

Translate this page into:
A DFT and TD-DFT study on emodin and purpurin and their functionalized molecules to produce promising organic semiconductor materials
⁎Corresponding author at: Department of Chemistry, Faculty of Science, University of Khartoum, Khartoum, Sudan. sahar.abdalla@uofk.edu (Sahar Abdalla)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Density functional theory (DFT) computations were done to explore the optical and electronic properties of two conjugated molecules, emodin and purpurin, as potential organic semi-conductors. The molecules were functionalized to explore the impact of functionalization on the electronic and optical properties. The properties calculated include reorganization energy (λh and λe), adiabatic ionization potential (IP), adiabatic electron affinity (EA), chemical hardness (n), HOMO and LUMO energies, and HOMO-LUMO energy gap (Eg) via B3LYP/6–3++G (d, p) method. In addition, the maximum absorption (λmax) and oscillator strength (f) at the excited states in vacuum and solvent (Ethanol) were investigated using time-dependent density functional theory (TD-DFT). The introduction of functional groups to emodin was considered to convert the molecule from a p-type into an n-type material, while purpurin is considered as an n-type material, its functionalization with NO2 and 2F resulted in a slight increase in λe values, which is considered detrimental for the process of charge-transport. However, the functionalized molecules have shown an increase in EA and a decrease in LUMO energy level, indicating their potential use as n-type materials. Furthermore, to have an understanding of the intermolecular interactions in emodin and purpurin molecules, Hirshfeld surface analysis and energy framework were studied.
Keywords
Organic semiconductors
Optical properties
Electronic properties
DFT
TD-DFT

- HOMO
-
Highest Occupied Molecular Orbital
- LUMO
-
Lowest Occupied Molecular Orbital
- λ
-
Reorganization energy
- λint
-
Internal reorganization energy
- λext
-
External reorganization energy
- IE
-
Adiabatic Ionization energy
- EA
-
Adiabatic Electron affinity
- Eg
-
Energy gap
- DFT
-
Density functional theory
- TD-DFT
-
Time Dependent – Density functional theory
- Η
-
Chemical Hardness
- λmax
-
Maximum Absorption
- f
-
Oscillator Strength
- ˚A
-
Bond lengths
- ◦
-
Bond angles
- B3LYP
-
Becke, 3-parameter, Lee-Yang-Parr
Abbreviations
1 Introduction
The Earth receives a total quantity of radiant energy from the sun of 1370 W/m2/s, with 343 W/m2/s received per unit area of the Earth's surface (Zaidi, 2018). The abundance and non-polluting characteristics of solar energy have made it a widely recognized renewable source of energy. The photovoltaic effect of solar cells can covert this solar energy into electrical energy. Organic solar cells are being increasingly attractive due to their low production cost (Bagher et al., 2015), electronic features such as light absorption and emission, charge generation and transportation (Cardia et al., 2014), tuning of molecular properties by modifying the length and type of functional groups (Cardia et al., 2014; Oshi et al., 2017, 2018, 2019).
The basic operating principle of organic solar cells is the initial absorption of incident photons that leads to the generation of free electrons and holes (excitons) (Arbouch et al., 2014). The Coulomb forces of attraction between the generated excitons are high owing to the low dielectric constant of organic semi-conductors. Therefore, the excitons dissociation requires a heterojunction that is created by two dissimilar organic semi-conductor materials; donor (n-type) and acceptor (p-type). A donor is defined as a material that has a high ionization energy (IP) and an acceptor is a material that has a high electron affinity (EA). Finally, upon the dissociation of excitons, the electron created is transferred to the cathode, and the hole is transferred to the anode to initiate current (Arbouch et al., 2014).
The most essential features of organic semiconductors are inclusive of abundance and low production cost, optimum energy gap (Eg) between HOMO and LUMO, strong light absorption, thermal and photo stability (Scharber and Sariciftci, 2013). However, there remains the challenge of enhancing the proficiency of solar organic cells in comparison to inorganic solar cells. Several research efforts are being carried out on organic solar cell systems to improve their efficacy, processing and stability (Arbouch et al., 2014; Sahdane et al., 2017). One of the approaches used is to enhance the optical and electronic properties of organic semi-conductor molecules by using functional groups to tune their properties (Oshi et al., 2017). For instance, it was stated that the introduction of electron-deficient atoms or groups increases the molecule electron affinity by reducing the molecular LUMO and thus resulting in better n-type organic semi-conductors. These functional groups include imides, amides, carbonyls, quinones and halogen atoms (Arbouch et al., 2014). To increase the environmental stability and electron affinity of conjugated systems, imides and amides are introduced into the backbone of the molecule as strong electron-withdrawing groups (Meng and Hu, 2012). In a study conducted by Zhenan et al., a p-type material, metallopthalocyanine, was converted into an n-type air-stable material by functionalization with fluorine (Bao et al., 1998). Other research showed the pentacene perfluorination converted it into an n-type material (Sakamoto et al., 2004), while the chloro (Cl) and Nitro (N) functionalization of naphthalene and pentacene molecules provided a better material than perfluoropentacene and octafluoronaphthalene (Oshi et al., 2017; Chen and Chao, 2006). The addition of the F and CN groups to 1,3,5-tripyrrolebenzene (TPB) have stabilized frontier molecular orbital and improved air stability (Hu et al., 2016). In addition, the influence of functionalization on absorption was reported by Cardia et al. (2014) in functionalized triisopropylsilylethynyl (TIPS), resulting in better visible region absorption. This improvement of absorption in the visible region was also shown by different sensitizers functionalized by OH, NH2, OCH3, CF3, F, and CN (Wang et al., 2020).
Reorganization energy, which is known to influence the rate of charge transfer (Li et al., 2012), is a critical element that dictates the efficiency of organic solar cells. The reorganization energy is often considered as the summation of internal and external influences. The internal reorganization energy (λint) is attributed to the change in equilibrium geometry of the donor and acceptor sites due to electron transfer, while the external reorganization energy (λext) is attributed to the polarization effects that lead to the change in the surrounding media (Oshi et al., 2019; Li et al., 2012; Nan et al., 2009). The external reorganization energies values are considered to be much smaller than the inner reorganization energies, and thus negligible (Hu et al., 2016; Cheung and Troisi, 2010).
The effect of functionalization on the reorganization energy has been reported by many studies. For example, Hutchison et al. (2005) investigated the functionalization impact of oligomers of thiophene and furan in terms of reorganization energy. Others have revealed that the cyanation of pentacene gives smaller values of hole (75 meV) and electron reorganization energy (87 meV) than pentacene (94 and 133 meV, respectively) (Kuo et al., 2007). Oshi and co-workers discussed the increase of hole and electron reorganization energy in 7,7,8,8-Tetracyanoquinodimethane by electron-donating groups CH3, OCH3, and OH (Oshi et al., 2019). Thus, it is valuable to recognize the effect of functionalization on molecule’s reorganization energy as it provides perception into charge transfer rate in organic semiconductor materials. Charge transport (at low temperatures) in organic semiconductors is described as band-like motion in which the charge is delocalized across the system. The charge transport technique at high temperatures is represented by a hopping mechanism where the charge transporters are localized to a single molecule, and jump from a molecule to a nearby molecule as described by the Marcus equation, which is given as follows (Oshi et al., 2018; Mas-Torrent et al., 2004). where t, λ and T represent the charge transfer matrix element, reorganization energy and absolute temperature, respectively. While h and KB are the Planck and the Boltzmann constants respectively.
The charge transport rate from the equation is mostly affected by the reorganization energy and electronic coupling (transfer integral). The charge transport rate is determined by the transfer integral, which is influenced by the orientation and distance of the molecules. A lower reorganization energy land larger transfer integral lead to a faster charge transport rate (Chen et al., 2017).
Quantum mechanics and molecular modeling give an understanding of the structure, optical and electronic properties relationships that lead to systematic molecular design (Sahdane et al., 2017; Meng and Hu, 2012; Sun et al., 2016). In this investigation, the influence of functionalization on electronic and optical properties of two conjugated molecules, emodin (6-methyl-1,3,8-trihydroxyanthraquinone), and purpurin (1,2,4-Trihydroxyanthraquinone) are studied to understand their possible use in organic solar cells, Fig. 1. Also, some of the bond lengths (Å) along with the bond angles (◦) of emodin and purpurin and their derivatives are presented as supplementary. Both molecules belong to the anthraquinones, which are considered to be the largest occurring quinones that are naturally abundant. They are used as natural colorants as well as in variety of other applications (Sun et al., 2018).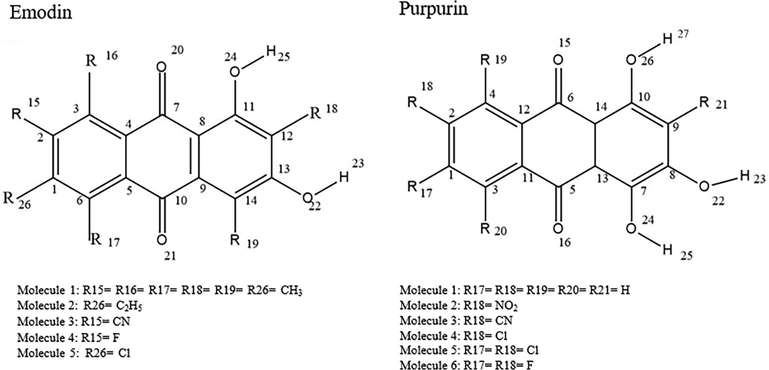
Structure and numbered atoms of emodin (6-methyl-1,3,8-trihydroxyanthraquinone) and purpurin (1,2,4-Trihydroxyanthraquinone).
2 Materials and methods
The structures of emodin and purpurin were optimized via DFT method (Hohenberg and Kohn, 1964) using Becke, 3-parameter, Lee–Yang–Parr (B3LYP) functional (Becke 1988; Lee et al., 1988; Becke, 1992) and the 6–31++G (d, p) basis set. The B3LYP has been used in the calculation of reorganization energy in several studies (Chen and Chao, 2006). The frequency calculations carried out at the same level of theory, reveal that all studied molecules have reached local minima with no imaginary frequencies. The performed calculations included λ for holes and electrons, adiabatic ionization potential (IP), and adiabatic electron affinity (EA), chemical hardness (n), HOMO and LUMO energy levels, and energy gap (Eg). The reorganization energies for holes (λh) and electrons (λe) are calculated as follows: where the ( ) represent the anionic (cationic) energy of the molecule in an optimized neutral structure, the ( ) is the anionic (cationic) energy of the molecule in an optimized anion (cation) structure, the ( ) is the neutral energy of the molecule in optimized anion (cation) structure and the is the neutral energy of the molecule in optimized neutral structure (Chen et al., 2017; Sun and Jin, 2017).
The IP and EA energies were calculated as per the following equation:
The E(M0), E(M−) and E(M+) represents the total energies of the neutral, anionic, and cationic state of the molecule (Oshi et al., 2017). The chemical hardness of the molecule, η, was calculated using η = (Scharber and Sariciftci, 2013; Prabha et al., 2017).
The excited states of the molecules were determined using time-dependent density functional theory (TD- DFT) (Stratmann et al., 1988) utilizing the B3LYP functional with 6–31++ (d, p) basis set from ground state optimized geometry in vacuum and solvent (Ethanol). This gives the absorption spectra and oscillator strength. All calculations were done using Gaussian 09 program (Frisch et al., 2009).
Furthermore, the intermolecular interaction of emodin and purpurin were investigated to determine the role of these interactions to the crystal lattice through Hirshfeld surface analysis (Spackman and Jayatilaka, 2009). To aid the interpretation of the interactions, d_ map, shape index and 2D fingerprint plots (Spackman and Jayatilaka, 2009) were demonstrated. The 3D energy framework was represented by computing the interaction energies, electrostatic energy, polarization energy, dispersion energy, exchange-repulsion energy, and total intermolecular energy (Jayatilaka and Grimwood, 2003) at B3LYP/6-31G (d,p), and a 3.8 Å cluster was generated around the molecule. The calculations were done using the CrystalExplorer17 program (Spackman et al., 2021). Emodin and hydrated purpurin was used in the representation of the 3D energy framework.
3 Results and discussion
3.1 Reorganization energy
The λh and λe values of emodin and its functionalized molecules are listed in Table 1. For emodin, it can be observed that the λh value is smaller than the value of λe, 0.22 eV and 0.41 eV, respectively. The λh values increased upon the introduction of functional groups, with the lowest value being 0.38 eV for molecules 4 and 5, and the highest value being 0.40 eV for molecules 2 and 3. The λe showed only a slight change in values with an increase of 0.03 eV on F-functionalized molecule 4 and a decrease for molecules 3 (0.03 eV) and 5 (0.01 eV). The changes in λh and λe are due to the changes in structure during oxidation and reduction that are attributed to the C-X (functional groups), bonds contribution. As mentioned in section 1, the charge transfers rate increases with the decrease of reorganization energy. As such, emodin without functionalization is predicted to have the highest charge transfer rate and serves as a good candidate as a hole transport material and electron transport material upon the introduction of functional groups.
Molecule
λh
λe
IP
EA
n
Emodin
1 – None
0.22
0.41
8.74
1.85
3.44
2 – (1) Ethylene
0.40
0.41
8.13
1.87
3.13
3 – (1) CN
0.40
0.38
8.42
2.33
3.05
4 – (1) F
0.38
0.43
8.24
1.98
3.13
5 – (1) Cl
0.38
0.40
8.30
2.13
3.09
Purpurin
1 – None
0.59
0.39
8.43
1.85
3.29
2 – (1) NO2
0.56
0.42
8.06
2.59
2.74
3 – (1) CN
0.56
0.37
8.01
2.37
2.82
4 – (1) Cl
0.60
0.39
7.81
2.04
2.88
5 – (2) Cl
0.58
0.39
7.88
2.19
2.85
6 – (2) F
0.58
0.42
7.93
2.12
2.91
Purpurin λh and λe values are also listed in Table 1. Unlike emodin, the value of λe is smaller than λh value, 0.39 eV and 0.59 eV, respectively. Accordingly, purpurin without functionalization may act as a good electron transport material. The addition of functional groups has led into a minor decrease of λh in molecules 2, 3, 5 and 6 with λh values of 0.56, 0.56, 0.58, and 0.58 eV, respectively. The only increase was in Cl-functionalized molecule 4, with λh to be 0.60 eV. The changes in λe values are also minor, with the largest increase for molecules 2 and 6 by 0.03 eV and the smallest decrease for molecule 3 by 0.02 eV. While no change occurred in the Cl-functionalized molecules, 4 and 5. Based on the given data, purpurin is inclined to have a high charge transport rate as an electron transport material and its functionalized molecules may also have a similar charge transfer rate as electron transport materials.
In general, for halogenated molecules, the values for F show higher reorganization energy than Cl, in emodin and purpurin as a result of the higher electronegativity of Fluorine. This is in agreement with results of halogenated naphthalene (Oshi et al., 2017) and tetracene (Sancho-García et al., 2010). It is suggested that the intra-ring functionalization of molecules may yield better results in minimizing the reorganization energy as it avoids the introduction of additional degrees of freedom for geometric relaxation (Li et al., 2012). Overall, the values of λh and λe are similar to those of proposed organic semiconductors (Oshi et al., 2017, 2018, 2019; Li et al., 2012). The values of chemical hardness for the two molecules are also presented in Table 1. The chemical hardness (η) is defined as the resistance of the chemical potential to change in the number of electrons (Sun and Jin, 2017), the functionalized molecules show similar values of η to that of emodin and purpurin, indicating their stability.
3.2 Adiabatic ionization potential and adiabatic electron affinity
The values IP and EA for emodin, purpurin, and their functionalized molecules are illustrated in Table 1. One of the concerns in n-type organic semiconductor systems is the lack of stability of their radical anions in the air, (De Leeuw et al., 1997) and functionalization of molecules may be used to tune IP and EA values for a more stabilized molecule. The IP value of emodin is calculated to be 8.74 eV and the EA is 1.85 eV, and because of the similarity in structure, the IP and EA of purpurin have shown similar values to that of emodin with values of 8.43 eV and 1.85 eV, respectively.
The functionalization of emodin and purp urin results in a decrease of IP and an increase in EA values. The values of emodin IP are in the sequence of molecules 1 > 3 > 5 > 4 > 2 and for EA values 3 > 5 > 4 > 2 > 1 and for purpurin the IP values are in the sequence of 1 > 2 > 3 > 6 > 5 > 4 and for EA 2 > 3 > 5 > 6 > 4 > 1. The NO2-functionalized purpurin has the highest IP and EA values due to the negative resonance effect and electron affinity of the NO2 group. Electrons are withdrawn from the rings, leading to electron deficit at all positions of the fused rings. It is followed by the CN-functionalized molecule, which is attributed to the interactions of the nitrogen lone pair of electrons (negative resonance effect) with the π-electron clouds of the fused rings, as well as the inductive effect of the electronegative nitrogen atom. These results are in agreement with the NO2 and CN functionalization of tetracene (Oshi et al., 2018).
Similarly, the CN-functionalized molecule in emodin has the highest EA value, 2.33 eV. The 2Cl-functionalized purpurin molecule has a higher EA value than the 2F-functionalized molecule, that is accounted for by the higher ability of Cl to withdraw electrons, taking into consideration resonance and inductive effects. This is in agreement with halogenated pentacene (Chen and Chao, 2006) and naphthalene (Oshi et al., 2017). In addition, the Cl-functionalized emodin molecule also has a higher EA than the F-functionalized molecule.
3.3 HOMO and LUMO energy, and optical properties
The values of Eg, HOMO and LUMO of emodin, purpurin and their functionalized molecules are shown in Table 2. The molecules Eg falls within the range of organic semiconductors, 1.4–4.2 eV (Chen et al., 2017). The functionalization of emodin and purpurin contributed to lowering the energy gap that predicts kinetic stability and an increase in conductivity.
Molecule
Eg(eV)
λabs
HOMO(eV)
LUMO(eV)
f
Emodin
1 – None
3.52
413.28
−6.69
−3.18
0.1139
2 – (1) Ethylene
3.51
414.72
−6.69
−3.18
0.1173
3 – (1)CN
3.40
427.31
−7.01
−3.62
0.091
4 – (1)F
3.51
411.77
−6.81
−3.30
0.1063
5 – (1)Cl
3.45
421.43
−6.89
−3.44
0.1183
Purpurin
1 – None
3.12
452.09
−6.31
−3.19
0.1274
2 – (1)NO2
2.84
509.51
−6.68
−3.84
0.0797
3 – (1)CN
2.97
480.52
−6.63
−3.66
0.1138
4 – (1)Cl
3.09
455.57
−6.44
−3.35
0.1349
5 – (2)Cl
3.06
461.24
−6.53
−3.47
0.135
6 – (2)F
3.09
456.44
−6.54
−3.45
0.1241
The energy gap of emodin and its functionalized molecules ranged between 3.40 and 3.52 eV, the smallest Eg value was for CN-functionalized molecule 3, 3.40 eV, followed by Cl-functionalized molecule 5, 3.45 eV, and accordingly is predicted to have better conductivity. In addition, the LUMO energy levels and λe of CN and Cl-functionalized molecules also have the lowest values, which results in improved emodin functionality as an n-type material.
The overall LUMO values decreased except for the functionalized C2H2 molecule. The LUMO values ordered as molecule 1 = 2 > 4 > 5 > 3. The CN-functionalized molecule showed the lowest value, leading to the smallest energy gap value. The HOMO levels also decreased in value with the order of molecule 1 = 2 > 4 > 5 > 3. The HOMO and LUMO values of purpurin and its functionalized molecules are in the sequence of molecule 1 > 4 > 5 > 6 > 3 > 2 and 1 > 4 > 6 > 5 > 3 > 2, respectively. The Eg values range from 2.84 − 3.12 eV, emodin showed the highest Eg value of 3.12 eV with NO2- functionalization. The value decreased into 2.84 eV to become the smallest Eg among the molecules, followed by the CN-functionalized molecule 3.
The decrease in Eg values of emodin and purpurin upon the addition of functional groups is in consistent with the results obtained for F, CN Cl, and NO2 functionalized tetracene and anthracene by (Oshi et al., 2018). Overall, the LUMO values of the molecules are in the range of typical n-type materials between −3.0 and −4.0 eV, lowering of LUMO also results in an increase in stability (Li et al., 2012) and a higher ability to accept electrons (Prabha et al., 2017). Hence, the studied molecules are proposed to be good n-type materials. The study of these frontier molecular orbitals (FMO), in qualitative manner, are insightful in predicting the electron transition (Sun and Jin, 2017). Therefore, the HOMO and LUMO qualitative molecular representations of emodin, purpurin and their functionalized molecules in S0 are represented in Fig. 2. For emodin and its functionalized molecules, the HOMO and LUMO are delocalized on one of the benzene rings and slightly on the functionalized benzene ring. This indicates a strong overlap between the HOMO and LUMO that leads to strong optical absorption due to electron excitation S0-S1. For purpurin, the HOMO and LUMO are delocalized on one of the benzene ring, the transition is mainly within the non-functionalized benzene ring. The electrostatic potential (ESP) is useful in predicting the reactive sites of the molecule. The ESP of the molecules increases in the order of red > orange > yellow > green > blue. The reactive sites of the emodin, purpurin and their functionalized molecules are also shown in Fig. 3. In general, the addition of the functionalization resulted in an increase in reactivity in the site except for molecule 2 in emodin.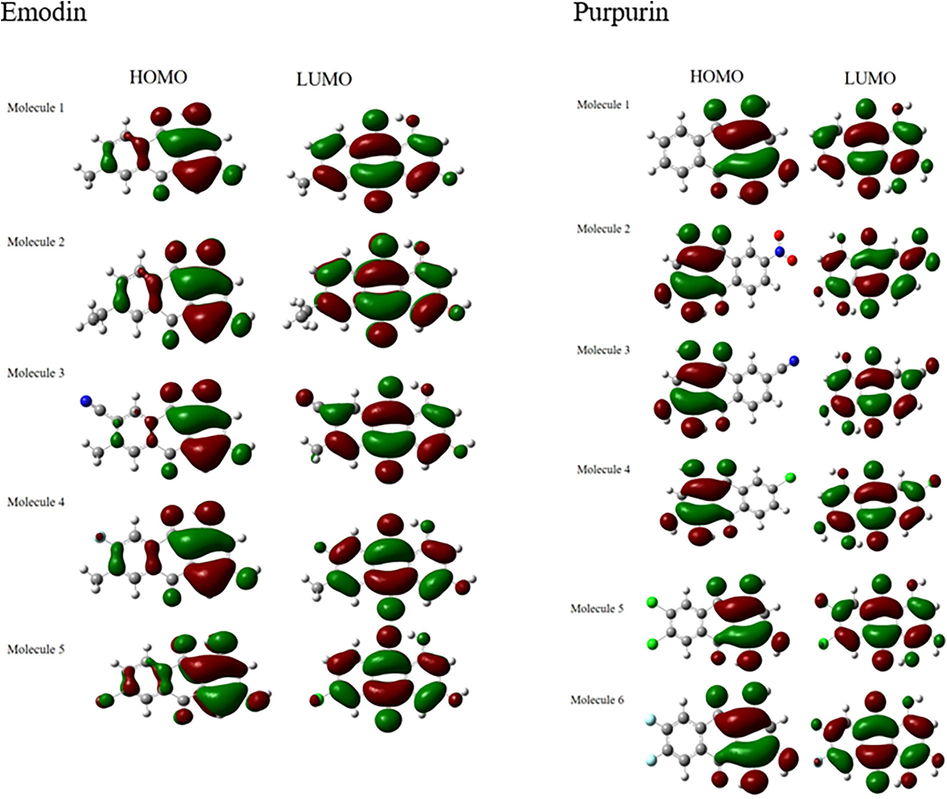
HOMO and LUMO of Emodin (molecule 1 – 5) and purpurin (molecule 1 – 6).
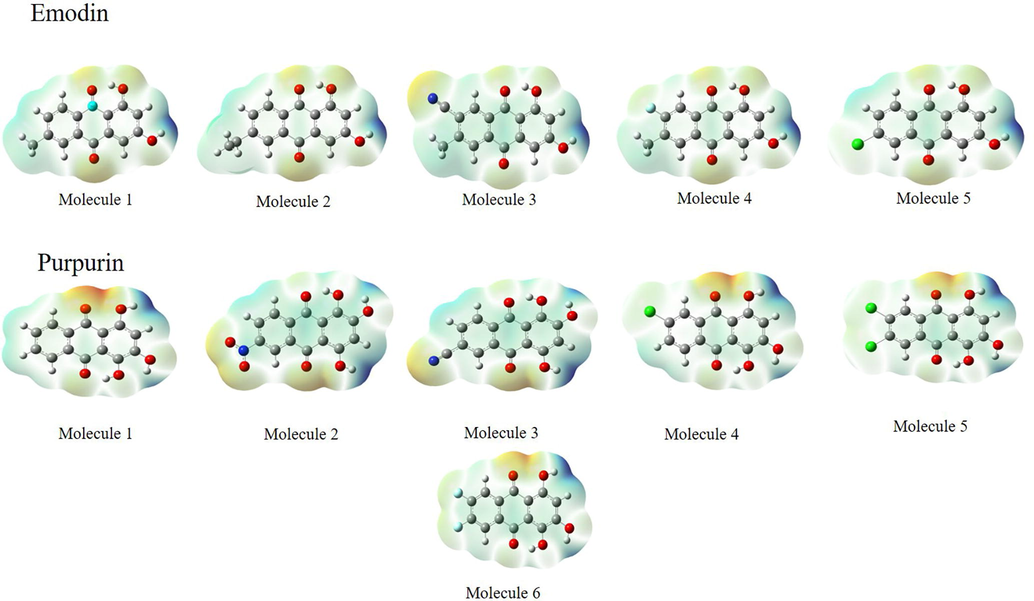
ESP of emodin and purpurin.
The oscillator strengths of emodin, purpurin and their functionalized molecules are presented in Table 3. Oscillator strength expresses the probability of absorption of electromagnetic radiation, the higher the oscillator strength is for a molecule the higher the expected absorption. In this study, the strongest absorption for emodin corresponds to excited state S1 with a value of 413.28 nm (f = 0.1139) in a vacuum. For molecule 2, the strongest absorption was at excited state S1 with a value of 429.11 nm (f = 0.1427) in the solvent. The strongest absorption in molecule 3 corresponds to 439.71 nm (f = 0.1168) in the solvent at excited state S1. The excited state S1 shows the strongest absorption for molecule 4 with a value of 425.47 nm (f = 0.1283) in the solvent. Finally, molecule 5 also shows the strongest absorption at excited state S1 with a value of 434.66 nm (f = 0.146) in the solvent.
Excited State 1
Excited State 2
Excited State 3
Molecule
λmax(nm) f
λmax(nm) f
λmax(nm) f
λmax(nm) f
λmax(nm) f
λmax(nm) f
Emodin
Without Solvent
With Solvent
Without Solvent
With Solvent
Without Solvent
With Solvent
1 – None
413.28
0.1139
419.83
0
388.39
0
407.63
0.175
381.58
0.1036
380.18
0
2 – (1) Ethylene
414.72
0.1173
429.11
0.1427
413.11
0.0003
403.01
0
365.56
0.012
378.55
0.0356
3 – (1) CN
427.31
0.091
439.71
0.1168
417.29
0
408
0
362
0
386.1
0.039
4 – (1) F
411.77
0.1063
425.47
0.1283
409.61
0
399.56
0
356.28
0
380.94
0.0464
5 – (1) Cl
421.43
0.1183
434.66
0.146
412.82
0
402.54
0
360.14
0
383.27
0.026
Purpurin
1 – None
452.09
0.1274
461.81
0.194
449.76
0
425.89
0
350.37
0
370.06
0.0317
2 – (1) NO2
509.51
0.0797
541.1
0.0944
467.25
0
442.21
0
374.59
0.0096
401.81
0.0597
3 – (1) CN
480.52
0.1138
1022.69
0.0297
457.45
0
1011.86
0.001
357.24
0
693.33
0
4 – (1) Cl
455.57
0.1349
466.58
0.2031
446.72
0
423.55
0
366.79
0.0651
379.2
0.0608
5 – (2) Cl
461.24
0.135
473.32
0.2028
447.96
0
424.82
0
373.14
0.0318
382.31
0.0322
6 - (2) F
456.44
0.1241
708.01
0
444.04
0
478.79
0
365.57
0.0265
465.55
0
For purpurin, the excited state (S1) corresponds to the maximum absorption with values of 461.81 nm (f = 0.194) in the solvent. For NO2- functionalized molecules, the strongest absorption value is 541.1 nm (f = 0.0944) in the solvent at excited state S1. For molecules 3, 1022.69 nm (f = 0.297) in the solvent at excited state S1. For the 1Cl-functionalized molecule at excited state S1 with a value of 466.58 nm (f = 0.2031) in the solvent. As for the 2Cl-functionalized molecule, which also corresponds to excited state S1 with a value of 473.32 nm (f = 0.2028) in the solvent. For molecule 6, the strongest absorption takes place in a vacuum with a value of 456.44 nm (f = 0.1241). It can be noticed from the given values in Table 3, that the solvent resulted in a redshift of the maximum absorption. Moreover, the decrease in the Eg values also resulted in a red shift in the maximum absorption, as listed in Table 2.
3.4 Hirshfeld surface analysis
The Hirshfeld surface analysis aids in the quantification and visualization of intermolecular interactions by utilizing different colors and intensities in graphical representation. Hirshfeld surface analysis has been used to investigate various intermolecular interactions in the crystal structure of organic molecules and complexes (Van Thong et al., 2022; Al-Resayes et al., 2020). The dnorm (normalized contact distance) maps of emodin and purpurin are depicted in Fig. 4. The contacts with shorter distances (close contacts) than the Van der Waals radii are represented by red surfaces, while the blue surfaces represent contacts with longer distances (distance contact). The white surfaces denote distance equivalent to the sum of Van der Waals radii. The red regions in emodin and purpurin appears to originate from the –OH groups in the molecules. The red regions of the molecules may act as donors in intermolecular reactions. Thereby, it is expected that their functionalized molecules with electronegative groups would adhere more sites that act as donors. The shape-index of emodin and purpurin on the Hirshfeld surface is presented in Fig. 4. The red and blue regions represent the donor and acceptor groups of the molecule, the adjacent red and blue triangles denote π-π stacking interaction among the structures.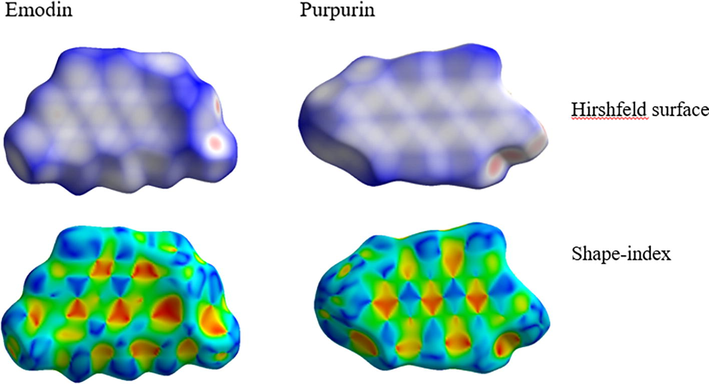
Hirshfeld surface of emodin and purpurin plotted over dnorm in the range of −0.4160 to 1.2016 and −1.7662 to 0.8459 a.u, respectively. Shape-index of emodin and purpurin on Hirshfeld surface.
The 2D fingerprint plots and fragment patches of emodin and purpurin are illustrated in Figs. 5 and 6, respectively. The de and di on the plots represent the distances to the nearest atom center, external and internal to the surface. Points on the plot without contribution are gray colored and points with small contribution is blue colored through green to red for the points with the largest contribution. The most significant interaction in emodin is O--H/H--O that contributes 31.8% to the overall surface. The second largest interaction is H--H with contribution of 30.1% as pair wings, followed by C--H/H--C (15.9%), C--C (10.9%) and O--C/C—O (9.1%). In purpurin, the O--H/H--O interactions dominated with a contribution of 41%, while the H--H and C--C interactions were 26.6% and 22.3%, respectively. Both molecules have other small contributions; emodin O--O (2.2%) and purpurin C--H/H--C (5.5%), O--O (2.8%) and C--O/O--C (1.9%).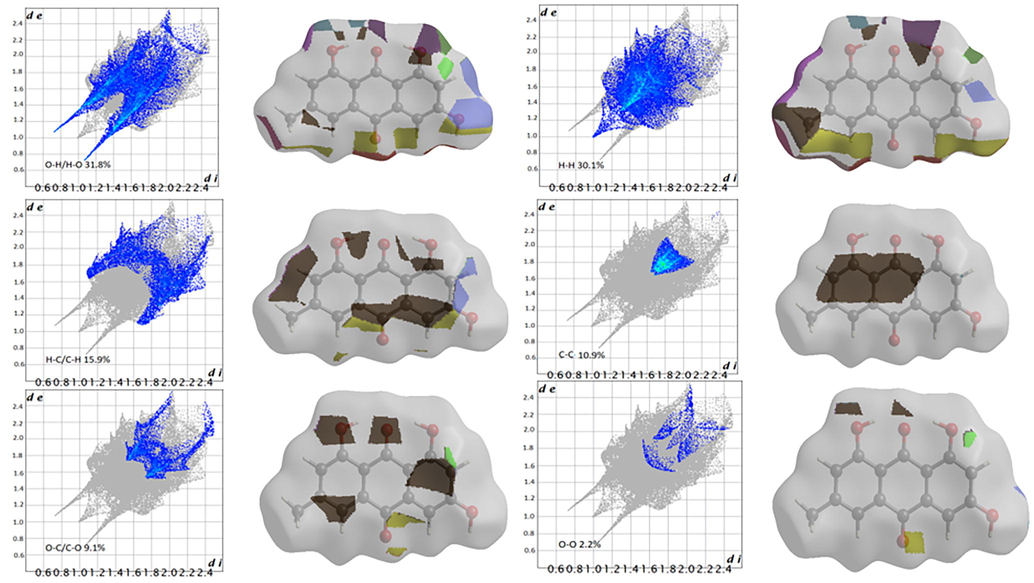
Emodin two-dimensional fingerprint plots and fragment patches (surface patches adjacent to neighboring surfaces are colored separately) for intermolecular interactions.
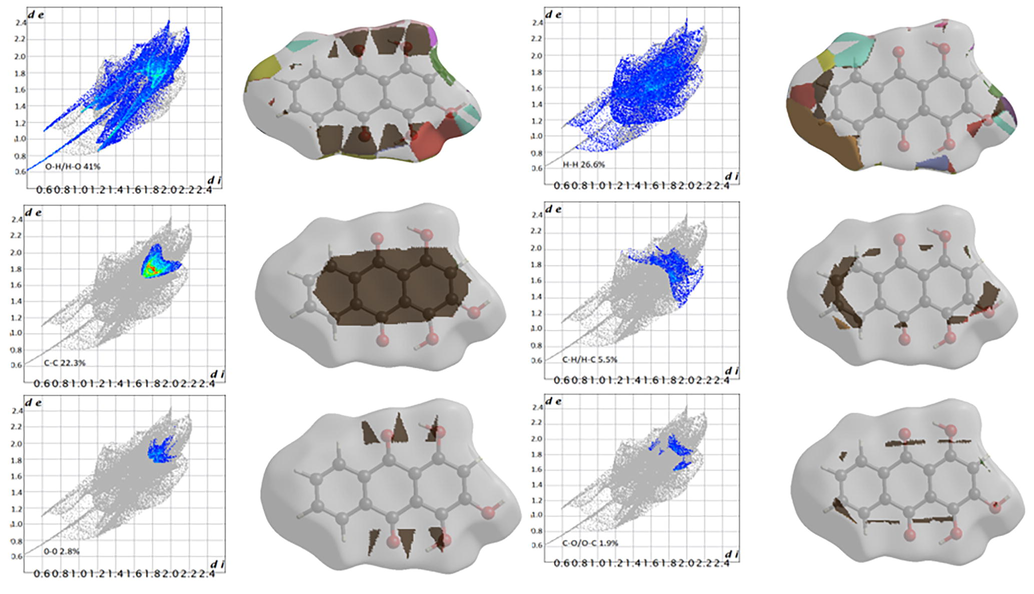
Purpurin two-dimensional fingerprint plots and fragment patches (surface patches adjacent to neighboring surfaces are colored separately) for intermolecular interactions.
3.5 Energy frameworks
The calculated interaction energies energy Etot (kJ mol−1) namely, electrostatic (Eelec), polarization (Epol), dispersion (Edis), exchange-repulsion (Erep) are used to generate 3D representation of the major interactions in the form of energy frameworks, Fig. 7. The interaction energies relative strength in individual directions are represented by exhibited cylinder-shaped energy frameworks, the size of the interactions that are less than 5 kJ mol−1 have been excluded. It can be seen that the intermolecular interactions are dominated by dispersion forces in emodin and purpurin, Fig. 7.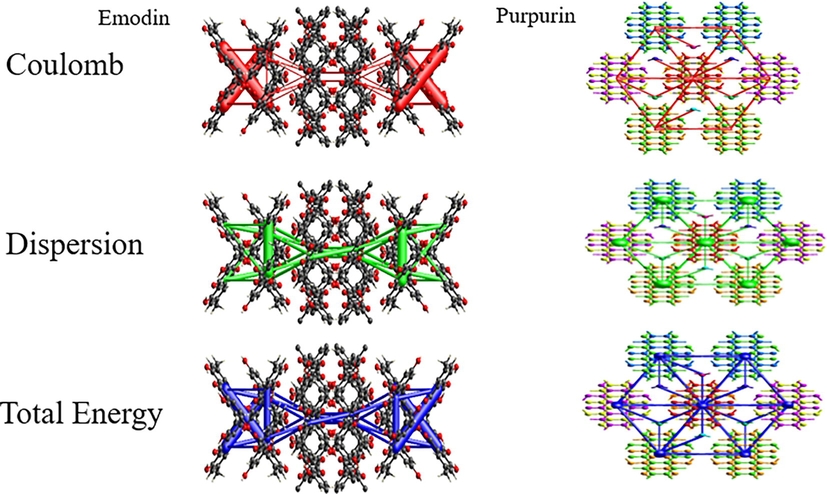
Emodin and purpurin energy frameworks, coulomb energy (red), dispersion energy (green) and total energy (blue).
4 Conclusion
In the present paper, the electronic and optical properties of emodin and purpurin were investigated utilizing DFT and TD-DFT methods, to learn about their possible use as organic semiconductors. The molecules were also functionalized by electronegative groups to appreciate the influence of functionalization on the optical and electronic properties of the molecules. There was an overall increase in reorganization energy due to the geometrical changes of the molecule upon oxidation and reduction processes. However, the decrease in Eg and LUMO energy levels of the molecules predicts the increase in stability and conductivity. Furthermore, an increase in EA values was also noted to reinforce the molecule’s ability to accept electrons. Hence, the functionalization of emodin and purpurin strengthened the n-type properties of the molecules. The energy gaps of the molecules are within the range of semi-conductors (1.4–4.2 eV) and the absorption ranges of the molecules are within the visible range. Based on the given data, we have concluded that emodin and purpurin along with their functionalized molecules are good candidates for organic semi-conductors. Also, the intermolecular interactions and 3D energy framework of emodin and purpurin molecules were studied through Hirshfeld surface analysis. It has been shown that the main interaction in the crystal structure of emodin and purpurin is O--H/H--O and dispersion energy was dominant in the 3D energy framework.
Acknowledgement
We would like to thank all our colleagues from Sudan University, Khartoum University and Jubail Industrial College for all their support. This study received no specific grant from public, private, or non-profit funding agencies.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Structural and theoretical investigations, hirshfeld surface analyses, and cytotoxicity of a naphthalene-based chiral compound. ACS Omega. 2020;5(42):27227-27234.
- [Google Scholar]
- Organic photovoltaic cells: operating principles, recent developments and current challenges–review. Phys. Chem. News. 2014;72(4):73-84.
- [Google Scholar]
- New air-stable n-channel organic thin film transistors. J. Am. Chem. Soc.. 1998;120(1):207-208.
- [Google Scholar]
- Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1988;38(6):3098-3100.
- [Google Scholar]
- Density-functional thermochemistry. i. the effect of the exchange-only gradient correction. J. Chem. Phys.. 1992;96(3):2155-2160.
- [Google Scholar]
- Effects of tipsfunctionalization and perhalogenation on the electronic, optical, and transport properties of angular and compact dibenzochrysene. J. Phys. Chem. A. 2014;118(28):5170-5177.
- [Google Scholar]
- Toward the rational design of functionalized pentacenes: reduction of the impact of functionalization on the reorganization energy. ChemPhysChem. 2006;7(9):2003-2007.
- [Google Scholar]
- Density functional theory calculations of charge transport properties of ‘plate-like’coronene topological structures. J. Chem. Sci.. 2017;129(9):1341-1347.
- [Google Scholar]
- Theoretical study of the organic photovoltaic electron acceptor pcbm: Morphology, electronic structure, and charge localization. J. Phys. Chem. C. 2010;114(48):20479-20488.
- [Google Scholar]
- Stability of n-type doped conducting polymers and consequences for polymeric microelectronic devices. Synth. Met.. 1997;87(1):53-59.
- [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A., Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam M., Klene M., Kno J. E. x, Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas O., Foresman J. B., Ortiz J. V., Cioslowski J., and Fox D. J.. Gaussian 09 Revision A.1. Gaussian Inc. Wallingford CT 2009.
- Theoretical investigation on charge transfer properties of 1, 3, 5-tripyrrolebenzene (tpb) and its derivatives with electron-withdrawing substituents. Croat. Chem. Acta. 2016;89(1):81-90.
- [Google Scholar]
- Hopping transport in conductive heterocyclic oligomers: reorganization energies and substituent effects. J. Am. Chem. Soc.. 2005;127(7):2339-2350.
- [Google Scholar]
- Tonto: a fortran based object-oriented system for quantum chemistry and crystallography. Springer; 2003. p. :142-151.
- Cyanation: Providing a three-in-one advantage for the design of n-type organic field-effect transistors. Chemistry–A European Journal. 2007;13(17):4750-4758.
- [Google Scholar]
- Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37(2):785-789.
- [Google Scholar]
- Theoretical study of the effects of different substituents of tetrathiafulvalene derivatives on charge transport. Chin. Sci. Bull.. 2012;57(31):4049-4056.
- [Google Scholar]
- Correlation between crystal structure and mobility in organic field-effect transistors based on single crystals of tetrathiafulvalene derivatives. J. Am. Chem. Soc.. 2004;126(27):8546-8553.
- [Google Scholar]
- Meng Q. and Hu W. 2012. Recent progress of n-type organic semiconducting small molecules for organic field-effect transistors. Physical Chemistry Chemical Physics, 14(41):14152–14164, 2012.
- Charge transfer rates in organic semiconductors beyond first-order perturbation: From weak to strong coupling regimes. J. Chem. Phys.. 2009;130(2)
- [Google Scholar]
- Study of the influence of functionalization on the reorganization energy of naphthalene using DFT. Comput. Theor. Chem.. 2017;1099:209-215.
- [Google Scholar]
- Theoretical study on functionalized anthracene and tetraceneas starting species to produce promising semiconductor materials. Comput. Theor. Chem.. 2018;1128:60-69.
- [Google Scholar]
- The impact of functionalization of organic semiconductors by electron donating groups on the reorganization energy. Eur. Phys. J. D. 2019;73(6):1-8.
- [Google Scholar]
- A complete theoretical study of indirubin-a blue dye for dye sensitized solar cells (DSSCs) applications. Int. J. Sci. Eng. Res.. 2017;8(10):13-25.
- [Google Scholar]
- Photovoltaic energy conversion and optical properties of organic molecules based on aceanthraquinoxaline. Der Pharma Chem.. 2017;9(1):37-42.
- [Google Scholar]
- Perfluoropentacene: High-performance p-n junctions and complementary circuits with pentacene. J. Am. Chem. Soc.. 2004;126(26):8138-8140.
- [Google Scholar]
- Molecular packing and charge transport parameters in crystalline organic semiconductors from first-principles calculations. PCCP. 2010;12(32):9381-9388.
- [Google Scholar]
- Efficiency of bulk-heterojunction organic solar cells. Prog. Polym. Sci.. 2013;38(12):1929-1940.
- [Google Scholar]
- Crystalexplorer: A program for hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr.. 2021;54(3):1006-1011.
- [Google Scholar]
- An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys.. 1988;109(19):8218-8224.
- [Google Scholar]
- Optical and electrical properties of purpurin and alizarin complexone as sensitizers for dye-sensitized solar cells. J. Mater. Sci.: Mater. Electron.. 2016;27(8):8027-8039.
- [Google Scholar]
- An experimental and theoretical investigation of the electronic structures and photoelectrical properties of ethyl red and carminic acid for dssc application. Materials. 2016;9(10):813.
- [Google Scholar]
- Dft and TD-DFT study on the optical and electronic properties of derivatives of 1, 4-bis (2-substituted-1, 3, 4-oxadiazole) benzene. Arabian J. Chem.. 2017;10:S2988-S2993.
- [Google Scholar]
- Influence of the drying method on the bioactive compounds and pharmacological activities of rhubarb. J. Sci. Food Agric.. 2018;98(9):3551-3562.
- [Google Scholar]
- NMR investigations on a series of diplatinum (ii) complexes possessing phenylpropenoids in CDCl3 and CD3CN: Crystal structure of a mononuclear platinum complex. Polyhedron. 2022;212:115612.
- [Google Scholar]
- Enhancing charge transfer and photoelectric characteristics for organic solar cells. J. Nanomater.. 2020;2020:1-12.
- [Google Scholar]
- Introductory chapter: Introduction to photovoltaic effect. London, UK: Solar Panels and Photovoltaic Materials; InTech Open; 2018. p. :1-8.
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102117.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




