.
- [CrossRef] [Google Scholar]
- Standards and guidelines for validating next-generation sequencing bioinformatics pipelines. J. Mol. Diagn.. 2018;20(1):4-27.
- [CrossRef] [Google Scholar]
- Rusk, N., 2010. Torrents of sequence. Nature Methods, 8(1), 44–44. doi: 10.1038/nmeth.f.330.
- A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol.. 1975;94(3):441-448.
- [CrossRef] [Google Scholar]
- DNA sequencing with chain-terminating inhibitors. PNAS. 1977;74(12):5463-5467.
- [CrossRef] [Google Scholar]
- Immunoassays with rolling circle DNA amplification: a versatile platform for ultrasensitive antigen detection. PNAS. 2000;97(18):10113-10119.
- [CrossRef] [Google Scholar]
- Evaluation of a modified salt-out method for DNA extraction from whole blood lymphocytes: A simple and economical method for gene polymorphism. Pharmaceutical Biomed. Res. 2018
- [CrossRef] [Google Scholar]
- NASBA: A detection and amplification system uniquely suited for RNA. Nat. Biotechnol.. 1995;13(6):563-564.
- [CrossRef] [Google Scholar]
- A strategy of DNA sequencing employing computer programs. Nucl. Acids Res.. 1979;6(7):2601-2610.
- [CrossRef] [Google Scholar]
- From Sanger sequencing to genome databases and beyond. Biotechniques. 2019;66(2):60-63.
- [CrossRef] [Google Scholar]
- What are biomarkers? Current Opinion in HIV and AIDS. 2010;5(6):463-466.
- [CrossRef] [Google Scholar]
- DNA, RNA, and protein extraction: the past and the present. J. Biomed. Biotechnol.. 2009;2009:574398
- [CrossRef] [Google Scholar]
- Thakur, N., Shirkot, P., Pandey, H., Thakur, K., Kumar, V., Rachappanavar., 2018. Next generation sequencing – techniques and its applications. J. Pharmacognosy Phytochem. E-ISSN: 2278-4136 P-ISSN: 2349-8234.
- DNA/RNA preparation for molecular detection. Clin. Chem.. 2015;61(1):89-99.
- [CrossRef] [Google Scholar]
- Thompson, J. F., Steinmann, K. E., 2010. Single molecule sequencing with a HeliScope genetic analysis system. Curr. Protocols Mol. Biol., Chapter 7, Unit7.10. doi: 10.1002/0471142727.mb0710s92.
- Gene expression profiling by massively parallel sequencing. Genome Res.. 2008;18(1):172-177.
- [CrossRef] [Google Scholar]
- Helicase-dependent isothermal DNA amplification. EMBO Rep.. 2004;5(8):795-800.
- [CrossRef] [Google Scholar]
- World Health Organization (WHO). 2001. Regional Office for the Eastern Mediterranean. Health and medical informatics in the Eastern Mediterranean Region. https://apps.who.int/iris/handle/10665/121939.
- Detection of multiple allergen-specific iges on microarrays by immunoassay with Rolling Circle Amplification. Clin. Chem.. 2000;46(12):1990-1993.
- [CrossRef] [Google Scholar]
- The ligation amplification reaction (lar)—amplification of specific DNA sequences using sequential rounds of template-dependent ligation. Genomics. 1989;4(4):560-569.
- [CrossRef] [Google Scholar]
- Yourgenome, 2021. Discover more about DNA, genes, and genomes, and the implications for our health and society. Retrieved from www.yourgenome.org (accessed 12 oct 2021).
- Visualization of oligonucleotide probes and point mutations in interphase nuclei and DNA fibers using rolling circle DNA amplification. PNAS. 2001;98(7):3940-3945.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102264.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




