

Translate this page into:
Brefeldin A: A newly identified cell death inducer selectively targets radio-resistant colorectal cancer cells by direct interacting with caspase-3
⁎Corresponding authors. lifeng823@tutanota.com (Li Feng)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Colorectal cancer is one of the most occurring cancers and is a cause of many deaths worldwide. Treatment of cancer is much dependent on the stage of the disease. The patients at the late stages of colorectal cancer show a very low rate of survival (about 10%). One of the challenges for successful treatment is the resistance to radiotherapy, which is a common intervention in combination with chemotherapy. In this study, we screened a panel of natural products aiming to identify small molecules that selectively kill radio-resistant colon cancer cells. For this, we established a radio-resistant colorectal cancer cell model which displayed overexpression of AMPK and TPP1, two markers of radio-resistant. We showed that these cells are resistant to radiation at doses as high as 12 Gy. The library was screened by measuring the caspase-3 activity. The hit compounds were then tested in cell death assay and titrated to calculate their potency and efficacy. Those compounds with the highest ability to kill radio-resistant HCT116 cells were then tested on radio-sensitive cells. We identified Brefeldin A as a potent inducer of death in radio-resistant cells. Brefeldin A induced death in 90% of radio-resistant cells while it affects 40% of the sensitive cells. Brefeldin A targeted radio-resistant colorectal cancer cells by apoptosis and necrosis. We showed that Brefeldin A directly interacts to caspase-3 but not caspase-9 and -6. Brefeldin A didn’t alter the transcription of apoptotic components. This finding opens new windows toward the treatment of resistant colon tumors and re-sensitizing them for adjuvant therapeutic approaches.
Keywords
Brefeldin A
Colorectal cancer
Radio-resistant cells
Natural product
High throughput screening
Caspase activity

1 Introduction
Colorectal cancer is the third most occurring type of cancers in both men and women worldwide and is a cause of 600,000 death annually (Siegel et al., 2014; Siegel et al., 2016). The five-year survival prognosis is variable ranging from 90% to 10% among the patients depending on the stages of the disease (Brenner et al., 2014). As colorectal cancer is usually diagnosed at the late stages the successful treatment is difficult. The common strategies for colorectal cancer treatment consists of surgery, targeted therapy, neoadjuvant radiotherapy and adjuvant chemotherapy (Andre and Schmiegel, 2005; Carrato, 2008). However, the treatment failure is the cause of 15% to 70% cases that are associated with local recurrence and distant metastasis (Geng and Wang, 2017; Huerta et al., 2009). Drug-resistance is one of the challenges toward successful treatment (Van der Jeught et al., 2018). To overcome this, the combination of radiotherapy and chemotherapy showed promising results in the survival rate. Radiotherapy can be used prior to or following surgical removal of cancerous tumors to diminish the risk of recurrence. However, the use of radiotherapy is limited due to the cellular radio-resistance in a large population of patients (Pajonk et al., 2010).
Therefore, it is of great interest to elucidate the molecular events that induce resistance to radiotherapy aiming to re-sensitize the cells to radiotherapy. Radiation treatment induces DNA damage like DNA double strand break by producing free radicals and reactive oxygen species (ROS) (Galanty et al., 2009). The major source of ROS generated is the mitochondrial oxidation respiratory chain (Larosa and Remacle, 2018). The role of glycolysis-related metabolic reprogramming has been shown before in development of radio-resistance tumor cells (Bhatt et al., 2015; Bol et al., 2015; Tang et al., 2018; Yu et al., 2017). In addition, negative regulatory roles of fatty acid oxidation mechanisms were also reported to be a potent target in this process (Kim et al., 2018; Tan et al., 2018).
At the molecular level ACD (Adrenocortical dysplasia protein homolog, or TPP1) and AMPK (Adenosine monophosphate-activated kinase) are recognized as the markers of radio-resistant colon cancer cells. AMPK is highly activated in the radio-resistant patient samples and thus inhibition of the AMPK may reverse radiation resistance (Jin et al., 2016). Furthermore, TPP1 overexpression (ACD gene) was shown to be correlated with radio-resistance and longer telomere length in colorectal cancer cells. TPP1 overexpression was correlated with a decreased apoptosis after irradiation (Yang et al., 2013).
Telomere length was inversely correlated to radio-sensitivity and thus telomere elongation was associated with radio-resistance in human carcinomas (Zhong et al., 2008). Telomere homeostasis is mainly regulated by shelterin, a complex of six telomere-associated proteins TRF1, TRF2, RAP1, TIN2, TPP1 and POT1. TPP1 is a key component of shelterin which heterodimerizes with POT1 and promotes the binding to telomere ssDNA. The POT1-TPP1 complex induces telomerase activity and thus regulates telomere length.
Overexpression of telomere-related proteins including TPP1 was shown to be correlated with radio-resistance in human laryngeal cancer (Tang et al., 2009). Overexpression of TPP1 is associated with radio-resistance and increases the length of telomere in human colorectal cancer cells. TPP1 overexpression also decrease the sensitivity of the cells to X-rays. TPP1 overexpression in radio-resistant colorectal cancer cells was associated with prolonged G2/M arrest mediated by ATM/ATR-Chk1 signaling pathway. In addition, TPP1 overexpression blocked radiation-induced apoptosis by enhancing the repair kinetics of DNA damage TPP1 defective mice showed the importance of the telomere-related proteins in cancer. TPP1 deficient animals showed reduced TERT binding to telomeres. TPP1 deficiency can be associated with perinatal mortality, severe skin hyperpigmentation, defective hair follicle morphogenesis and widespread epithelial dysplasia (Tejera et al., 2010). TPP1 inhibition was shown to induce the telomere shortening in the skin. Previous data suggested that TPP1 deficiency in mouse models indicated that TPP1 plays a dual role in telomere protection and elongation.
In this study, we made a colorectal cancer cells resistant to radiation. Then, the radio-resistant cells were used to screen a panel of natural compounds to identify those that are able to induce caspase activation. Eight compounds were identified in the first round of screening which were then tested in a second round of screening using cell death assay to see if this high caspase activation kills the cells. The hit compounds were then tested using sensitive cells to identify those that specifically kill resistant cells rather than the sensitive cells. Here, we report Apigenin, Deoxyshikonin and Brefeldin A as compounds that are more potent to induce death in radio-resistant cells than sensitive cells.
2 Material and methods
2.1 Cell culture
The radioresistant colorectal cancer HCT116 cells was established by fractional radiation as describe before (Afshar et al., 2018; Su et al., 2014). Radio-resistant HCT116 cells were cultured in DMEM (Sigma), containing 10% FBS (Gibco), and 1% Penicillin/Streptomycin (Sigma). Cells were incubated at 37 °C in a humidified atmosphere with 5% CO2.
2.2 Development of radio-resistant model of colorectal cancer cells
Cells were irradiated using a Faxitron cabinet X-ray system 43855D. Parental cells were weekly exposed to single fractions of radiation. The starting dose of 2 Gy was followed by weekly incremental doses of 0.5 Gy for six months. Cells were then kept in culture with further weekly doses of 4 Gy.
2.3 Cell death assay
The cells were treated with the hit compounds at 0.3, 1, 3, 10, 30, 100, and 300 µM and microscopy was used to visualize the Propidium Iodide (1 µg mL−1) and Hoechst 33,342 (1 µg mL−1) double stained cells. Cell death analyses were completed using Operetta high-content imaging system and the percentage of cell death was calculated as the number of PI positive cells/number of Hoechst positive cells) × 100. The cell survival was calculated as the number of PI negative cells/number of Hoechst positive cells) × 100.
2.4 Colony formation assay
Radio-resistant and the sensitive parental cells were plated into a six well plate (1 × 103 cells/well) and incubated for three weeks. The cells were then washed and fixed with 1,9-dimethyl-methylene blue zinc chloride double salt. The wells were washed and left at room temperature to air-dry and number of visible colonies were counted.
2.5 Caspase-3 like activity
The cell permeable caspase substrate (ApoBrite™ V570 caspase 3/7 substrate Cell-Permeable, AABio and ALX-260–032, Enzo Life Science) was used for screening according to manufacturer’s instruction. Briefly, the substrate were added to each well and the fluorescence was measured at >610 nm wavelength. The activity was calculated as arbitrary fluorescence units per min (AFU min−1).
2.6 Immunoblotting
The sensitive and resistant HCT116 cells were harvested and lysed for 1 h on ice in lysis buffer containing 1% CHAPS, 50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 150 mM KCl, 1 mM EDTA, 1 mM EGTA, 0.1 mM PMSF. The lysate was spinned at 4 °C, 16,000 g and the protein concentration was determined using Bradford reagent (Bio-Rad Laboratories). The proteins were loaded in polyacryl amide gel, electrotransferred to a nitrocellulose membrane, and incubated overnight with the primary antibodies including Anti-TPP1, Anti-AMPK alpha 1 (Abcam) and β-Actin (Cell Signaling). The membranes were then washed and incubated with the relevant secondary antibodies.
2.7 Screening
A library of known natural products with 2500 compounds including some from the classes of terpenoids, peptolides, flavones, coumarins, alkaloids, macrolides, isoflavones, and etc was tested initially at 100 µM (HY-L021, MedChemExpress). The top 10% compounds that increased caspase-3 activity was picked for further analysis.
2.8 Thermal shift assay
Fifteen micrograms of recombinant proteins (ALX-201-059, Enzo Life Science) was incubated with either Brefeldin A or DMSO for 30 min and heated for 5 min at temperatures from 55 to 90 °C in a 96-well Thermal Cycler as described before (Tashakor et al., 2019). The reactions were then spin at 13,000 g for 30 min. The supernatant was then used for SDS-PAGE.
3 Results
3.1 Establishment of radio-resistant colon cancer cells
Radio-resistant HCT-116 cells were made as described in Material and Method. To validate the radio-resistant HCT116, the cells were exposed to a range of radiation doses and cell death was measured. The sensitive cells all died at the highest dose of radiation (12 Gy) while only 45% of the resistant cells died (Fig. 1A). The IC50 was calculated as 4 Gy for wild type and >12 Gy for the resistant cells. However, the maximum survival response was massively higher in resistant cells as 55% of the cells survived in comparison with no survival in wild type HCT116. This level of resistance is consistent with previous reports on the HCT116 as well as other colorectal cancer cells (Endo et al., 2018; Tan et al., 2020; Yang et al., 2013).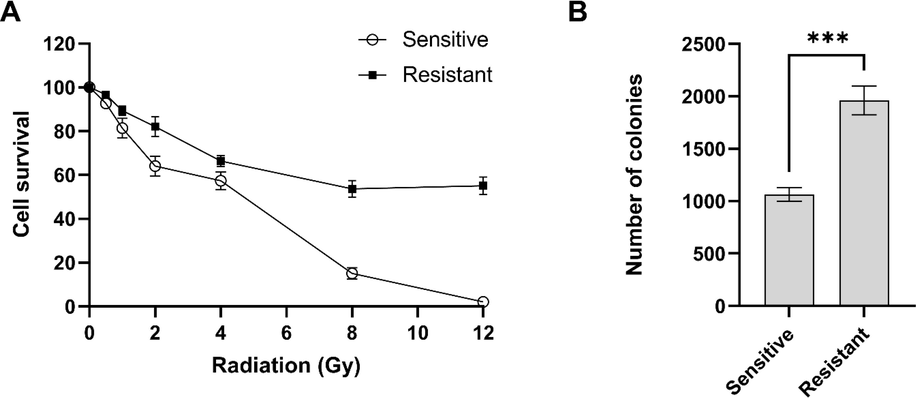
Validation of radio-resistant HCT116 cells. (A) The resistant and sensitive HCT116 cells were exposed to different doses of radiation and cell death was measured. (B) Radio-resistant cells showed higher proliferation in colony formation assay. Error bars represent mean ± SD of three independent experiments.
To further validate the phenotype of radio-resistant HCT116 cells, we looked at the expression of two markers. It has been shown that AMPK and TPP1 (ACD gene) are both overexpressed in radio-resistant patient samples (Jin et al., 2016; Yang et al., 2013). Therefore, the immunoblotting was performed to detect the level of proteins. As expected, HCT116 cells exposed to radiation showed higher level of both proteins (data not shown). These data suggests that the radio-resistant HCT116 cell model is consistent with physiological conditions and can be used as a reliable experimental model.
Colony formation assay was performed to examine the long-term proliferation of the resistant cells. The radio-sensitive and radio-resistant cells were exposed to Brefeldin Afor 48 h and then the cells incubated for three weeks. Counting the number of colonies showed that radio-resistant cells has higher rate of proliferation than the sensitive cells (p-value 0.0005) (Fig. 1B). The higher rate of proliferation was reported before in breast and head and neck cancer cells and this data was consistent with previous reports (Gray et al., 2019; Todorovic et al., 2019).
3.2 Screening a natural product library identified eight hit compounds to increase caspase-3 activity
The in cellulo screening was performed in 384-well format. The cells treated with the library compounds and caspase-3 like activity was measured using cell-permeable fluorescent caspase-3 substrate. The etoposide was used as a positive control to induce caspase activation (Zou et al., 2012). The inhibitor of caspase-3, QVD-OPh, was used as a negative control. These positive and negative controls were used to identify the range of caspase activity (Fig. 2A). The Z-score was 0.62 which shows the well-separation of the different experimental groups. The screening was performed and the lowest and the highest 10% of the compounds were picked (Fig. 2B). This exclude the compounds with 3SD in caspase-3 like activity. For further studies the eight compounds that increased the caspase activation were selected.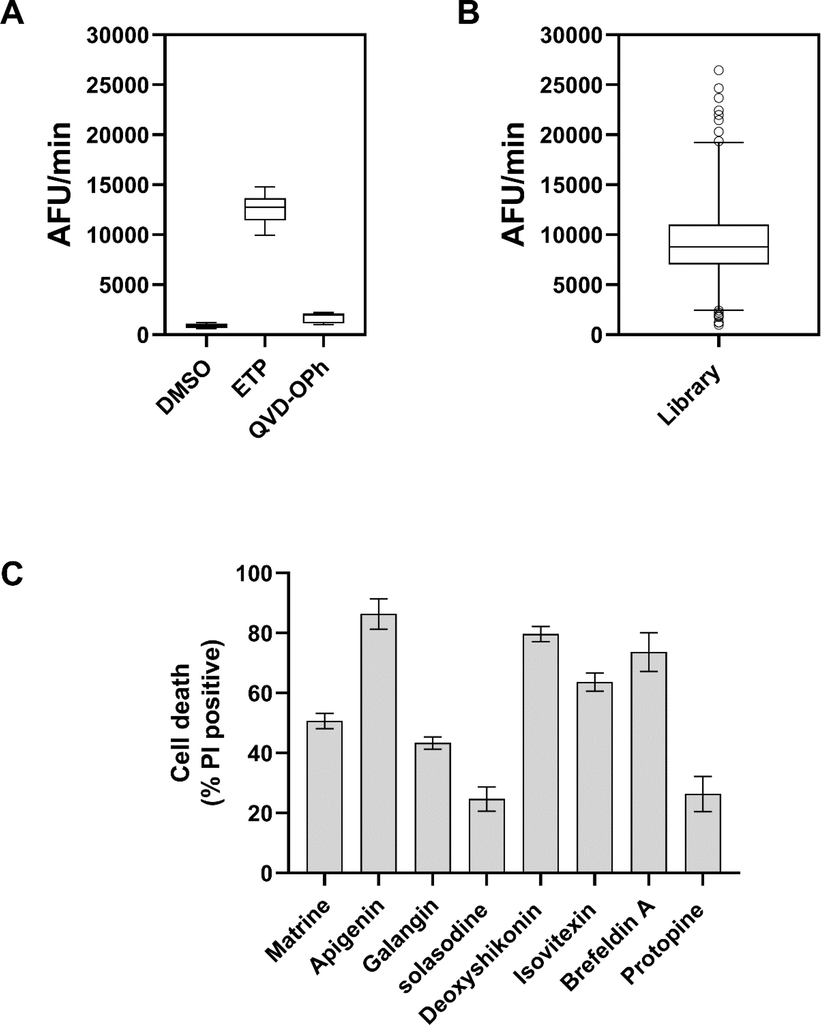
Screening of a natural product library. (A) Untreated, etoposide 15 µM and QVD-Oph treated radio-resistant cells were used in caspase activity assay to identify the range of caspase activation. (B) The screening was performed and the top 10% of the compounds that increased the caspase-3 activity was selected as hit compounds. Box and whiskers represent the mean and 10–90 percentile. (C) The radio-resistant cells were treated with eight hit compounds and cell death was measured after 24 h. Error bars represent mean ± SD of three independent experiments.
3.3 Six compounds showed the high cell death in radio-resistant HCT116 cells
In the second round of screening, the cell death assay was performed to ensure that the increased caspase-3 activation leads to death of radio-resistant cells. The radio-resistant cells were treated with hit compounds at 100 µM and cell death was measured by PI staining. The results showed that three of the compounds including Apigenin, Deoxyshikonin and Brefeldin A induced massive cell death all >70%. Three of the compounds induced 40%-60% death (Matrine, Galangin, and Izovitexin). However, Solasodine and Protopine failed to induce massive death (20%) in radio-resistant cells (Fig. 2C). Therefore, those six compounds with the ability to kill approximately 50% of the cells were selected for further investigation.
3.4 Hit compounds were titrated to calculate the potency and efficacy
The screening and the following experiments were performed using 100 µM of the compounds. To better understand the potency and efficacy, the hit compounds were titrated to see how they impact the cell death at lower or higher concentrations. We tested a range of concentrations from 0.3 to 300 µM. The IC50 values (potency) and the maximum responses (efficacy) were summarized in Table 1.
Compounds
IC50 (µM)
Maximumresponse
Matrine
36
68
Apigenin
42
100
Galangin
27
60
Deoxyshikonin
50
100
Izovitexin
45
85
Brefeldin A
45
87
Apigenin and Deoxyshikonin killed all the treated radio-resistant cells with the IC50 about 50 µM (Fig. 3A–F). Ixovitexin and Brefeldin A induced 85% cell death with the IC50 of 45 µM. Matrine and Galangin induced 68% and 60% death with IC50 of 36 and 27 µM, respectively.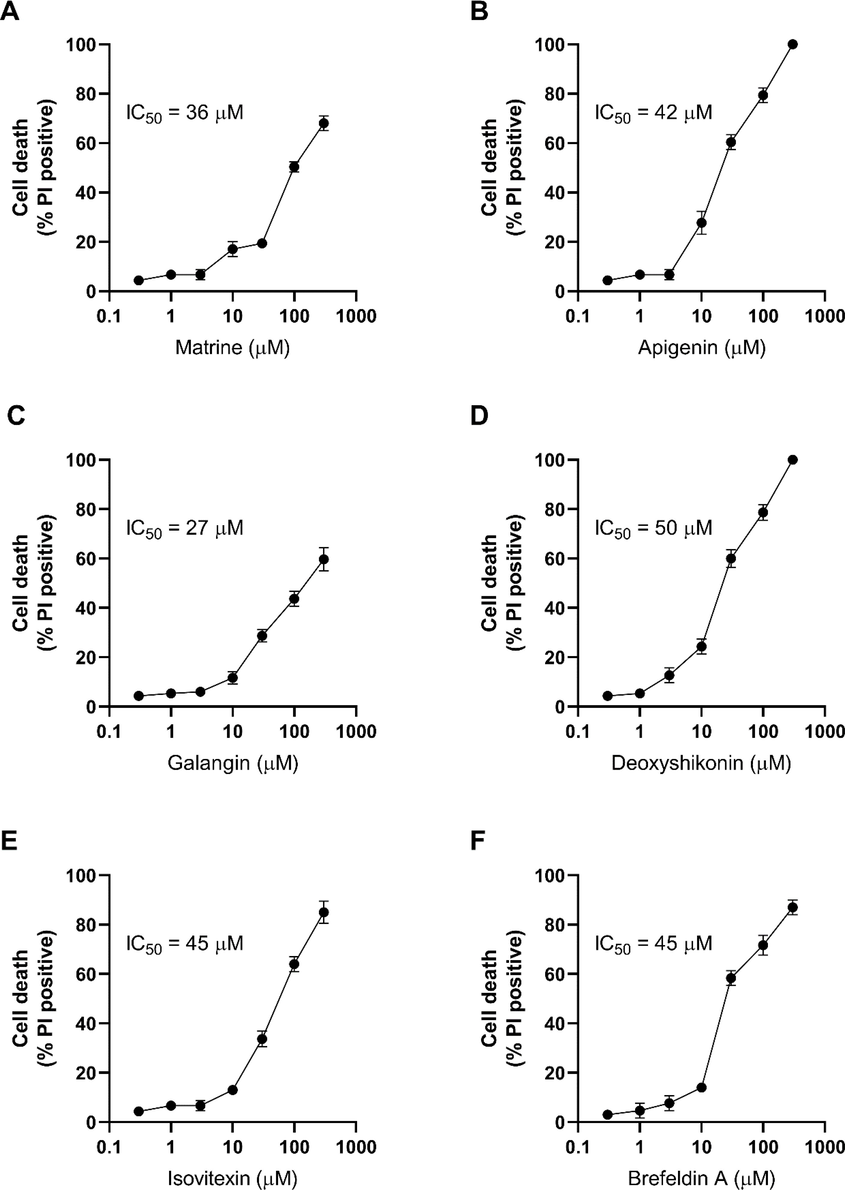
Titration of the hit compounds on radio-resistant HCT116 cells. The radio-resistant cells were treated with a range of concentrations from 0.3 to 300 µM of (A) Matrine, (B) Apigenin, (C) Galangin, (D) Deoxyshikonin, (E) Izovitexin and (F) Brefeldin A and cell death was measured 24 h. Error bars represent mean ± SD of three independent experiments.
3.5 Three of the hit compounds induced low level of death in radio-sensitive cells
As we were seeking to identify the compounds that specifically kill the radio-resistant cells not the sensitive cells, we tested the cell death induced by hit compounds in radio-sensitive HCT116 cells. Except for Izovitexin, all the other hits showed 40%-60% death in sensitive cells. However the impact of hit compounds was different on radio-resistant and -sensitive cells (Fig. 4A). Apigenin, Deoxyshikonin and Brefeldin A were selected to be titrated on radio-sensitive HCT116 cells as they showed high potency in killing the radio-resistant cells. Brefeldin A killed 87% of the resistant cells while it killed 41% of sensitive cells. The highest concentration of Deoxyshikonin tested in this study killed all the radio-resistant cells and also killed 50% of sensitive cells. Apigenin also induced 100% death in resistant cells while it killed 64% of sensitive cells (Fig. 4B–D).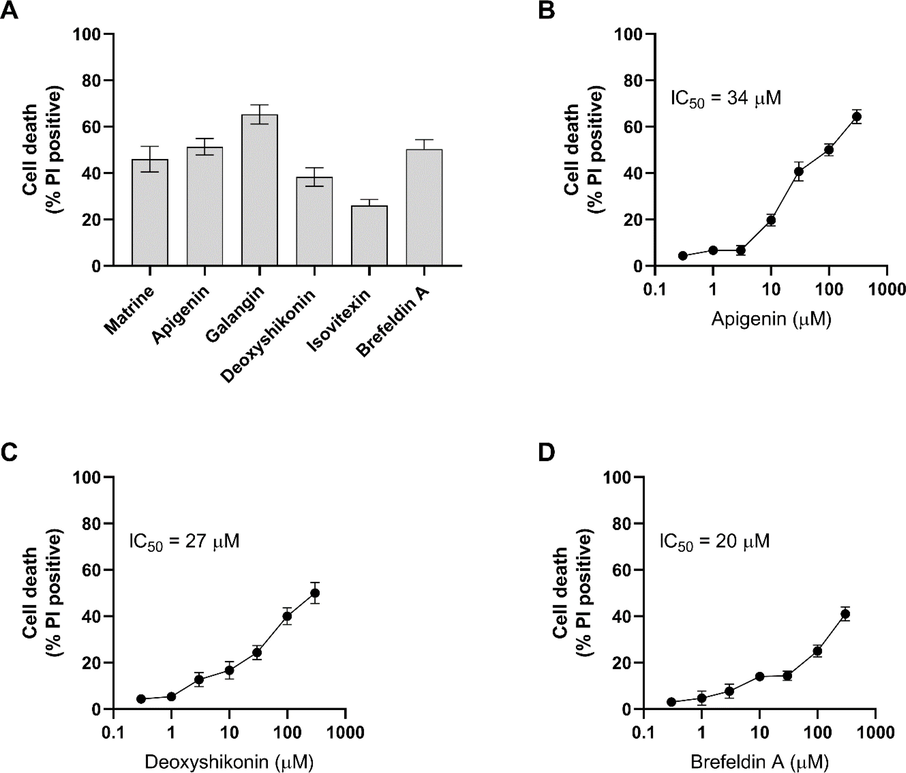
Cell death induced by hits on radio-sensitive cells. (A) The radio-sensitive HCT116 cells were treated with 100 µM and cell death was measured 24 h. (B-D) Three of the compounds with the highest impact on radio-resistant cells and lowest effect on sensitive cells were titrated. Error bars represent mean ± SD of three independent experiments.
3.6 Brefeldin A inhibits the long-term proliferation of radio-sensitive cells
In order to examine the ability of Brefeldin A to alter the proliferation of radio-resistant cells, colony formation assay was performed using sub-IC50 and IC50 concentrations of Brefeldin A. The number of colonies in radio-resistant cells was twice that of sensitive cells. 10 µM Brefeldin A significantly inhibited the colony formation and 50 µM Brefeldin A induced even more inhibition (Fig. 5) suggesting that this inhibition in dose-dependent. This finding was very interesting as showed that even very low concentrations of Brefeldin A was able to inhibit the proliferation of the radio-resistant cells in a long-term, although low cell death was induced in short-term exposure.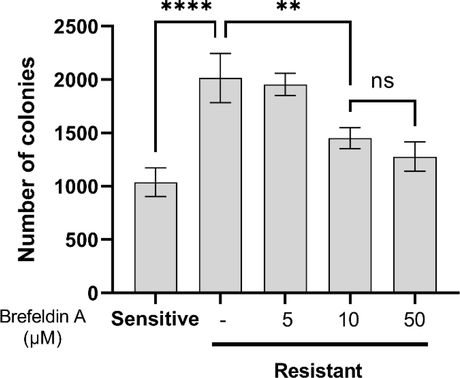
Brefeldin A inhibits the colony formation of radio-resistant cells in a dose-dependent manner. Error bars represent mean ± SD of three independent experiments.
3.7 Brefeldin A kills the radio-resistant cells mainly by apoptosis and stabilizing caspase-3
To further evaluate the mode of death induced by Brefeldin A in radio-resistant colorectal cancer cells, the cells were treated with M50054 (Apaf-1 inhibitor) to block intrinsic apoptosis, ferrostatin-1 to inhibit ferroptosis, necrostatin-1 to inhibit necroptosis and IM-54 to block necrosis and cell survival was tested in response to Brefeldin A. We showed that M50054 blocked the death induced by Brefeldin A (Fig. 6A). This data was consistent with previous reports about the apoptotic function of Brefeldin A (Lee et al., 2013). Ferrostain-1 and necrostatin-1 failed to rescue the cells exposed to Brefeldin A suggesting that ferroptosis and necroptosis were not the mode of death induced by Brefeldin A. However, IM-54 rescued some cells treated with Brefeldin A but to a lower level to M50054 (p-value 0.0011). This data suggested that Brefeldin A was able to induce mainly apoptosis and also necrosis in radio-resistant cells. To ensure that both this modes of death are responsible for Brefeldin-induced death, we co-treated the radio-resistant cells with both M50054 and IM-54 and we found that Brefeldin A totally failed to induce death (Fig. 6B).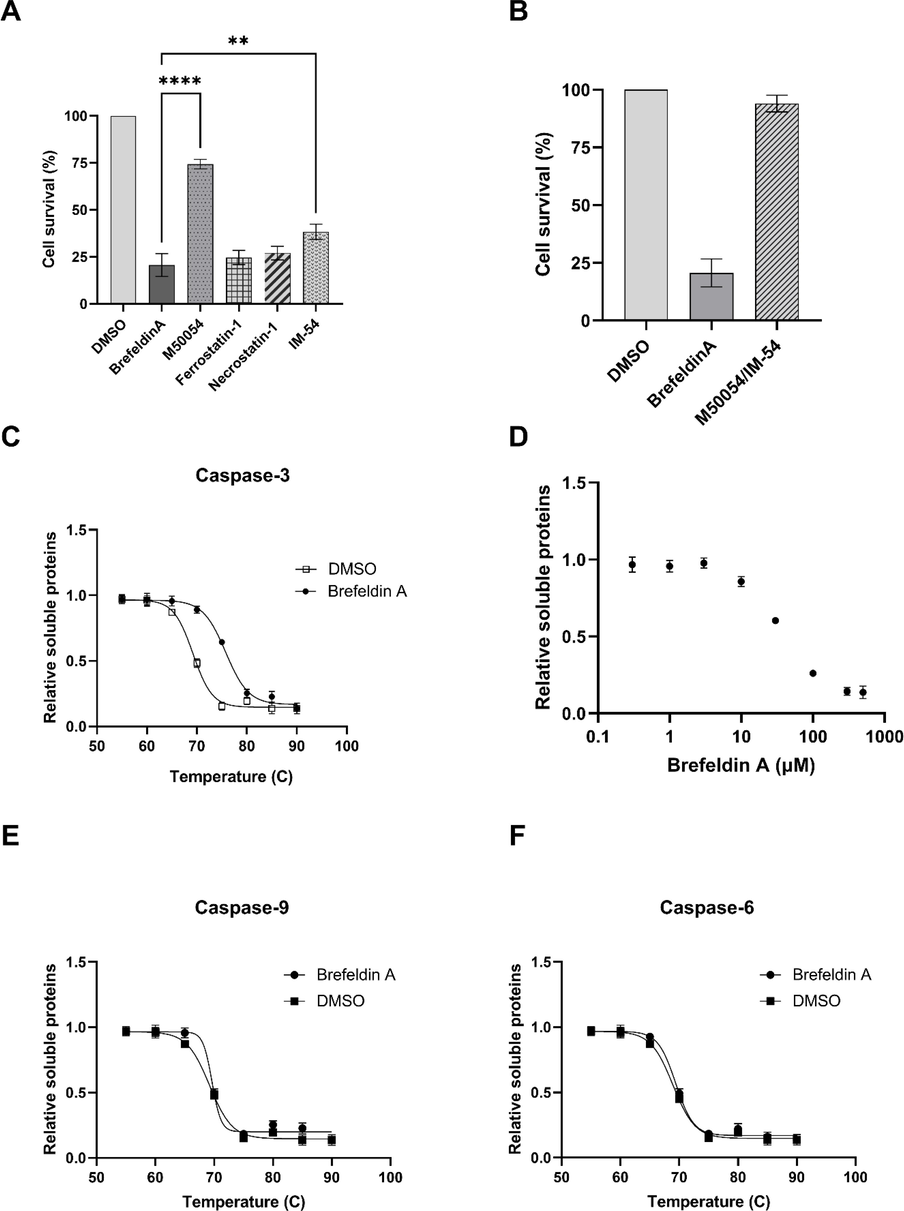
Mechanism of Brefeldin induced death. (A) Cell death modalities induced by Brefeldin A. The cells treated with specific inhibitors of apoptosis, ferroptosis, necroptosis and necrosis and cell death was measured. (B) Co-treatment of apoptosis and necrosis inhibitors totally blocked the death induced by Brefeldin A. Thermal shift assay of Brefeldin A with (C) Caspase-3, (E) Caspase-9 and (F) Caspase-6. (D) Caspase-3 treated with Brefeldin A concentrations and the amount of soluble proteins were determined. Error bars represent mean ± SD of three independent experiments.
As Brefeldin A was identified by measuring the caspase-3 activity (Fig. 2), we aimed to investigate if Brefeldin A interacted to caspase-3. We used recombinant caspase-3 and performed thermal shift assay in the presence of Brefeldin A. thermal shift assay showed that 50% of caspase-3 was denatured at 70 °C and treatment with Brefeldin A stabilized the caspase-3, as shown by 50% denaturation at 76 °C (Fig. 6C). This thermal shift suggested that Brefeldin A directly binds to and stabilizes caspase-3. Then thermal shift assay was performed using brefeldin concentrations and we found that Brefeldin A binds to caspase-3 with the EC50 of about 50 µM (Fig. 6D). This concentration is close to the one that induce death in radio-resistant cells, suggesting that binding to caspase-3 was the main cause of death. We also looked at the Brefeldin A binding to other apoptotic caspases like caspase-9 and caspase-6. Thermal shift assay failed to detect any interactions (Fig. 6E,F). This is an interesting finding as it suggests that Brefeldin A specifically interacts to caspase-3 not caspase-6 and -9. Natural products have been reported to regulate the gene expressions. We measured the transcripts of key components of intrinsic apoptosis and identified that Brefeldin A didn’t alter the transcription of Apaf-1, caspase-3, caspase-9, PARP and XIAP (supplementary Fig. 1).
4 Discussion
Radioresistance is a major challenge for further improvement of therapeutic effects of chemotherapy and neoadjuvant radiotherapy. However, the underlying mechanisms that lead to radio-resistance are not fully understood, although DNA repair system, apoptosis and cell survival, epithelial-mesenchymal transition, tumor stem cell, and the generation of ROS have been proposed to be involved (Baptistella et al., 2019; Ji et al., 2018; Wang et al., 2019; Yu et al., 2014). Radiation resistance have been reported in many cancers like gastrointestinal cancer where it occurs in 70–96% of cases and colorectal cancer (Buckley et al., 2020). To date, not a single compound has been identified to fully re-sensitize the radio-resistant cells. In this study, we screened a large panel of natural product library aiming to identify compounds to selectively target radio-resistant colorectal cancer cells.
We here established a radio-resistant model of colorectal cancer. We exposed HCT116 cells to increasing doses of radiation. The cell death analysis confirmed the resistance to radiation as radiated HCT116 cells survived at high doses of radiation, unlike the control group. Furthermore, to further validate our radio-resistant model, the expression of AMPK and ACD was studied. In radio-resistant cells both AMPK and ACD overexpressed. This is consistent with previous studies that showed the overexpression of these two proteins in colorectal patients with radio-resistant tumor cells (Jin et al., 2016; Yang et al., 2013). However, there are some other factors that differ in radio-resistant cells such as overexpression of phosphatidylinositol transfer protein, cytoplasmic 1 (PITPNC1) and HIF-1α (Nagaraju et al., 2015; Tan et al., 2020).
Then we used radio-resistant cells to screen the inducers and inhibitors of caspase-activation. We identified eight compounds which massively increased caspase-3 activation. In this study we focused in the inducers of caspase activation. The inhibitors are being studied to see if they directly target caspases or it is a consequence of upstream events. These compounds then tested in cell death assay to ensure they kill the radio-resistant cells. This step was essential as in some cases caspase activation does not lead to cell death, instead it functions in non-apoptotic roles such as muscle differentiation, neuroplasticity and neurogenesis (Dehkordi et al., 2020; Tzeng et al., 2013).
The six compounds with the highest ability to kill radio-resistant cells were then titrated on radio-resistant HCT116 cells. Among all the compounds Apigenin, Deoxyshikonin and Brefeldin A killed all the resistant cells at the highest concentration tested in this study. We also showed that these compounds kill the sensitive cells but at much lower level than resistant cells. Brefeldin A can induce apoptosis in human tumor cells independent of p53 and by activating caspase-3 (Lee et al., 2013). Apigenin has been shown before to induce apoptotic cell death in a dose-dependent manner and elevates ROS. In HepG2 cells Apigenin induced ROS and activated NADPH oxidase and thus induced apoptosis. Deoxyshikonin was shown to decrease the tumor mass and downregulated PI3K, p-PI3K, Akt, p-Akt308 and mTOR (Zhu et al., 2019). All our screening data is consistent with the apoptosis inducing function of the hit compounds reported earlier. In addition, here we showed that not only these compounds are able to induce caspase-dependent apoptosis, but also able to kill radio-resistant colorectal cancer cells through a different mechanism.
Using natural products to identify pharmacological small molecules is a powerful source due to their diversity (Wilson et al., 2020). In this study, we found the compounds that kills radio-resistant cells, however, there are some effects on sensitive cells. These natural compounds need to be further studied aiming to identify their direct targets in radio-resistant cells.
Brefeldin A was shown to induce death in radio-resistant cells. The mode of Brefeldin A induced death was mainly apoptosis and necrosis, to the lower extend. This was confirmed by co-treatment of the cells with M50054 and IM-54, intrinsic apoptosis and necrosis inhibitor respectively and restoring the survival of Brefeldin A treated cells to the control level. We also showed using recombinant apoptotic caspases that caspase-3 is the direct target for Brefeldin A and this interaction stabilizes the caspase-3. However, other caspases like caspase-6 and caspase-6 did not show interaction with Brefeldin A. Many of the natural compounds modulate the expression of cellular proteins at the transcription level. We here showed that Brefeldin A did not at least alter the transcription of key components of intrinsic apoptosis including Apaf-1, caspase-9, caspase-3, PARP and XIAP. Our data here would open a new window toward re-sensitizing the radio-resistant cells by modifying the structure of the compounds to improve the affinity. In addition, identifying the direct target for other hit compounds can provide a novel therapeutic candidate which enable us to modulate the signaling of radio-resistant cells.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- MiR-185 enhances radiosensitivity of colorectal cancer cells by targeting IGF1R and IGF2. Biomed. Pharmacother.. 2018;106:763-769.
- [CrossRef] [Google Scholar]
- Rab5C enhances resistance to ionizing radiation in rectal cancer. J. Mol. Med.. 2019;97(6):855-869.
- [CrossRef] [Google Scholar]
- Transient elevation of glycolysis confers radio-resistance by facilitating DNA repair in cells. BMC Cancer. 2015;15:335.
- [CrossRef] [Google Scholar]
- Reprogramming of tumor metabolism by targeting mitochondria improves tumor response to irradiation. Acta Oncol.. 2015;54(2):266-274.
- [CrossRef] [Google Scholar]
- Targeting hallmarks of cancer to enhance radiosensitivity in gastrointestinal cancers. Nat. Rev. Gastroenterol. Hepatol.. 2020;17(5):298-313.
- [CrossRef] [Google Scholar]
- Fra-1 enhances the radioresistance of colon cancer cells to X-ray or C-ion radiation. Oncol. Rep.. 2018;39:1112-1118.
- [CrossRef] [Google Scholar]
- Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009;462(7275):935-939.
- [CrossRef] [Google Scholar]
- Molecular effectors of radiation resistance in colorectal cancer. Prec. Radiat. Oncol.. 2017;1(1):27-33.
- [CrossRef] [Google Scholar]
- Development and characterisation of acquired radioresistant breast cancer cell lines. Radiat. Oncol.. 2019;14:64.
- [CrossRef] [Google Scholar]
- Apoptosome-dependent myotube formation involves activation of caspase-3 in differentiating myoblasts. Cell Death Dis.. 2020;11(5)
- [CrossRef] [Google Scholar]
- Mechanisms of resistance to ionizing radiation in rectal cancer. Expert. Rev. Mol. Diagn.. 2009;9(5):469-480.
- [CrossRef] [Google Scholar]
- Enhancement of sensitivity to chemo/radiation therapy by using miR-15b against DCLK1 in colorectal cancer. Stem Cell Rep.. 2018;11(6):1506-1522.
- [CrossRef] [Google Scholar]
- Re-sensitization of radiation resistant colorectal cancer cells to radiation through inhibition of AMPK pathway. Oncol. Lett.. 2016;11:3197-3201.
- [CrossRef] [Google Scholar]
- Targeting the enzymes involved in arachidonic acid metabolism to improve radiotherapy. Cancer Metastasis Rev.. 2018;37(2-3):213-225.
- [CrossRef] [Google Scholar]
- Insights into the respiratory chain and oxidative stress. Biosci. Rep.. 2018;38
- [CrossRef] [Google Scholar]
- Brefeldin a induces apoptosis by activating the mitochondrial and death receptor pathways and inhibits focal adhesion kinase-mediated cell invasion. Basic Clin. Pharmacol. Toxicol.. 2013;113:329-338.
- [CrossRef] [Google Scholar]
- Hypoxia inducible factor-1α: Its role in colorectal carcinogenesis and metastasis. Cancer Lett.. 2015;366(1):11-18.
- [CrossRef] [Google Scholar]
- Radiation resistance of cancer stem cells: the 4 R’s of radiobiology revisited. Stem Cells. 2010;28(4):639-648.
- [CrossRef] [Google Scholar]
- Colorectal cancer statistics, 2014. CA Cancer J. Clin.. 2014;64(2):104-117.
- [CrossRef] [Google Scholar]
- Identification of microRNAs involved in the radioresistance of esophageal cancer cells. Cell Biol. Int.. 2014;38(3):318-325.
- [CrossRef] [Google Scholar]
- PITPNC1 fuels radioresistance of rectal cancer by inhibiting reactive oxygen species production. Ann Transl Med. 2020;8:126.
- [CrossRef] [Google Scholar]
- Targeting CPT1A-mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy. Theranostics. 2018;8(9):2329-2347.
- [CrossRef] [Google Scholar]
- Role of metabolism in cancer cell radioresistance and radiosensitization methods. J. Exp. Clin. Cancer Res.. 2018;37:87.
- [CrossRef] [Google Scholar]
- Increased expression of telomere-related proteins correlates with resistance to radiation in human laryngeal cancer cell lines. Oncol. Rep.. 2009;21:1505-1509.
- [CrossRef] [Google Scholar]
- A new split-luciferase complementation assay identifies pentachlorophenol as an inhibitor of apoptosome formation. FEBS Open Bio. 2019;9(7):1194-1203.
- [CrossRef] [Google Scholar]
- TPP1 is required for TERT recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice. Dev. Cell. 2010;18(5):775-789.
- [CrossRef] [Google Scholar]
- Mechanisms of different response to ionizing irradiation in isogenic head and neck cancer cell lines. Radiat. Oncol.. 2019;14:214.
- [CrossRef] [Google Scholar]
- Caspase 3 involves in neuroplasticity, microglial activation and neurogenesis in the mice hippocampus after intracerebral injection of kainic acid. J. Biomed. Sci.. 2013;20:90.
- [CrossRef] [Google Scholar]
- Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol.. 2018;24(34):3834-3848.
- [CrossRef] [Google Scholar]
- Genome-wide RNAi screening identifies RFC4 as a factor that mediates radioresistance in colorectal cancer by facilitating nonhomologous end joining repair. Clin. Cancer Res.. 2019;25(14):4567-4579.
- [CrossRef] [Google Scholar]
- Creating and screening natural product libraries. Nat. Prod. Rep.. 2020;37(7):893-918.
- [Google Scholar]
- Telomere-binding protein TPP1 modulates telomere homeostasis and confers radioresistance to human colorectal cancer cells. PLoS ONE. 2013;8(11) e81034
- [CrossRef] [Google Scholar]
- Silencing the Girdin gene enhances radio-sensitivity of hepatocellular carcinoma via suppression of glycolytic metabolism. J. Exp. Clin. Cancer Res.. 2017;36:110.
- [CrossRef] [Google Scholar]
- Multidrug resistance-associated protein 3 confers resistance to chemoradiotherapy for rectal cancer by regulating reactive oxygen species and caspase-3-dependent apoptotic pathway. Cancer Lett.. 2014;353(2):182-193.
- [CrossRef] [Google Scholar]
- Telomere length inversely correlates with radiosensitivity in human carcinoma cells with the same tissue background. Biochem. Biophys. Res. Commun.. 2008;367(1):84-89.
- [CrossRef] [Google Scholar]
- Deoxyshikonin isolated from Arnebia euchroma inhibits colorectal cancer by down-regulating the PI3K/Akt/mTOR pathway. Pharm. Biol.. 2019;57(1):412-423.
- [CrossRef] [Google Scholar]
- Interaction of caveolin-1 with Ku70 inhibits Bax-mediated apoptosis. PLoS ONE. 2012;7(6) e39379
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2021.101728.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




