Translate this page into:
Docking and molecular dynamics studies of human ezrin protein with a modelled SARS-CoV-2 endodomain and their interaction with potential invasion inhibitors
⁎Corresponding authors at: Nell Watson Ltd, c/o Mills Pyatt, 11 Kingfisher Business Park, Arthur Street, Redditch, Worcestershire B98 8LG, United Kingdom (E. Watson). selvaakumar.c@dypatil.edu (Selvaa Kumar Chellasamy), eleanorwatson@connect.glos.ac.uk (Eleanor Watson)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
Human ezrin protein interacts with SARS-CoV S endodomain and restricts virus fusion, entry, and early events of infection. In general, their binding strength and their structural stability determines their successful entry into the host cells. However, the binding affinity of these two endodomains with the ezrin protein has been elusive due to a paucity of knowledge on the 3D structure. This study modelled the endodomains of both SARS-CoV-1 and SARS-CoV-2 and then docked these models with human ezrin protein. This study establishes that the modelled endodomains of both SARS-CoV-1 and SARS-Cov-2 consisted of three disulphide bridges for self-stabilization. Protein-protein docking listed four salt bridges with a higher buried surface area between ezrin-SARS-CoV-1 endodomain compared to that of ezrin-SARS-CoV-2 with six salt bridges with lower buried surface area. Molecular simulation of the ezrin-SARS-CoV-1 endodomain showed better structural stability with lower Root Mean Square Deviation score compared to that of ezrin-SARS-CoV-2 endodomain due to the substitution of alanine with cysteine residue. Protein-ligand docking studies confirmed better ezrin-drug interaction for quercetin, minocycline, calcifediol, calcitriol, selamectin, ivermectin and ergocalciferol. However, protein–ligand simulation confirmed strong drug-protein interaction during simulation for all the above-listed drugs except for ergocalciferol which could not establish its interaction with the protein during simulation. Strong drug binding within the active site pocket therefore restricts the interaction of viral endodomain and simultaneously stabilizes the ezrin protein. Furthermore, the higher stability between the ezrin after their interaction with the drug moiety could restrict the virus fusion and the infection. This study provides a basis for further development of these drug molecules to clinical trials aiming to identify potential drug molecules which can treat COVID-19 infection.
Keywords
Ezrin
SARS-CoV-1
SARS-CoV-2
Drug repurposing
COVID-19
Endodomain

1 Introduction
Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) is a single-stranded positive-sense RNA beta coronavirus of the coronaviridae family which include SARS-CoV-1 and the Middle Eastern respiratory syndrome coronavirus (MERS-CoV) (Paules et al., 2020; de Wit et al., 2016). This highly pathogenic member of the coronaviridae family targets the upper respiratory tract (URT) with pneumonia-like symptoms which was termed Coronavirus Disease 2019 (COVID-19) which was first detected in Wuhan in the Hubei Province of China (Zhu et al., 2019). The pandemic has so far involved 535 million infections with a death count of 6.3 million as on 16th of June 2022 (WHO, n.d.).
Genome annotation of SARS-CoV 2 lists twelve ORFs, namely, Orf1a, Orf1b, S, Orf3, E, M, Orf6, Orf7a, Orf7b, Orf8, N, Orf10 (Zhang and Holmes, 2020). Of these ORFs, the spike (S) glycoprotein received the widest attention from the scientific community due to its interactions with host cell targets such as ACE2, cyclophilins, Ezrin and CD26, etc. These targets are essential for envelope fusion with the host cell membranes and associated virulence factors (Millet et al., 2012). These viruses are commonly composed of spike protein (S), envelope protein (E), matrix protein (M), nucleoprotein and viral RNA (shown in Fig. 1) (Lu et al., 2020).
The spike membrane domain is 1255 amino acids long which is further classified into N-terminal ectodomain (1211 amino acids), along with smaller regions that form the transmembrane domain (TM) (23 amino acids), and the C-terminal endodomain (39 amino acids) (Li et al., 2005; Lu et al., 2013) (Fig. 1a). In particular, the endodomain is made up of both cysteine-rich and charge-rich motifs (DEDDSE) (Petit et al., 2007). The cysteine-rich motif is comprised of 4 cysteine clusters, namely, Cl1, Cl2, Cl3 and Cl4 which are principally involved in cell-cell membrane fusion and are accompanied by S protein-protein interaction (shown in Fig. 1b). The charge rich motif is involved in virion assembly, effected through the interaction of S and M proteins (Fehr and Perlman, 2015). It plays a crucial role in virus entry after the formation of the fusion pore (Millet et al., 2012). Based on the literature survey, the F1 lobe of the human ezrin protein interact with the endodomain of SARS-CoV-1 which in turn belongs to the ERM family (Millet et al., 2012). They are mainly involved in organizing the membrane domain through their association with actin (filamentous), transmembrane protein and lipid rafts (Fehon et al., 2010). However, the crystal structure of this part of the endodomain is still to be elucidated.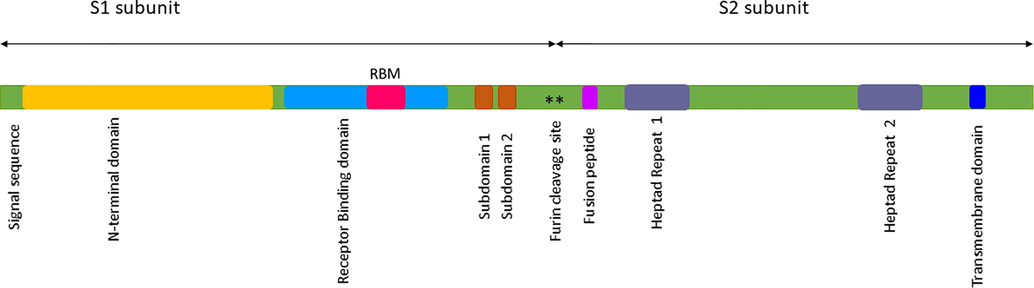
The Spike glycoprotein (S) of SARS-CoV-2 displaying the prominent subunits S1 and S2 with their respective domains in distinct colors.
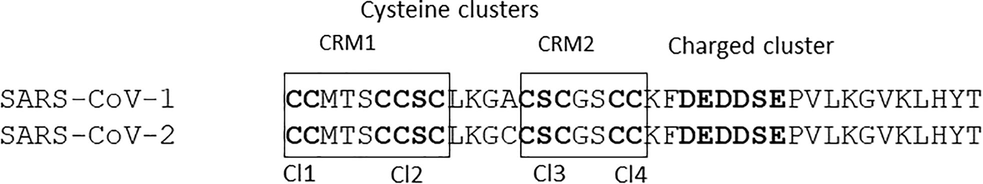
The pairwise alignment of SARS-CoV-1 and SARS-CoV-2 displaying the cysteine and charged clusters. Individual cysteine clusters like Cl1, Cl2, Cl3 and Cl4 are shown in bold text, broadly classified into CRM1 and CRM2.
Ezrin is comprised of three functional domains classified into the FERM domain (1–296), central domain (297–496), and C-terminal domain (C-ERMAD) (497–586) (Fiévet et al., 2007). The FERM domain with three lobes (F1, F2 and F3) prefers to interact with membrane and signaling proteins (Smith et al., 2003; Bretscher et al., 2002). The F1 lobe of the ezrin protein interacts with the SARS-CoV-1 endodomain (Millet et al., 2012). The coiled central domain interacts with p85 and the C-ERMAD interacts with F-actin (Gautreau et al., 1999). Such binding of ezrin with the SARS-CoV endodomain restricts the membrane fusion entry of the virus and S dependent early events of infection, which appears to alter the efficacy of fusion (Millet et al., 2012). Based on the literature review, a single amino acid substitution is observed between the SARS-CoV-1 and 2 endodomain wherein alanine at position 13 of SARS-CoV-1 is substituted by cysteine in SARS-CoV-2 (Xia, 2021). The impact of this substitution within the endodomain and their role in ezrin binding remains elusive till date.
A search for potential drugs to treat the patients with COVID-19 infection is underway (National Institute of Health., Singh et al., 2020; Kumar et al., 2020). Two bis-piperazine based inhibitors (DHHC9) were recently reported to inhibit the palmitoylation in SARS-CoV resulting in lower fusion and infection (Ramadan et al., 2022).
This in-silico based study examines the difference in binding affinity of the ezrin protein with SARS-CoV-1 and 2 endodomain and drug molecules.
2 Materials and methods
2.1 3D modelling of the protein complexes
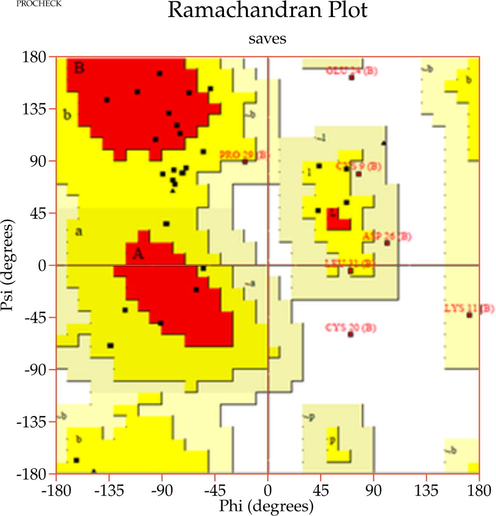
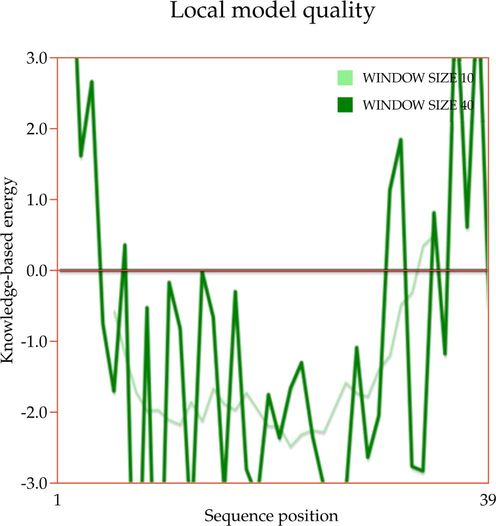
In this study, the 3D protein models of human actin, human ezrin, and the endodomain of SARS-CoV-1 and 2 were generated. Firstly, the actin protein sequence of Homo sapiens was downloaded from the Uniprot database with Accession Number P63267 (Bairoch and Apweiler, 1997). This sequence was submitted to the SWISS-MODEL server to identify a potential template for generating actin protein (Schwede et al., 2003). A potential template with better query coverage and resolution was selected for model building and energy minimization processes using the GROMACS standalone software (Berendsen et al., 1995). Secondly, the ezrin protein sequence of Homo sapiens was retrieved from the Uniprot database (Accession Number: P15311) and subjected to similar approaches. Finally, an endodomain sequence for SARS-CoV-1 and 2 of 39 residues was retrieved from the National Center for Biotechnology Information database (NCBI, 1988) (SARS-CoV-1: YP0098250, SARS-CoV-2 MT050493), and a structural search was initiated. With no reported hits from the Protein Data Bank, the I-TASSER online server was considered for model building (Berman, 2000; Yang and Zhang, 2015). The five largest structure clusters itemized five models, each with C- and TM-scores. The highest C-score confirms the highest confidence score, and the highest TM score ensures the accuracy of the topology of the model. Furthermore, the amino acid alanine in SARS-CoV-1 was replaced by cysteine to generate the endodomain of SARS-CoV-2. Finally, all the minimized 3D structures were subjected to structural validation using the PROCHECK online server (Laskowski et al., 1993) as well as the ProSA-Web server (Wiederstein and Sippl, 2007). These validated structures were further subjected to docking studies.
2.2 Optimization of the ligand molecules
Based on the literature review, drugs listed as being of interest for potential repurposing for the treatment of COVID-19 were retrieved from Protein Data Bank (Berman et al., 2000) and PubChem Database (Kim et al., 2016). Drug molecules retrieved from the PDB are listed in Supplementary Table 1. Data from PubChem include enoxaparin (772), 24,25-dihydroxycholecalciferol (6434253), moxidectin (9832912), terfenadine (5405), penduline (44259755), nystatin (6433272), niclosamide (4477), neomycin (8378), flubendazole (35802), fenbendazone (3334), cepharanthine (10206), moxidectin (9832912), selamectin (9578507), coclobine (5315989), hydroxychloroquine (3652), aspirin (2244), and berbamine (275182), promethazine (4927), nystatin (6433272), ergocalciferol (5280793), methylene blue (6099), and andrographolide (5318517). Retrieved SDF files were submitted to the FROG2 online server for the generation of stable conformers (Miteva et al., 2010). Regarding the procedural settings, the “input type file” was 1D2D and the “input drug” description was SDF. The “output format” was in PDB formats. Regarding the produce setting, here we opted for “single” for a single energetically reasonable conformation taken from several passes for multi-conformer generations.
Human Ezrin
Human Actin
Arg569
Glu335
Glu582
Lys329
Thr533
Thr6
Ser536
Gln354
Arg542
Gln355
Arg559
Glu168
Gln560
Glu168‘
Arg562
Gly147
Gln570
Gly147
Glu525
Thr7
Arg559
Tyr144
Gln570
Arg148
Gln578
Lys329
Gln573
Lys329
Gln555
Thr352
Glu522
Gly24
Gln570
Gly147
2.3 Protein-protein docking simulations
The human ezrin protein was docked with actin protein using the HADDOCK online server (van Zundert et al., 2015) to identify their exact region of interaction. For the docking, the requisite active and passive residues were provided as needed. For the actin protein, the active residues include Gly24, Asp26, Ser349, Thr352, Thr149, and Glu168 (Chen et al., 2012). Residues of the passive were defined automatically through the checkbox. For the ezrin protein, based on the literature review, the last 34 amino acids of C-ERMAD from the C-terminal responsible for binding are considered to be the active site region (Turunen et al., 1994). Passive residues were again selected automatically. Next, the endodomains of SARS-CoV-1 and SARS-CoV-2 were docked with the ezrin protein to understand the effect of amino acid substitution on their binding affinity. Endodomain proteins were targeted against the F1 lobe (6–93 amino acid residues) of the ezrin protein which were also considered to be the active/passive residues. After each run, the HADDOCK score and the buried surface area were taken into consideration for clustering to derive a maximum score. All the docked poses were subjected to a PRODIGY online server analysis Xue et al., 2016). All the selected poses were visualized using Discovery Studio (BIOVIA, 2019).
2.4 Protein-ligand docking studies
To understand the overall drug binding affinity in ezrin protein, forty-six stable chemical compounds were docked with the ezrin protein using the AutoDock version 1.5.6 software (Morris et al., 2009). Docking studies were initiated with the addition of Kollman and Gasteiger charges for proteins and ligands respectively. The grid consists of twelve residues based on literature review29 (His48, Trp58, Tyr85, Gln105, Ile115, Tyr201, and Asn204, Trp278, Leu274, Phe250, Pro272, and Leu281) was placed within the binding sites. The size of the grid box was maintained at 74 Å, 60 Å and 58 Å for ×, y, and z respectively. The grid center was also set to −8.583 Å, 6.056 Å, and −1.056 Å for x, y, and z, respectively. The AutoGrid 4.0 and AutoDock 4.0 programs were used to generate grid maps, and 10 conformers were generated using a Lamarckian Genetic Algorithm (LGA) for each of the compounds. Additionally, the binding energy and inhibition constant for each compound were selected for visualization using Discovery Studio Visualizer.
2.5 Molecular dynamics simulation
The structural stability of the ezrin protein in association with the viral endodomains and the drug moieties were investigated under optimum thermobaric conditions using the Desmond v.3.6 package (Shaw, 2013). The TIP3P water model was applied for the simulation of water molecules. Furthermore, orthorhombic periodic conditions were considered to define the shape and size of the repeating unit at 10 Å distances. In total, 12 Cl− ions were added to neutralize the system. The overall concentration of Na was 50.9 mM and Cl was 66.2 mM. Later the solvated system was further subjected to energy minimization under NPT ensemble maintaining the default parameters. The simulation was performed with the periodic boundary conditions in the NPT ensemble using OPLS 2005 force field parameters (Jorgensen et al., 1983). The overall temperature was maintained at 300 K, and the atmospheric pressure was at 1 atmospheric pressure using Nose-Hoover temperature coupling and isotropic scaling (Nosé, 1984). After equilibration, the molecular dynamics was performed for 100 ns to analyze the trajectory of the protein-protein and protein-ligand complex through the system. The Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and the number of hydrogen bonds and other interactions between the ezrin-endodomain and ezrin-drug moieties were analyzed from the generated images.
3 Results and discussion
3.1 3D protein modelling
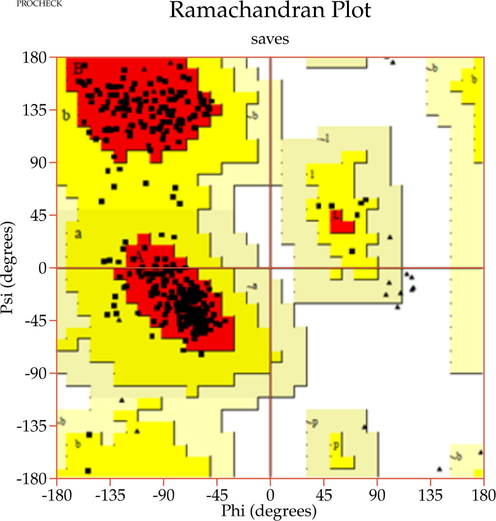
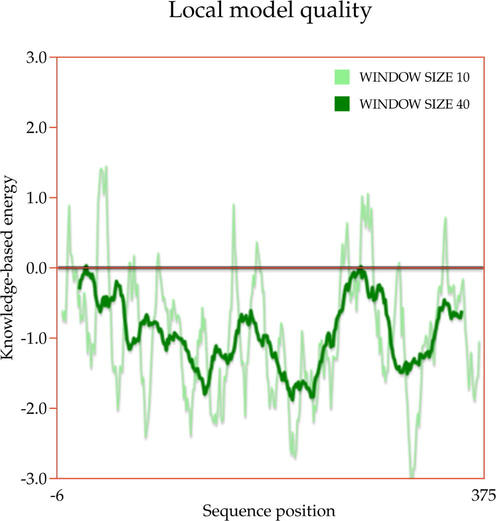
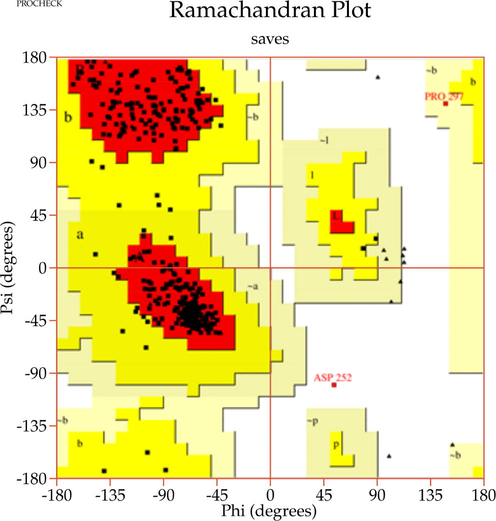
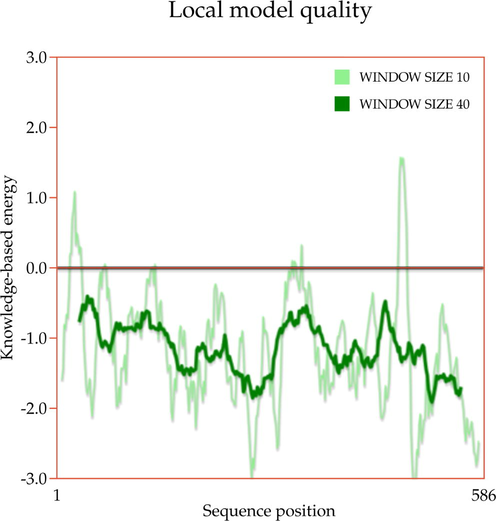
A template search for human actin protein through the SWISS-MODEL server listed 4EFH of Acanthamoeba Actin Complex with Spir Domain D, with a 92.5% amino acid sequence identity and 100% query coverage. The modelled structure (shown in Fig. 2a) was subjected to structural validation using a PROCHECK server Ramachandran plot analysis. As per the report, it showed 88.4% of residues in the most favored region, and 11.6% in the additional allowed regions. However, no residues were observed in the generously allowed and disallowed regions (shown in Supplementary Fig. 1a & b). Then, the partially available human ezrin protein (shown in Fig. 2b) from the Protein Data Bank with PDB ID: 4RM8 was energy minimized and submitted for Ramachandran Plot analysis. 92.5% of these residues were in the most favored region, 7.2% in the additional allowed region, no residues were in the generously allowed region, and 0.3% in the disallowed region. Based on a ProSA-Web server report, the local model quality of the actin and ezrin proteins was stable, with the graph running >0 value in both instances (shown in Supplementary Fig. 1c & d).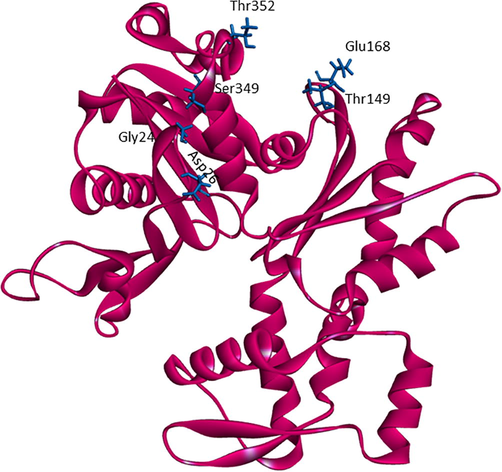
The modelled human actin protein in ribbon format showing the protein-protein interacting residues with labels.
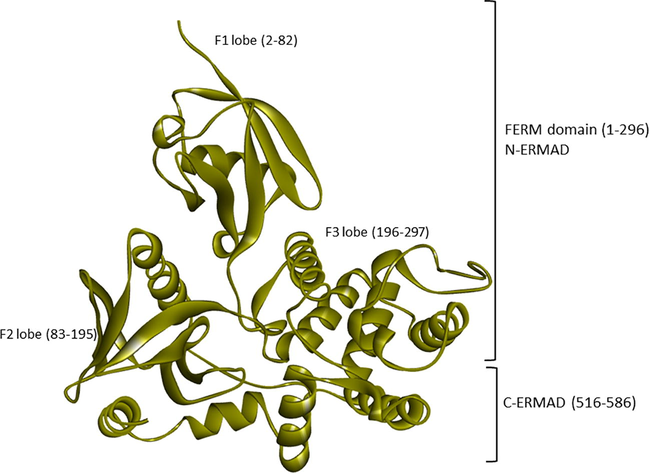
The crystal structure of the human ezrin protein with its distinct FERM (N-ERMAD) and C-ERMAD domains. Their lobes F1, F2 and F3 are labelled with their respective amino acids.
Next, the modelled endodomain of the SARS-CoV-1 using the I-TASSER with a C-score and an estimated TM-score of −2.13 and 0.46 ± 0.15 respectively, was selected for further analysis. The energy minimized modelled endodomain reported 21.2% residues in the most favored regions, 60.6% in additional allowed regions, 0.0% in the generously allowed region, and 3.0% in the disallowed region according to the Ramachandran plot. A ProSA-Web report also confirmed this short peptide region to be energetically stable (shown in Supplementary Fig. 1e & f). The 1–39 long generated endodomain model with its actual amino acid position of 1235–1273 was considered for bond analysis. As per the generated report, the 3D model of the viral endodomain displayed three disulphide bridges, between Cys9-Cys14, Cys2-Cys6 and Cys7-Cys19. Their images were generated using PyMol standalone software (PyMOL Molecular Graphics System, Schrödinger, LLC). These disulphide bridges were observed between Cl-1 and Cl-2 (Cys2-Cys6); Cl-2 and Cl-3 (Cys9 and Cys14) and Cl-2 and Cl-4 (Cys7 and Cys19). Thus, all 4 subdivisions of clusters interact with each other to give more stability to the endodomain from a structural perspective. Our 3D modelled viral endodomains of SARS-CoV-1 and SARS-CoV-2 are the first of their kind to be reported (shown in Fig. 2c). To confirm the disulphide bridge formation in the endodomains of SARS-CoV-1 and SARS-CoV-2 here we performed sequence search with the endodomains of viral protein. The listed hit was matured human hepcidin peptide involved in iron metabolism sequentially similar to the SARS-CoV-1 and SARS-CoV-2 endodomains, with conserved cysteine residues involved in disulphide bridge formation (Ehsani, 2020).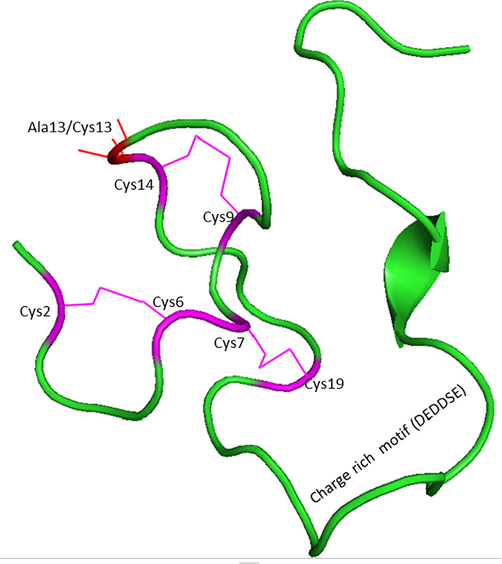
The modelled endodomain of the SARS-CoV-1 protein highlighting the three disulphide bridges (in pink) and the charge rich motif. The location of the substitution of Ala13 with Cys13 is shown with a red line. The substitution of Ala13 to Cys13 forms the endodomain of SARS-CoV-2.
3.2 Protein-protein docking
Based on the docking studies ezrin and actin proteins generated ten clusters with 4 poses each. The cluster was assessed with a HADDOCK score of −54.0 ± 6.1 and a buried surface area of 2602.8 ± 36.0 Å2. As per the PRODIGY report, the binding energy of ezrin with actin is −9.5 kcal/mol. The bond analysis revealed three salt bridges between ezrin and actin which include Arg569-Glu335, Glu582-Lys329, and Arg559-Glu168. Additionally, thirteen hydrogen bonds were observed between the ezrin and actin proteins which stabilized their interactions (shown in Fig. 3a and in Table 1). The ezrin-actin protein docking agrees with the literature wherein actin prefers to bind within the 34 amino acids (Ile553-Leu586) of the C-terminal region. Additionally, interactions expand further into C-ERMAD (Thr533, Ser536, Arg542, and Glu525). Three salt bridges assist in stabilizing these interactions which are located within the C-terminal region. With respect to the ezrin-SARS-CoV-1 docking, the HADDOCK score is −17.4 ± 15.6 with a buried surface area of 1934.4 ± 32.6 Å2. Furthermore, the HADDOCK score of the endodomain of SARS-CoV-2 with the ezrin protein is −24.5 ± 6.7 with a buried surface area of 1835.4 ± 74.5 Å2. To conclude, the binding affinity between the endodomains and ezrin revealed that the binding of the SARS-CoV-1 endodomain was stronger with a larger binding surface area than that of SARS-CoV-2. The 99 Å2 difference in surface area may assist in binding the host with the viral protein. According to the available literature, a containment in infection at the entry stage was reported by Millet et al., 2012. This could be due to the higher binding affinity and larger binding surface area, as concluded by these docking studies. Thus, the substitution of alanine with cysteine between SARS-CoV-1 and SARS-CoV-2 due to the mutation plays a vital role in the overall change in the binding pattern, wherein ezrin prefers lesser interaction with the cysteine cluster residues in SARS-CoV-1, which could assist through a better protein-protein interaction with a larger surface area.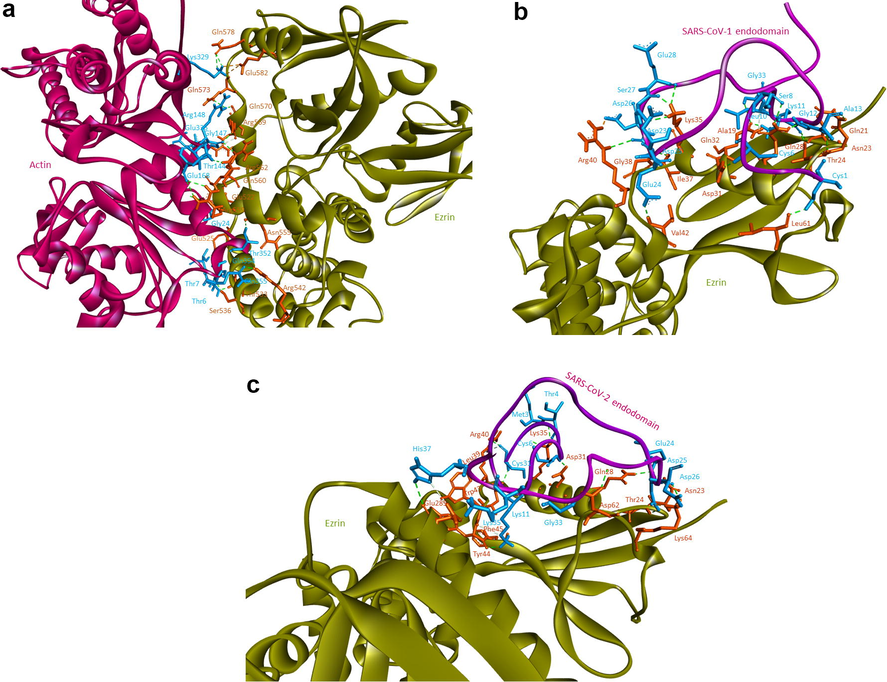
Protein-protein docking performed using HADDOCK software: 3(a) human ezrin and actin protein interacting with each other, 3(b) ezrin protein interacting with the endodomain of SARS-CoV-1, and (3c) ezrin protein interacting with the endodomain of SARS-CoV-2.
As per the PRODIGY report, the binding affinities of SARS-CoV-1 and SARS-CoV-2 were −14.2 kcal/mol and −13.7 kcal/mol respectively. Based on the docking report, the endodomain of SARS-CoV-1 exhibited a stronger affinity than that of SARS-CoV-2 (shown in Table 2). The substitution of Ala 13 in SARS-CoV-1 with Cys1247 in SARS-CoV-2 confers a considerable change in the binding pattern of viral endodomains with ezrin. From the analyses, it’s observed that the Ala13 prefers to interact only with Asn23, which after substitution with Cys13 in SARS-CoV-2 binds with Lys64 apart from Asn23 (shown in Fig. 3b and c). An overall change in interaction patterns between the SARS-CoV-1 and SARS-CoV-2 endodomains against the ezrin protein due to the amino acid substitution is established through their amino acids’ interaction via salt bridges and cysteine cluster interactions. Salt bridge analysis confirms that four salt bridges were observed between ezrin-SARS-CoV-1 and six salt bridges between ezrin-SARS-CoV-2. Additionally, cysteine clusters 1 and 2 of the endodomain of SARS-CoV-1 interact with the F1 lobe of the ezrin protein, whereas cysteine clusters 1, 2, and 3 of the endodomain of SARS-CoV-2 interact with the human ezrin protein. Compared to salt bridges, cysteine cluster interaction plays a crucial role in ezrin-endodomain interaction. Two cysteine clusters interacting between ezrin-SARS-CoV-1 and four cysteine clusters interacting between ezrin-SARS-CoV-2 have been observed, which may have positive effects on the interactions between the viral endodomain and the human ezrin protein (shown in Table 2). These additional cysteine cluster interactions and the ezrin protein may play an important role in reduced membrane fusions. In contrast, an increase in the number of cysteine cluster interactions between SARS-CoV-2 and ezrin has decreased the binding affinity between these two subunits, also considerably reducing their overall binding surface area. This lower level of affinity between the ezrin and the endodomain could negatively modulate the entry of the virus.
Human Ezrin
SARS-CoV-1 (Endodomain)
Human Ezrin
SARS-CoV-2 (Endodomain)
Lys35 (F1)
Asp23
Asn23 (F1)
Gly12
Arg40 (F1)
Asp26 (Charged cluster)
Arg40(F1)
Asp26 (Charged cluster)
Gln21 (F1)
Lys11
Arg40 (F1)
Asp25 (Charged cluster)
Asn23 (F1)
Ala13
Gln21 (F1)
Lys11
Thr24 (F1)
Lys11
Asn23 (F1)
Cys14 (Cysteine cluster 3)
Gln28 (F1)
Gly12
Thr24 (F1)
Lys11
Gln28 (F1)
Ser8
Thr25 (F1)
Cys6 (Cysteine cluster 2)
Lys35 (F1)
Ser27 (Charged cluster)
Lys35 (F1)
Ser27 (Charged cluster)
Lys35 (F1)
Glu28 (Charged cluster)
Gly38 (F1)
Asp25 (Charged cluster)
Ile37 (F1)
Asp25 (Charged cluster)
Gln21 (F1)
Leu36
Gly38 (F1)
Asp25 (Charged cluster)
Gln28 (F1)
Cys6 (Cysteine cluster 2)
Arg40 (F1)
Asp25 (Charged cluster)
Lys35 (F1)
Asp25 (Charged cluster)
Val42 (F1)
Glu24(Charged Cluster)
Lys35 (F1)
Glu24 (Charged cluster)
Leu61 (F1)
Cys1 (Cysteine cluster 1)
Lys35 (F1)
Glu28 (Charged cluster)
Asp31 (F1)
Cys6 (Cysteine cluster 2)
Gly38 (F1)
Asp25 (Charged cluster)
Ala19 (F1)
Lys11
Arg40 (F1)
Asp25 (Charged cluster)
Gln32 (F1)
Lys11
Lys64 (F1)
Cys13
Asn23 (F1)
Ala13
Leu61 (F1)
Cys1 (Cysteine cluster 1)
Gln32 (F1)
Gly33
Ala19 (F1)
Lys11
Gln28 (F1)
Leu10
Gln32 (F1)
Lys11
3.3 Protein-ligand docking
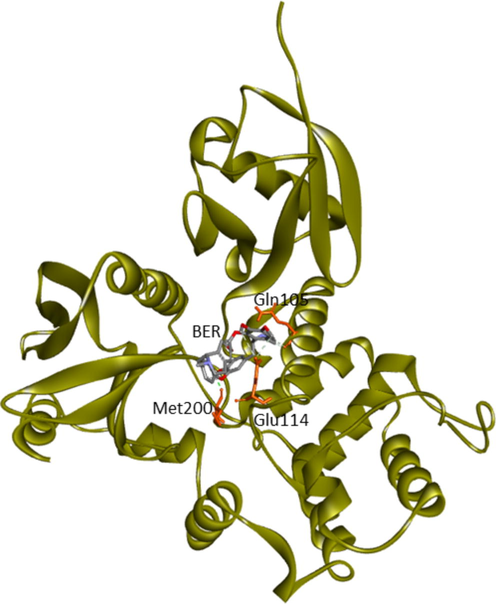
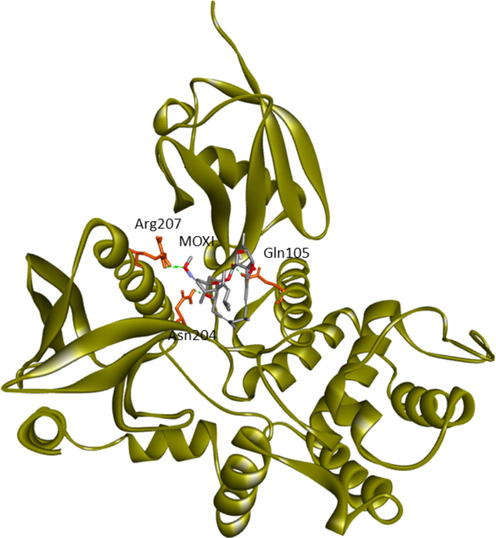
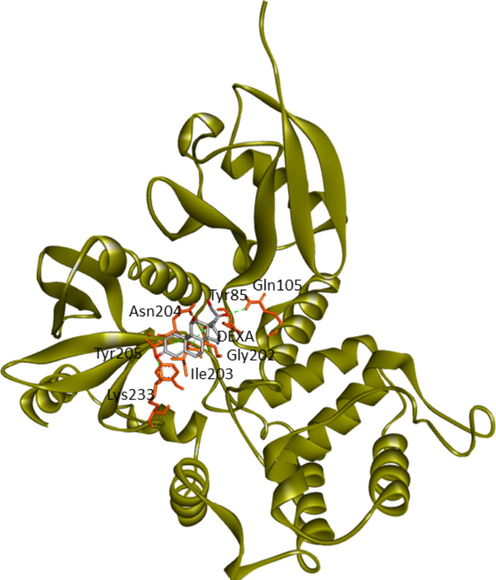
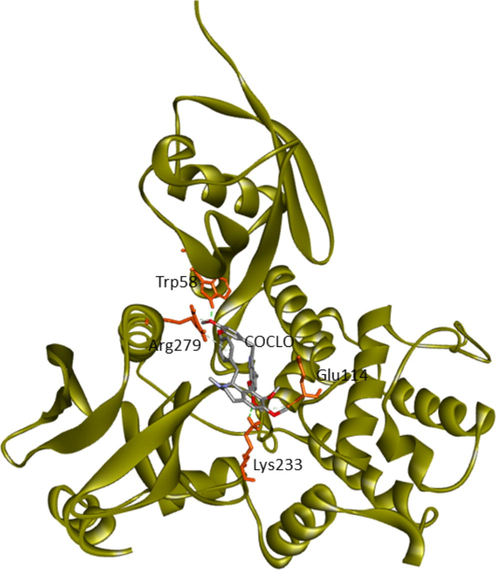
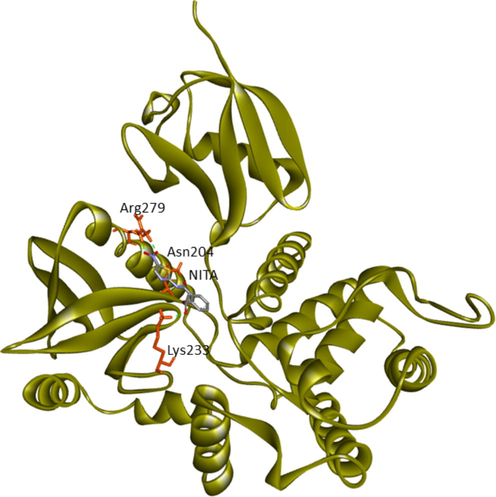
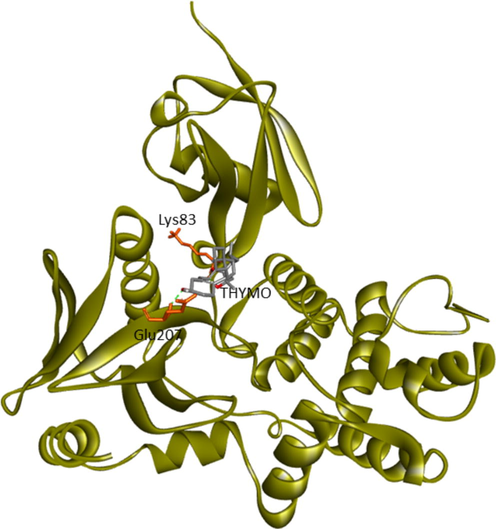
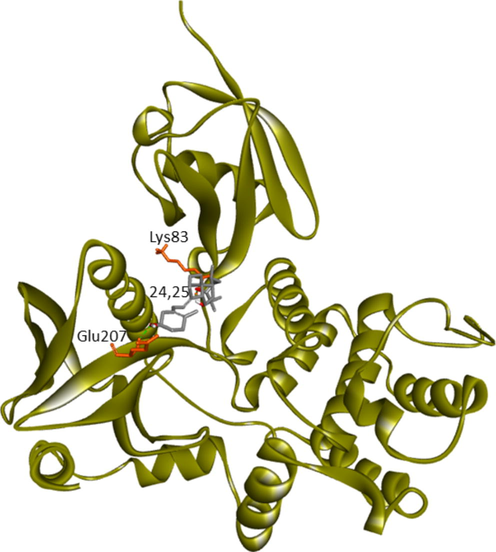
Current research provided further validation of the importance of ezrin regarding the infection processes in SARS-CoV-2, along with elucidation as to its interactions with drug molecules. It seemed prudent to attempt to discern the potential value of various drug repurposing candidates that may be applied as ezrin-targeted therapies, to assist further research and clinical validation. Forty-six targeted compounds were docked against ezrin, of which seven showed promising binding affinities in conjunction with lower inhibition constants. The poses with a better binding affinity and inhibition constant (µM) value of <2 µM include ivermectin, minocycline, selamectin, calcifediol, calcitriol, ergocalciferol, and quercetin, with a binding affinity of −8.12, −8.16, −7.81, −7.83, −7.91, −7.81, and −7.85 kcal/mol respectively (shown in Fig. 4a–g). Next, the chemical compounds with inhibition constant values (µM) of between >2 and <3 were selected. These include berbamine, moxidectin, dexamethasone, coclobine, nitazoxanide, thymoquinone, and 24,25-dihydroxycholecalciferol, with binding energies of −7.75, −7.69, −7.56, −7.61, −7.7, −7.68 and −7.56 kcal/mol respectively (shown in Supplementary Fig. S2 and in Table 3). Docked complexes were able to associate with these two distinct binding sites, site1 (F1, F2, and F3), and site2 (F3), observed in the ezrin protein. Site1 with lobes F1, F2, and F3 was preferred by coclobine, ivermectin, selamectin, and minocycline of which lobe F1 is the viral endodomain binding site. Dexamethasone, moxidectin, 24,25-dihydroxycholecalciferol, berbamine, quercetin, and thymoquinone restrict their interactions to within lobes F2 and F3. Nitazoxanide, calcifediol, calcitriol, and ergocalciferol interact only with lobe F3. 32 other drug molecules with lower binding affinities and higher inhibition constant values were not afforded further analysis in this study (refer to Supplementary Table 2).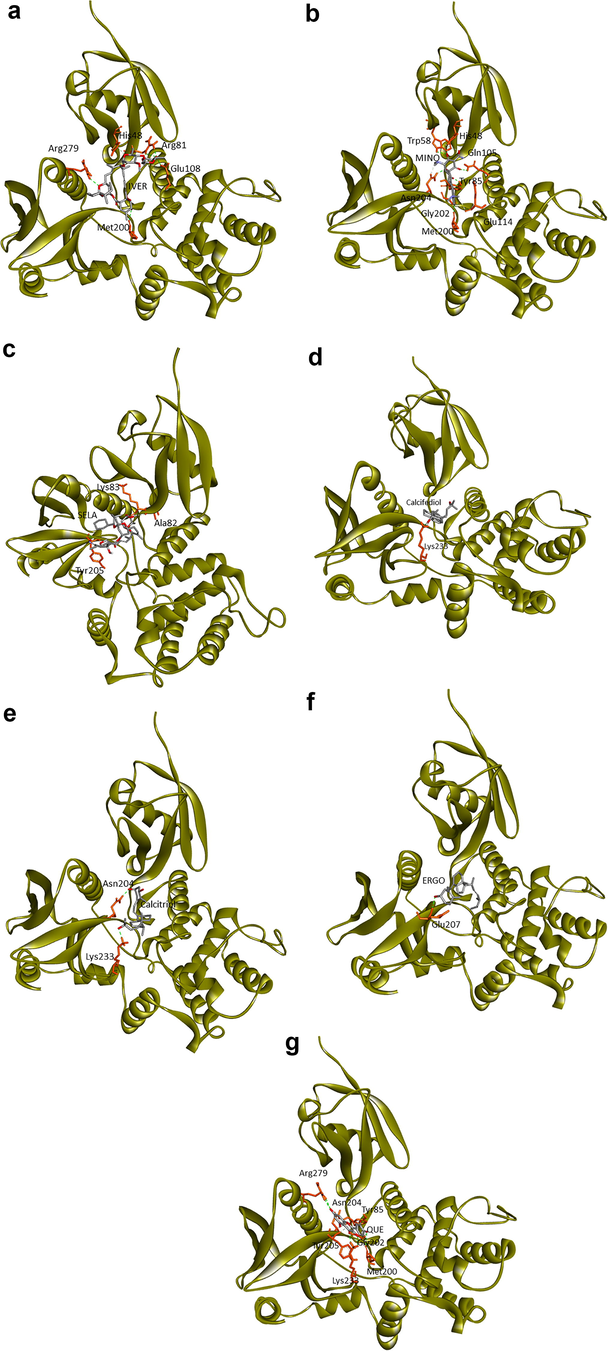
Protein-ligand docking of human ezrin protein with drug molecules: 4(a) ivermectin, 4(b) minocycline, 4(c) selamectin, 4(d) calcifediol, 4(e) calcitriol, 4(f) ergocalciferol, and 4(g) quercetin.
Drug
Binding affinity (kcal/mol)
Inhibition constant (μM)
Amino acids
Berbamine
−7.75
2.09
Gln105 (Lobe F2), Glu114 (Lobe F2), Met200 (Lobe F3)
Ivermectin
−8.12
1.11
His48, Arg81 (Lobe F1), Glu108 (Lobe F2), Arg279, Met200 (Lobe F3)
Minocycline
−8.16
1.04
His48, Trp58 (Lobe F1), Tyr85, Gln105, Glu114 (Lobe F2), Gly202, Asn204, Met 200 (Lobe F3)
Moxidectin
−7.69
2.29
Gln105 (Lobe F2), Asn204, Arg279 (Lobe F3)
Selamectin
−7.81
1.89
Ala82, Lys83 (Lobe F1), Tyr205 (Lobe F3)
Dexamethasone
−7.56
2.85
Tyr85, Gln105 (Lobe F2), Gly202, Ile203, Asn204, Tyr205, Lys233 (Lobe F3)
Coclobine
−7.61
2.66
Trp58 (Lobe F1), Glu114 (Lobe F2), Lys233, Arg279 (Lobe F3)
Calcifediol
−7.83
1.81
Lys233 (Lobe F3)
Calcitriol
−7.91
1.59
Asn204, lys233 (Lobe F3)
Ergocalciferol
−7.81
1.87
Glu207 (Lobe F3)
Thymoquinone
−7.68
2.34
Lys83, Glu207 (Lobe F2, F3)
Nitaoxanide
−7.7
2.28
Asn204, Lys233, Arg279 (Lobe F3)
24,25-Dihydroxycholecalciferol
−7.56
2.88
Lys83, Glu207 (Lobe F2, F3)
Quercetin
−7.85
1.77
Tyr85 (Lobe F2), Met200, Gly202, Asn204, Tyr205, Lys233, Arg279 (Lobe F3)
Quinoline is considered as a reference drug which has experimental approval as inhibitor for ezrin protein (Bulut et al., 2012). However, it showed a binding score of -5.76 kcal/mol with an inhibition constant of 59.7 (µM), much lower than that of the top seven drug molecules like ivermectin, minocycline, selamectin, calcifediol, calcitriol, ergocalciferol, and quercetin. Clinical studies and validation should follow up on these findings to further validate the potential efficacy of these ezrin-interacting drug candidates in treating COVID-19 infection.
3.4 Molecular dynamics simulation
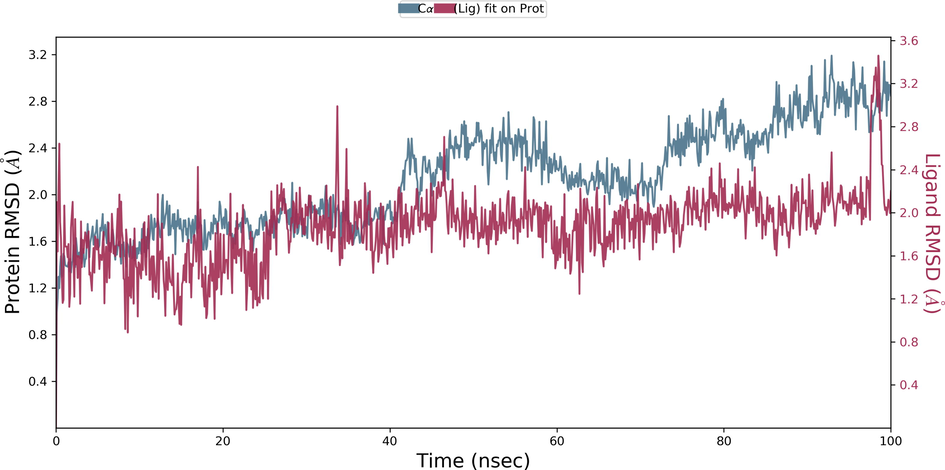
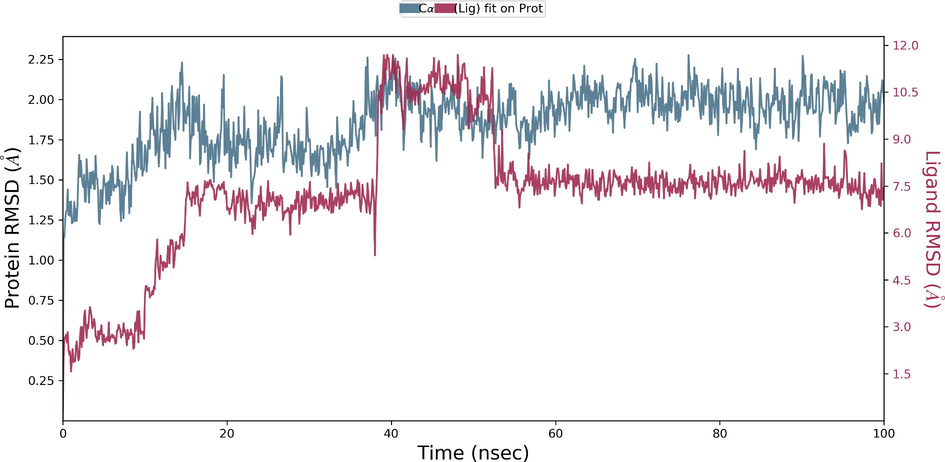
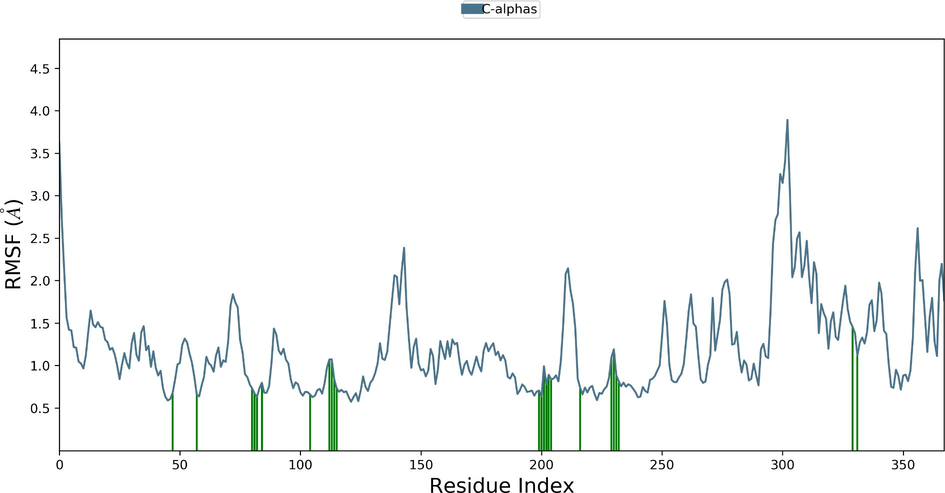
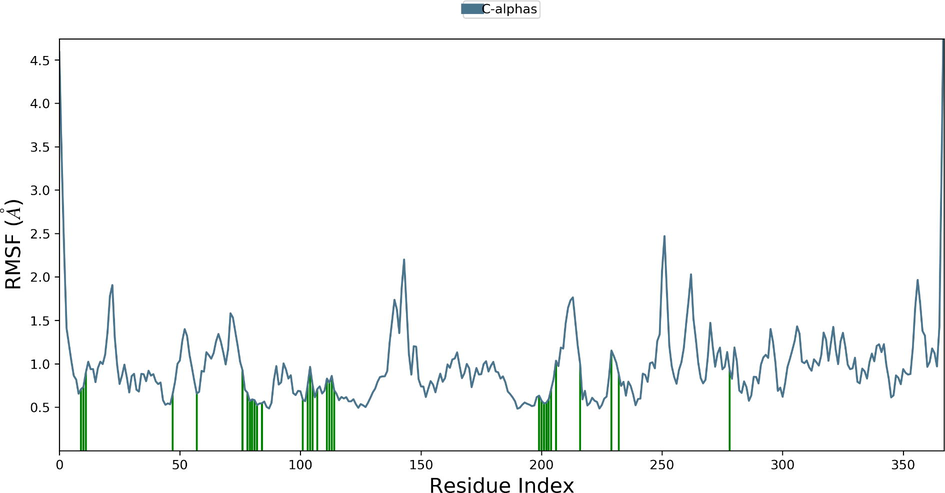
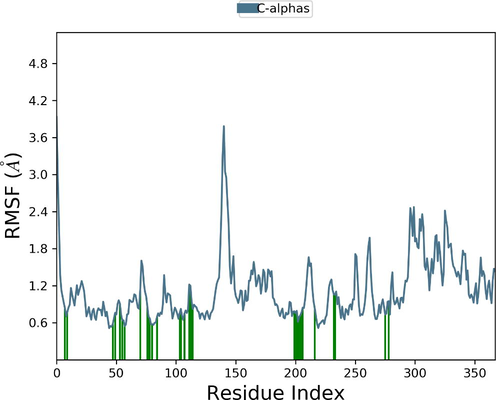
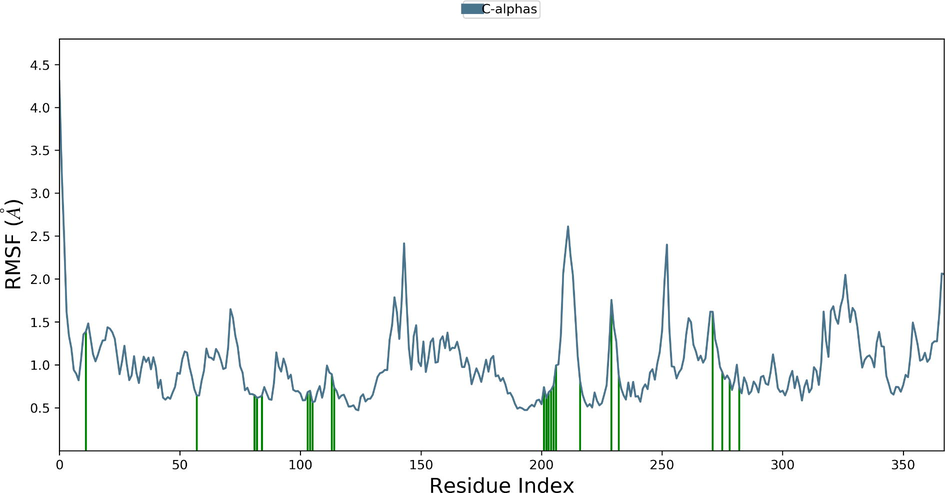
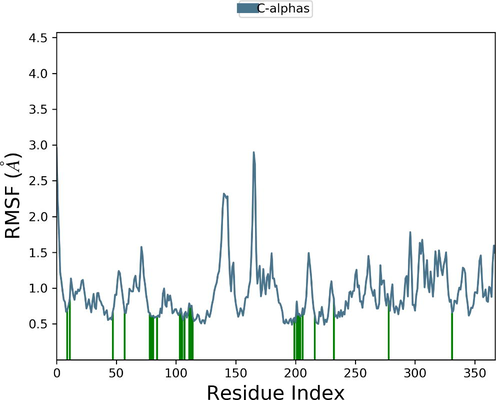
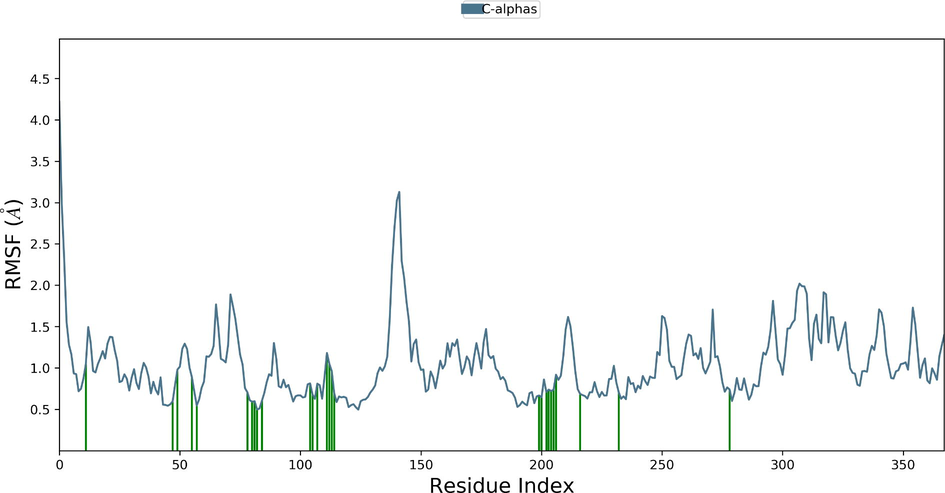
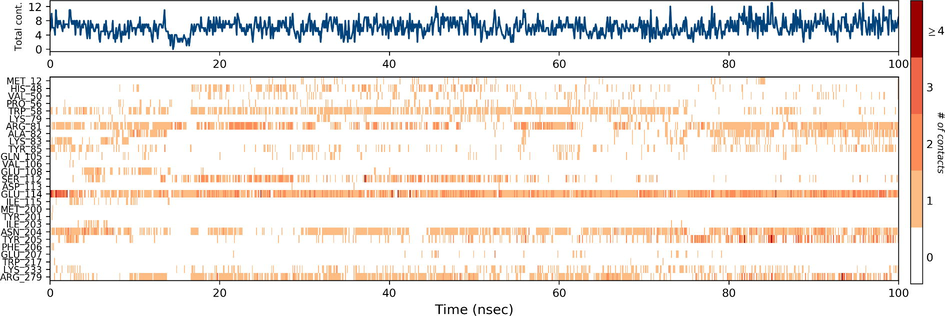
Desmond molecular dynamics package was used to validate the structural stability of the docked complex of the ezrin-SARS-CoV-1 for the time scale of 100 ns. The overall RMSD of the SARS-CoV-1 endodomain-ezrin complex is maintained between 3.0 Å and 3.5 Å (Fig. 5a). The RMSD of the SARS-CoV-2 endodomain ezrin complex showed the RMSD score of 3.0–4.5 Å (Fig. 5b). The RMSF plot analysis of SARS-CoV-1 confirms the higher stability of the ezrin protein and higher flexibility of the endodomain (Fig. 5c). The endodomain of SARS-CoV-2 confirms a higher flexibility of the ezrin protein with the rigidity of the endodomain (Fig. 5d). The substitution of Ala with Cys in SARS-CoV-2 therefore conveys a dramatic effect on their stability. This is reflected in their protein-protein interaction wherein the ezrin-SARS-CoV-1 scored better with a larger interface area compared to that of the ezrin-SARS-CoV-2.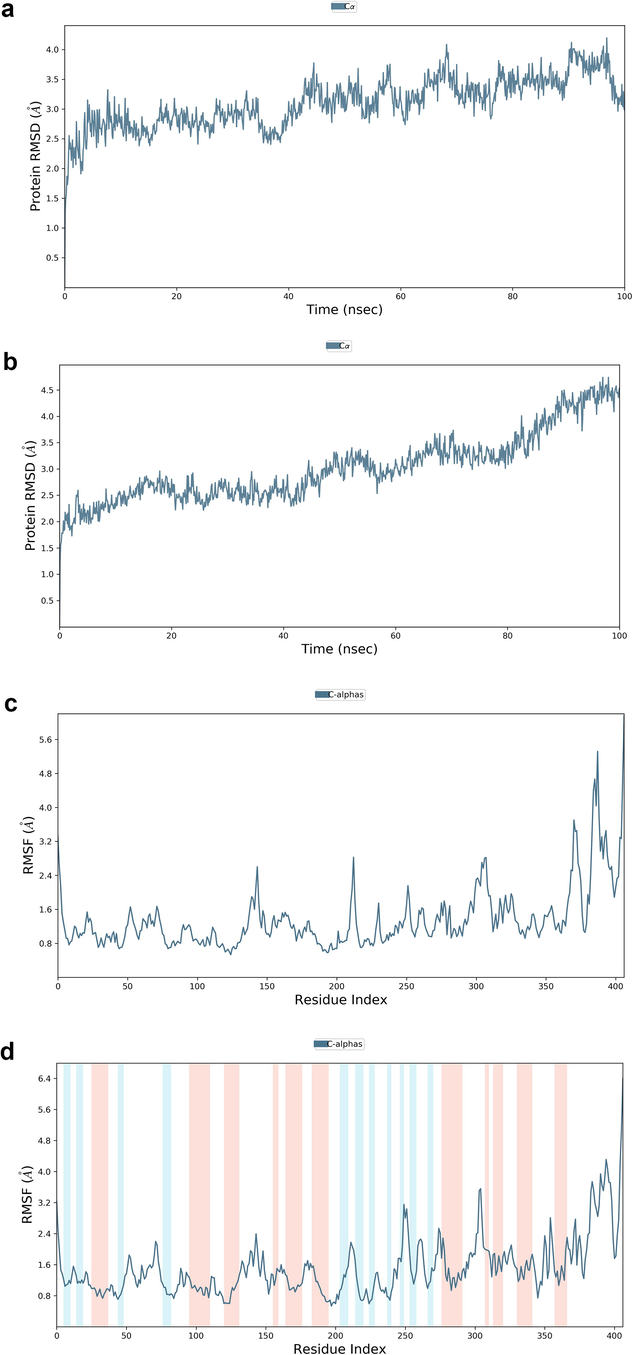
Root Mean Square Deviations and Root Mean Square Fluctuations: 5(a) The RMSD of SARS-CoV-1, 5(b) the RMSD of the SARS-CoV-2, 5(c) RMSF of SARS-CoV-1, 5(d) RMSF of SARS-CoV-2.
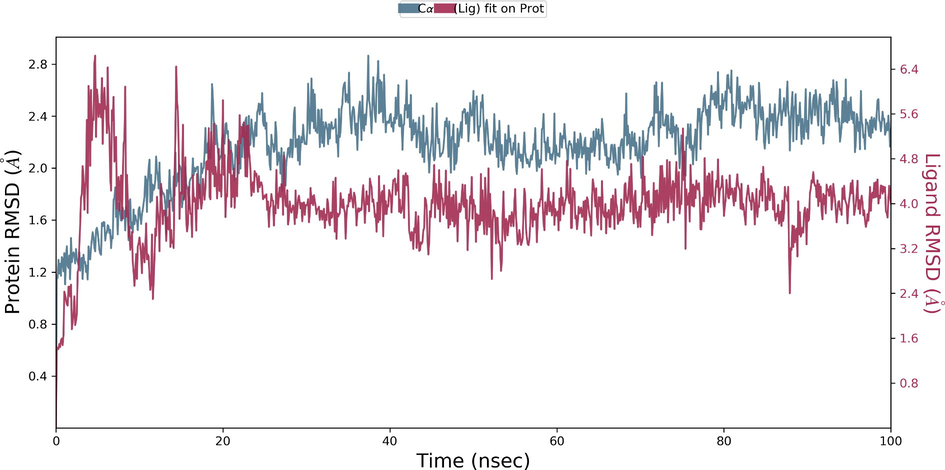
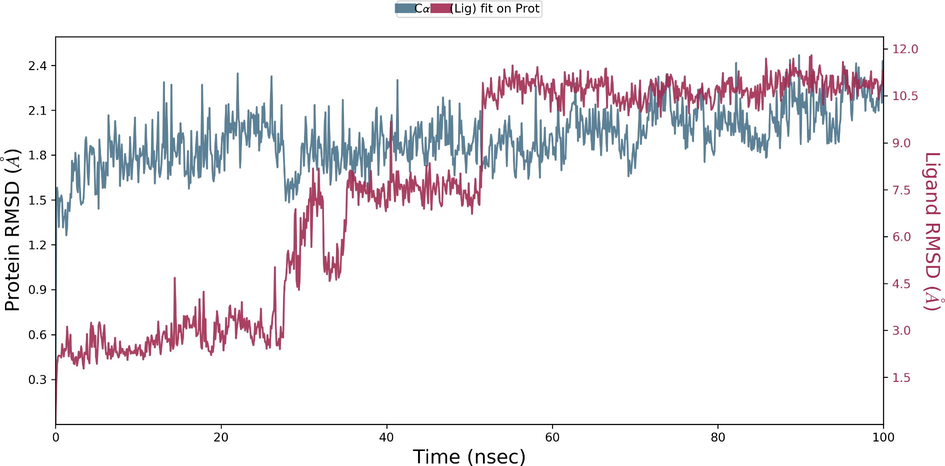
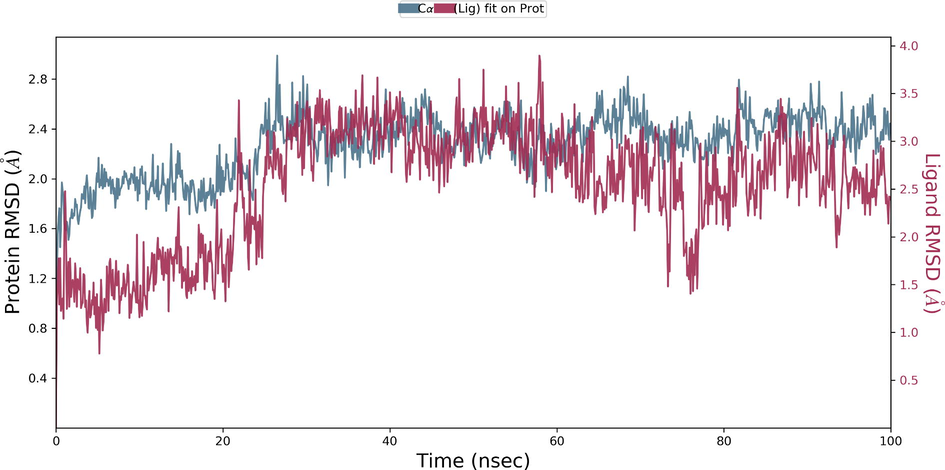
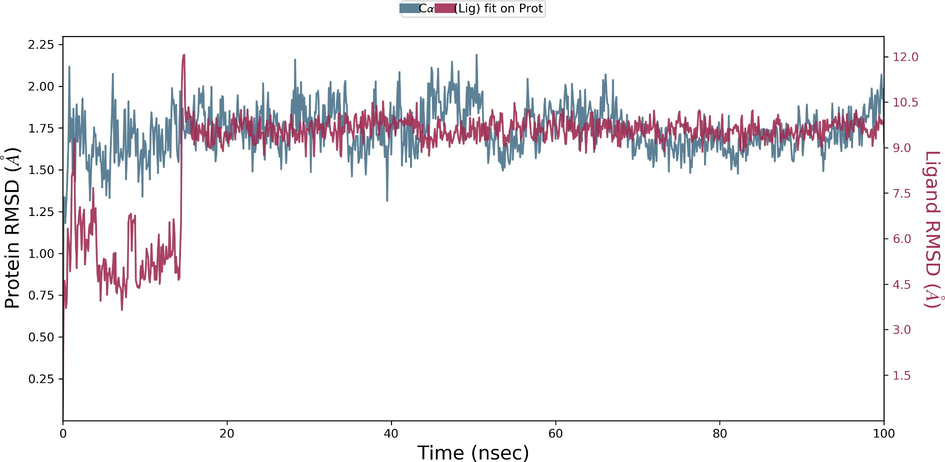
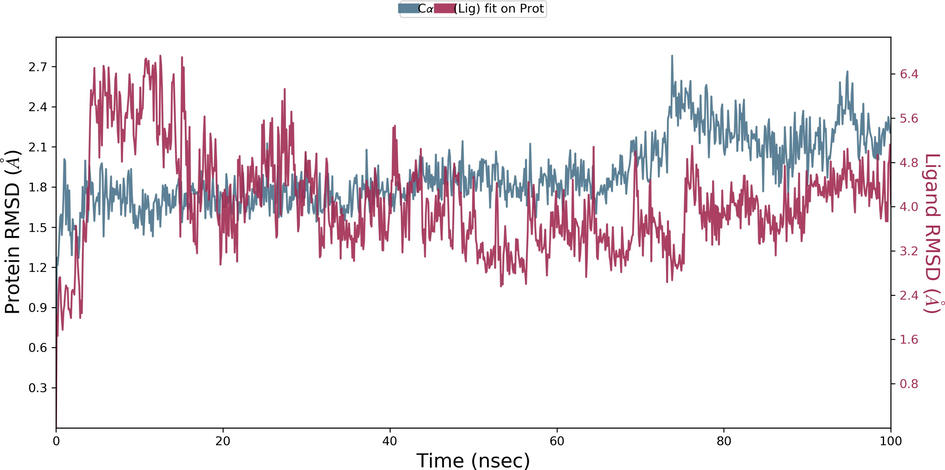
During the protein-ligand simulation, the RMSD score of selamectin in complex with human ezrin protein maintained the stability of the protein starting from 5 ns to 70 ns at 1.8 Å. Calcitriol scored between 1.75 Å and 2.00 Å indicating their tightly bound state within the cavity till the end of the simulation. Ivermectin maintains 2.0 Å-2.5 Å. Similarly, minocycline maintains 2.4 Å with their strong binding starting from 25 ns till the end of the simulation. Even though, calcifediol fluctuates between 2.0 Å −2.8 Å whereas the drug maintains its stability within 2.0 Å from 25 ns till the end. Larger conformational change was observed with the binding of ergocalciferol. As a result, the drug could not maintain contact within the binding site. Thus, they showed a higher RMSD value compared to that of the ezrin protein. Quercetin exhibits stable conformation throughout the course of simulation at 1.75 Å (Supplementary Fig. S3a–g). Based on the RMSF plot of ezrin protein, the F1 lobe displayed rigidity after the binding of ivermectin which becomes highly flexible after the interaction of minocycline. Moderate flexibility of the F1 lobe is reported after the interaction of selamectin, calcitriol, calcifediol, quercetin, and ergocalciferol. F2 lobe shows higher flexibility after the interaction of selamectin, quercetin, and ivermectin. Regarding the F3 lobe, ergocalciferol binding enhances their flexibility which is followed by the binding of the calcifediol interaction. However, the binding of ivermectin, quercetin, and calcitriol restricts the mobility of the F3 lobe (Supplementary Fig. S4a–g).
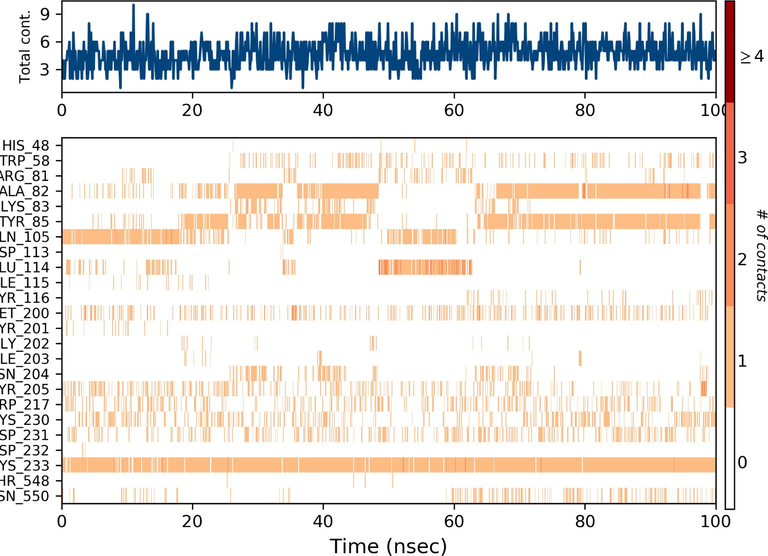
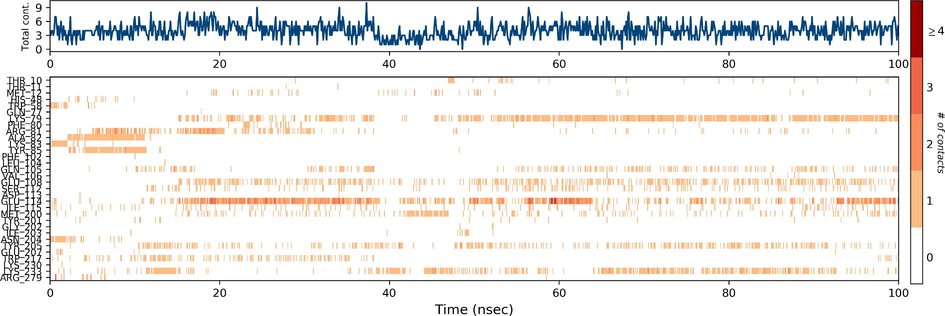
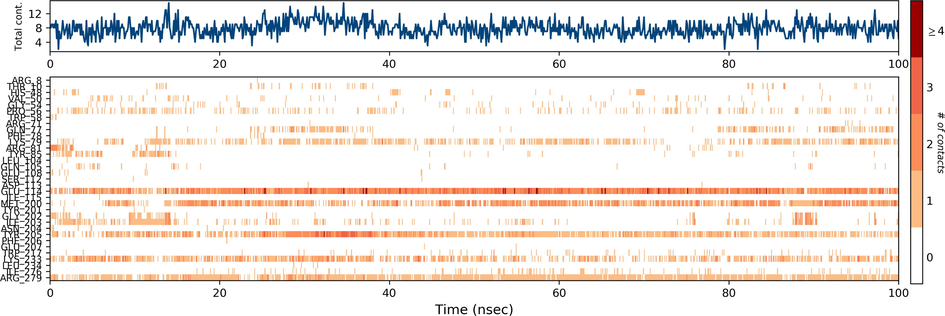
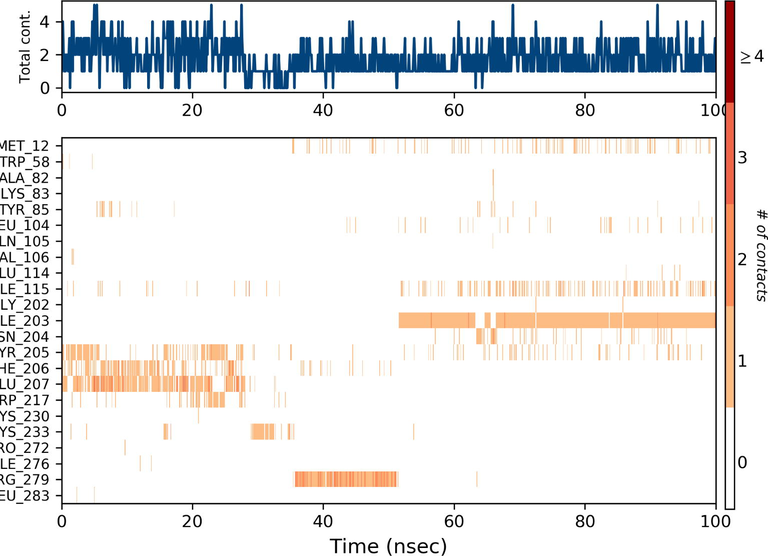
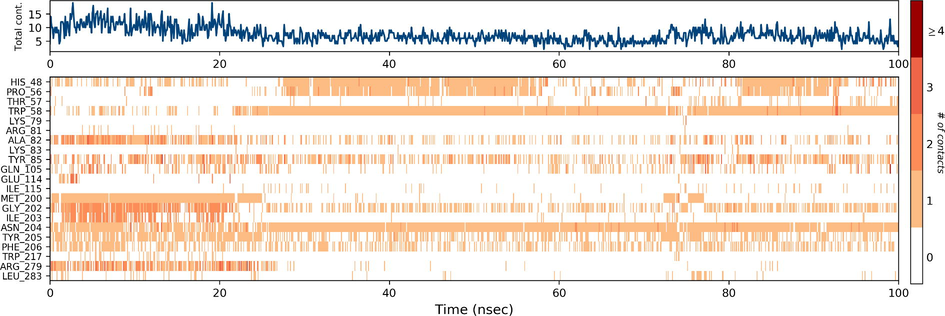
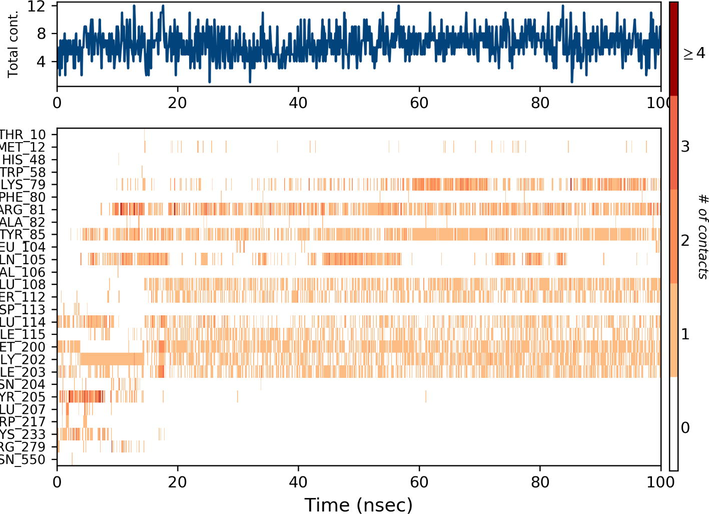
Ezrin interaction with the seven drug molecules can be monitored through the simulation. To begin with, calcifediol displays a positively charged interaction of Lys233 with ezrin. Their interaction with the protein during simulation is mentioned in percentage (Fig. 6a–g). For Lys233 it is 94%. Two hydrophobic interactions of Ala82 and Tyr85 scored 49% and 41% respectively. Calcitriol shows a positive and negative charge-based interaction of Lys79 (41%) and Glu114 (26%). Ivermectin displayed a negative interaction wherein Glu114 (69%) followed by Arg279(56%) and Lys233(40%). The protein-ligand contact report of minocycline with ezrin confirm two polar interactions of His48 (37%) and Asn204 (80%). Two hydrophobic interactions viz. Trp58 (73%) and Pro56 (34%) were observed. Also, a water bridge between Gly202 (35%) and ezrin was observed. Negatively charged interaction between quercetin and ezrin was observed between Glu108 (46%) and ezrin. Arg81(35%) showed a positive interaction with ezrin. Three hydrophobic interactions (Tyr85 (56%), Met200 (55%) and Ile203 (42%)) was observed. A single water bridge Gly202 (56%) was also observed. Selamectin showcased two positive charged interactions of Arg81 (51%) and Arg279 (51%). A negative interaction of Glu114 (71%) was observed. A polar interaction of Asn204 (45%) was also observed during the simulation. Ergocalciferol displayed a single hydrophobic interaction of Ile203 (45%)(Fig. 7a–g) (Supplementary Fig. S5a–g). The average total energy of the protein-ligand complex was calculated using the Simulation Quality Analysis tool which confirm quercetin to have a total average energy score of −134124.289 kcal/mol followed by minocycline to have the average total energy score of −134089.225 kcal/mol followed by calcifediol with an average total energy score of −134024.699 kcal/mol. Calcitriol shows a score of −133997.913 kcal/mol followed by ergocalciferol with a score of −133984.27 kcal/mol. Both selamectin and ivermectin showed an average energy score of −133741.2 kcal/mol and −133613.193 kcal/mol respectively. Drugs like quercetin, minocycline, calcifediol, and calcitriol showed better total average scores as per their order.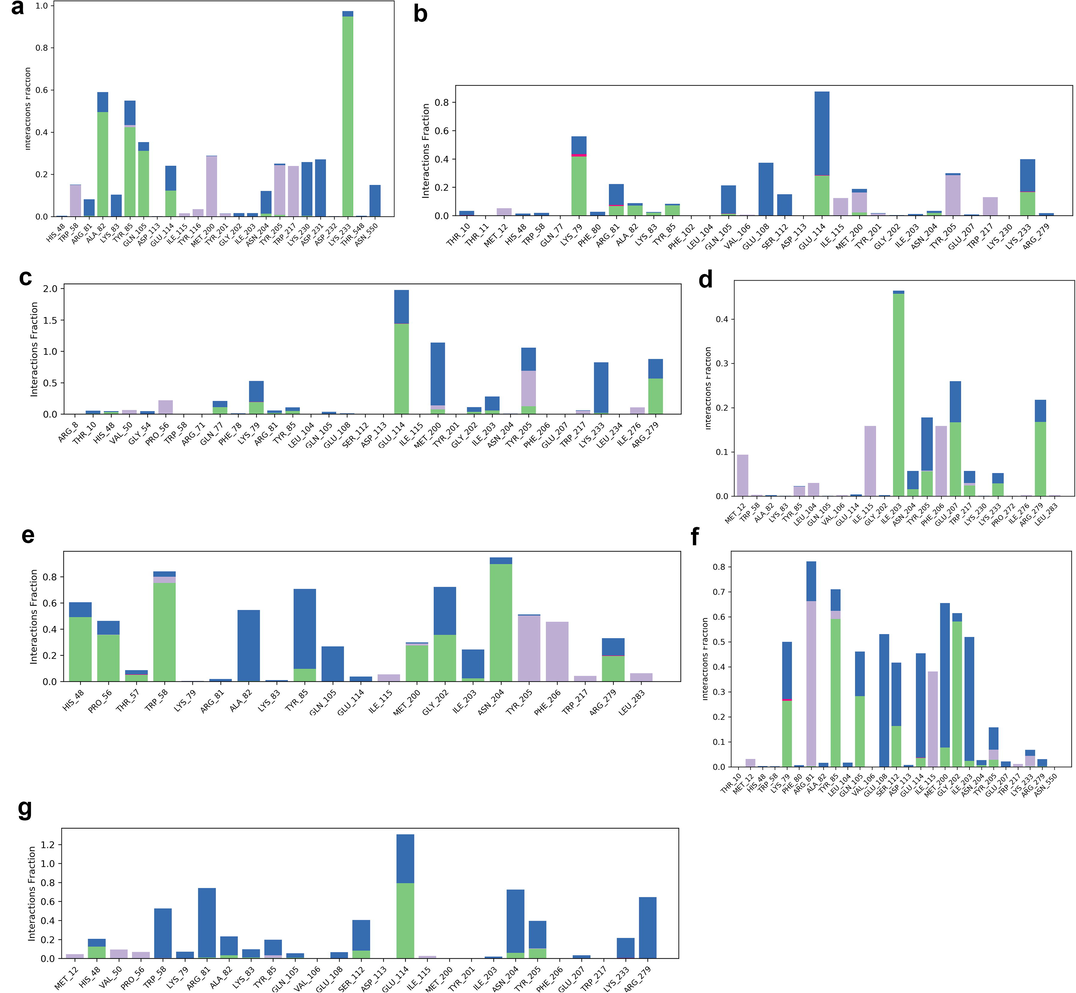
Protein-Ligand contact histogram: 6(a) Calcifediol, 6(b) Calcitriol, 6(c) Ivermectin, 6(d) Ergocalciferol, 6(e) Minocycline, 6(f) Quercetin, 6(g) Selamectin.
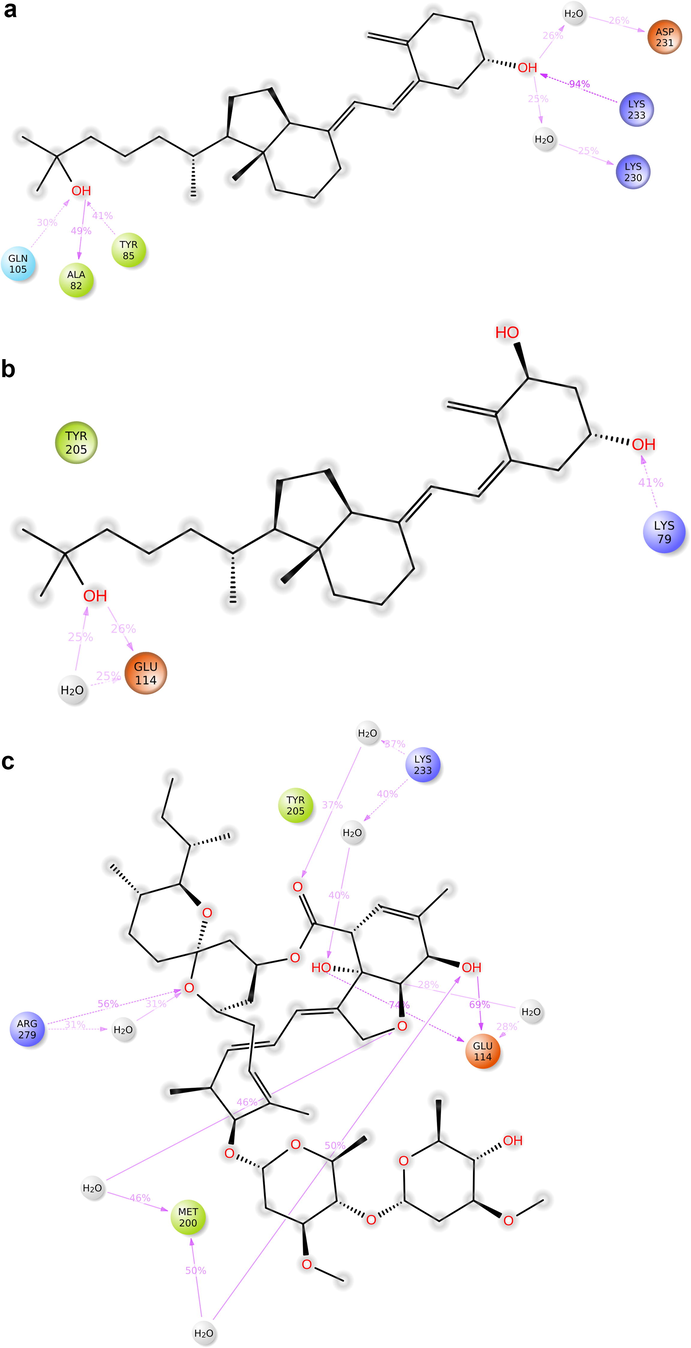
Protein-ligand contact 2D: 7(a) Calcifediol, 7(b) Calcitriol, 7(c) Ivermectin, 7(d) Ergocalciferol, 7(e) Minocycline, 7(f) Quercetin, 7(g) Selamectin.
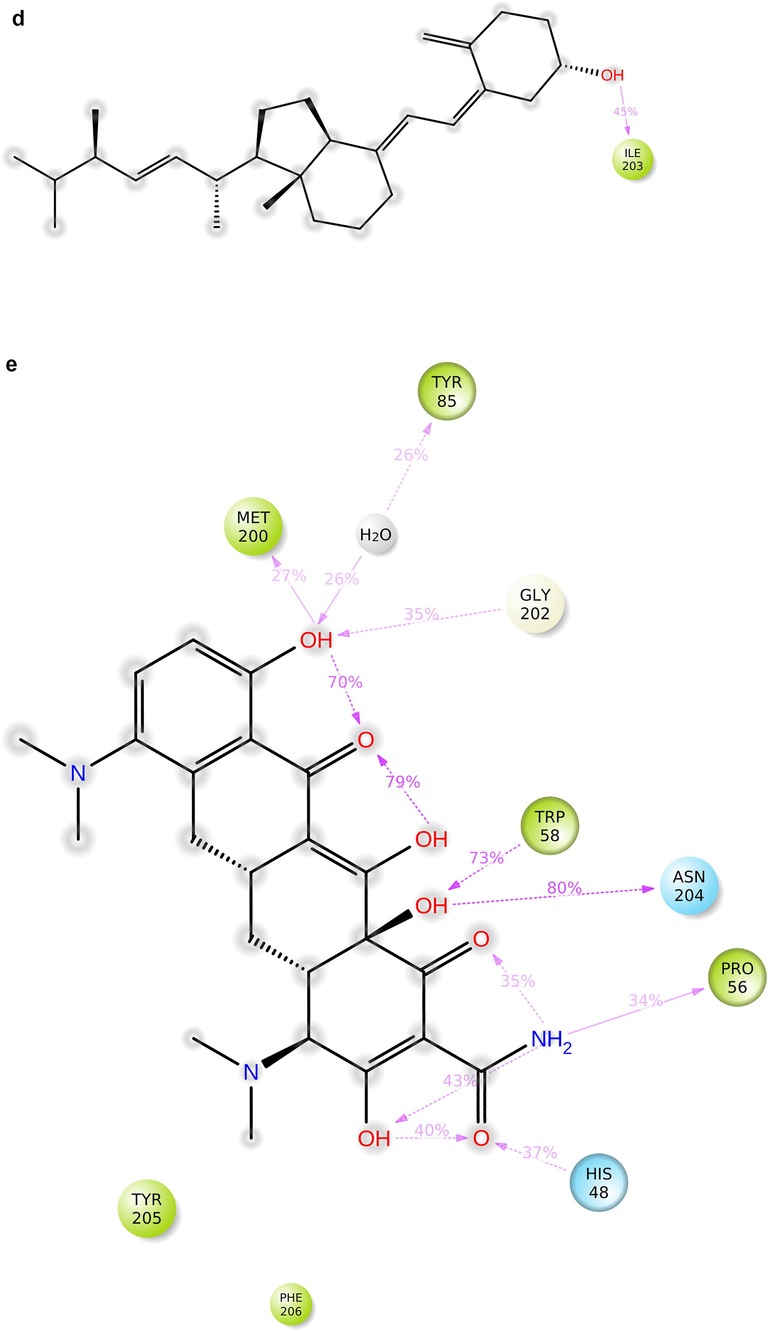
Protein-ligand contact 2D: 7(a) Calcifediol, 7(b) Calcitriol, 7(c) Ivermectin, 7(d) Ergocalciferol, 7(e) Minocycline, 7(f) Quercetin, 7(g) Selamectin.
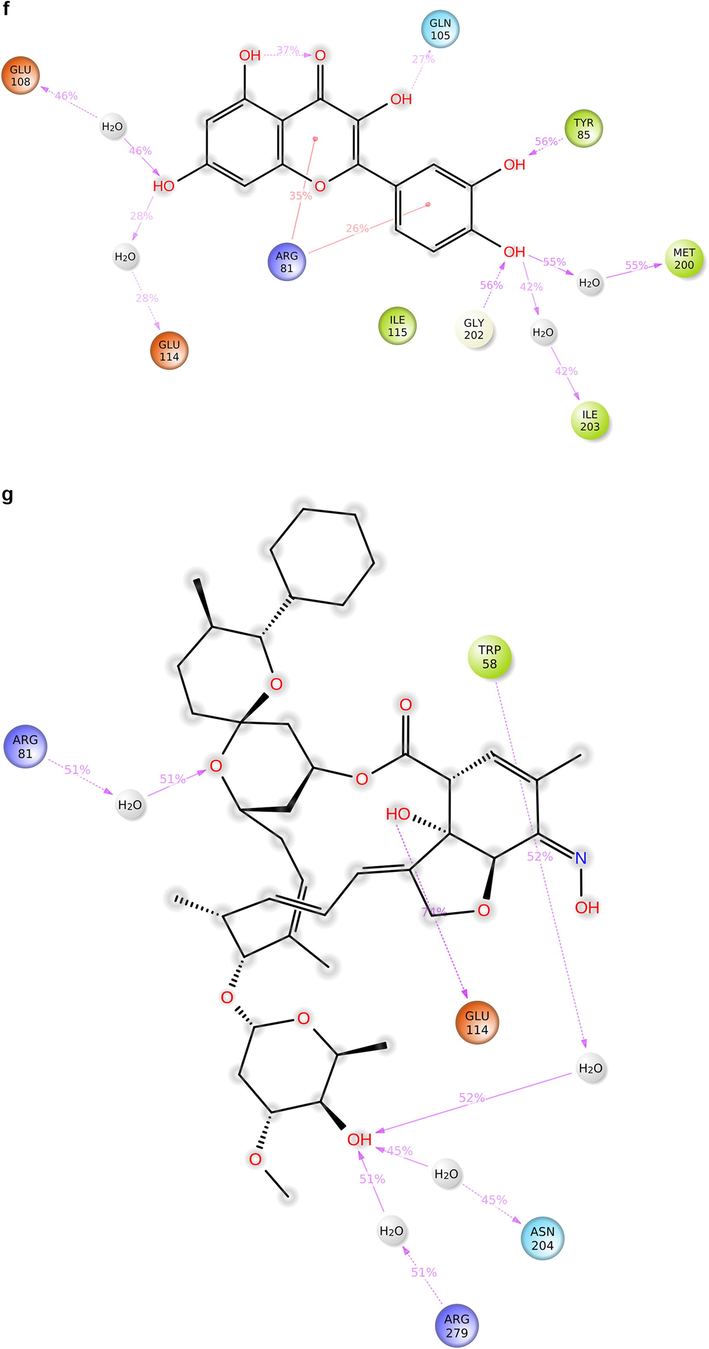
Protein-ligand contact 2D: 7(a) Calcifediol, 7(b) Calcitriol, 7(c) Ivermectin, 7(d) Ergocalciferol, 7(e) Minocycline, 7(f) Quercetin, 7(g) Selamectin.
4 Conclusions
A paucity of structural details of the SARS-CoV-1 and SARS-CoV-2 viral endodomains warranted the modelling and docking of these viral peptides with ezrin protein. Three disulphide bridges stabilize the endodomain. The SARS-CoV-1 endodomain demonstrates strong interaction with ezrin protein compared to SARS-CoV-2 due to the presence of alanine. This study establishes the role of a cysteine cluster of viral endodomain in interaction with ezrin protein, a property that likely assists SARS-CoV-2 with its observed unrestricted entry of fusion and S dependent infection. Finally, seven drugs were able to show strong binding with ezrin of which six of them bind within the pocket during simulation. This mechanism assists the stability of the ezrin protein and may block the entry of the viral endodomain. Future studies may further proceed to clinical trials aiming to identify potential drug molecules which can treat COVID-19 infection.
CRediT authorship contribution statement
Selvaa Kumar Chellasamy: Conceptualization, Validation. Eleanor Watson: Conceptualization, Investigation, Writing – review & editing.
Acknowledgements
Overall technical support was provided by The School of Biotechnology and Bioinformatics, D.Y. Patil Deemed to be University, CBD Belapur. Publication support was provided by the University of Gloucestershire. Media and dissemination support has kindly been offered by The Atlantic Council. Both the authors would like to acknowledge these elements of support. The authors also wish to express their gratitude to Vladimir Arabadzhi for assistance in remastering several figures to reach a higher standard of visual clarity, and Dr. Mahmud T.H. Khan for formatting and proofreading guidance.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- The Swiss-Prot protein sequence database: its relevance to human molecular medical research. J. Mol. Med.. 1997;75:312-316.
- [Google Scholar]
- GROMACS: A message-passing parallel molecular dynamics implementation. Comp. Phys. Comm.. 1995;91(1-3):43-56.
- [Google Scholar]
- BIOVIA. Dassault Systèmes, Discovery Studio, San Diego: Dassault Systèmes. 2019.
- ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol.. 2002;3(2002):586-599.
- [Google Scholar]
- Small molecule inhibitors of ezrin inhibit the invasive phenotype of osteosarcoma cells. Oncogene. 2012;31(3):269-281.
- [Google Scholar]
- Multiple Forms of Spire-Actin Complexes and their Functional Consequences. J. Biol. Chem.. 2012;287:10684-10692.
- [Google Scholar]
- COVID-19 and iron dysregulation: distant sequence similarity between hepcidin and the novel coronavirus spike glycoprotein. Biol Direct. 2020;15(1):19.
- [CrossRef] [Google Scholar]
- Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol.. 2010;11:276-287.
- [Google Scholar]
- ERM proteins in epithelial cell organization and functions. Biochim Biophys Acta. 2007;1773(5):653-660.
- [Google Scholar]
- Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol.. 2015;1282(2015):1-23.
- [Google Scholar]
- Ezrin, a plasma membrane-microfilament linker, signals cell survival through the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA. 1999;96(13):7300-7305.
- [Google Scholar]
- Comparative docking studies to understand the binding affinity of nicotine with soluble ACE2 (sACE2)-SARS-CoV-2 complex over sACE2. Toxicology Reports. 2020;7(2020):1366-1372.
- [Google Scholar]
- Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79(2):926-935.
- [Google Scholar]
- PROCHECK – A program to check the stereochemical quality of protein structures. J. App. Cryst.. 1993;26:283-291.
- [Google Scholar]
- Structure of SARS coronavirus spike receptor binding domain complexed with receptor. Science. 2005;309(5742):1864-1868.
- [Google Scholar]
- Genomic characterization and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565-574.
- [Google Scholar]
- Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor. CD26. Nature. 2013;500(7461):227-231.
- [Google Scholar]
- PubChem Substance and Compound databases. Nucleic Acids Res.. 2016;44(Database issue):D1202. D1213
- [Google Scholar]
- Millet, J.K., Kien, F., Cheung, C.Y., Siu, Y.L., Chan, W.L., Li, H., Leung, H.L., Jaume, M., Bruzzone, R., Peiris, J.S.M., Altmeyer, R.M., Nal, B. 2012. Ezrin interacts with the SARS coronavirus spike protein and restrains infection at the entry stage. PLoS One. 7(11). e49566. (doi: 10.1371/journal.pone.0049566, Epub 2012 Nov 21).
- Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Research. 2010;38(Web Server):W622. W627
- [Google Scholar]
- Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Computational Chemistry. 2009;30(16):2785-2791.
- [Google Scholar]
- A unified formulation of the constant temperature molecular dynamics methods. The Journal of Chemical Physics. 1984;81(1):511-519.
- [Google Scholar]
- Palmitoylation of the cysteine-rich endodomain of the SARS-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology Apr 10. 2007;360(2):264-274.
- [Google Scholar]
- Identification of SARS-CoV-2 Spike Palmitoylation Inhibitors That Results in Release of Attenuated Virus with Reduced Infectivity. Viruses. 2022;14:531.
- [Google Scholar]
- SWISS-MODEL: an automated protein homology-modelling server. Nucleic Acids Res. Jul 1. 2003;31(13):3381-3385.
- [Google Scholar]
- Structure of the Active N-terminal Domain of Ezrin. Journal of Biological Chemistry. 2003;278(7):4949-4956.
- [Google Scholar]
- Ezrin has a COOH-terminal actin-binding site that is conserved in the ezrin protein family. J. Cell Biol.. 1994;126:1445-1453.
- [Google Scholar]
- ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research. 2007;35(2007):W407. W410
- [Google Scholar]
- WHO Dashboard https://covid19.who.int.
- Domains and Functions of Spike Protein in Sars-Cov-2 in the Context of Vaccine Design.. 2021;Viruses,13(1):109.
- [CrossRef]
- PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics (Oxford, England). 2016;32(23):3676-3678.
- [Google Scholar]
- A genomic perspective on the origin and emergence of SARS-CoV 2. Cell. 2020;181(2):223-227.
- [Google Scholar]
- Integrative Modeling of Biomolecular Complexes: HADDOCKing with Cryo-Electron Microscopy Data. Structure. 2015;23(5):949-960.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102277.
Appendix A
Supplementary data
The following are the Supplementary data to this article: