

Translate this page into:
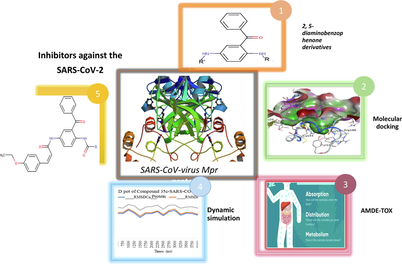
Combined docking methods and molecular dynamics to identify effective antiviral 2, 5-diaminobenzophenonederivatives against SARS-CoV-2
⁎Corresponding author. prof.belaidi@gmail.com (Salah Belaidi),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
The aim of this work is to contribute to the research in finding lead compounds for clinical use, to identify new drugs that target the SARS-CoV-2 virus main protease (Mpro). In this study, we used molecular docking strategies to analyze 2.5-diaminobenzophenone compounds against Malaria and to compare results with the Nelfinavir as a FDA-approved HIV-1 protease inhibitor recommended for the treatment of COVID-19. These efforts identified the potential compounds against SAR-COV-2 Mpro with the docking scores ranges from −6.1 to −7.75 kcal/mol, which exhibited better interactions than the Nelfinavir. Among thirty-six studied, compounds 20c, 24c, 30c, 34c, 35c and 36c showed the highest affinity and involved in forming hydrophobic interactions with Glu166, Thr24, Thr25, and Thr26 residues and forming H-bonding interactions with Gln189, Cys145, and His41residues. Pharmacokinetic properties and toxicity (ADMET) were also determined for identified compounds. This study result in the identification of two compounds 35 and 36 having high binding affinity, good pharmacokinetics properties and lowest toxicity. The structural stability and dynamics of lead compounds within the active site of 3CLpro was also examined using molecular dynamics (MD) simulation. Essential dynamics demonstrated that the two complexes remain stable during the entire duration of simulation. We have shown that these two lead molecules would have the potential to act as promising drug-candidates and would be of interest as starting point for designing compounds against the SARS-CoV-2.
Keywords
SARS-CoV-2
Inhibitor
Nelfinavir
Benzophenone
Molecular Docking

1 Introduction
At the end of 2019 in Wuhan, COVID-19 was first reported in China and has since spread abundantly in China and around the world (Huang et al., 2020) is a new strain of the coronavirus species SARS -CoV.
The progression of this disease, led the World Health Organization to declare it a public health emergency of international scope on January 30, 2020, then pandemic on March 11, 2020. According to the World Health Organization, Corona viruses are a large family of viruses that can be pathogenic in humans and animals. In humans, several corona virus scans cause respiratory infections ranging from colds to more illnesses that are serious. (Zhu et al., 2020)
The complete virus particle made of four major structural proteins, namely, spikes (S), nucleocapsid (N), membrane (M) and envelope (E) encoded by the virus genome. The virus's S protein shows similarity to SARS-CoV's S protein. COVID-19 spreads from person to person, and this makes them more likely to cause infection. Many efforts are being made to find strategies for preventing COVID-19. (Forni et al. 2017)
Coronavirus replication (CoV) is a highly coordinated process that includes complex transcription mechanisms to protect the virus genome, which is the largest known RNA genome, and viral proteins from the host's antiviral defense mechanisms (Brian and Baric, 2005).
Coronary proteases, papain-like proteases (PLpro) and 3C-like proteases (3CLpro) are attractive targets for antiviral drugs because they are essential for the reproduction of the coronavirus. (Báez-Santos et al., 2015)
Then there is a need to quickly develop and approve a vaccine, which is not yet available. Nonetheless, Chang et al., Suggested that some drugs against the same type of virus approved by the United States Food and Drug Administration (FDA) may offer promising results.
An investigation performed by (Xu et al. (2020)), indicated that among 4 tested drugs (nelfinavir, pitavastatin, perampanel, and praziquantel), Nelfinavir was identified as the best potential inhibitor against SARS-CoV-2main protease, based on binding energy calculations using the molecular mechanics with generalized Born and surface area solvation (MM/GBSA) model and solvated interaction energy (SIE) methods (Khaerunnisa et al. 2020).
Molecular docking is the most used method of computational chemistry in structure-based drug design because of its ability to predict, with reliable accuracy (Meng et al., 2011).
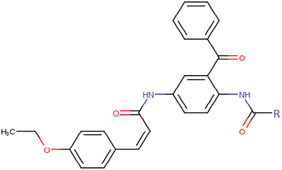
Since it has been reported that antimalarial drugs can be used as anti SARS drugs by targeting 3CLpro (3-chymotrypsin-like cysteine protease) of SARS-CoV-2. We have selected thirty-six 2,5-diamino benzophenone derivatives have been antimalarial activity to explore their binding interactions with 3CLpro and to evaluate their potential against coronavirus pneumonia (SARS-CoV-2) infection by means of computational methods using docking tool using MOE software.
We believe that this study should provide a good overview of the binding and interaction of antimalarial agents with the SARS-CoV-2Mpro.
2 Materials and methods
2.1 Ligand identification
The thirty-six2.5-diaminobenzophenones that were considered for the study were obtained from literature (Wiesner et al., 2001).as it is a class it suppresses the growth of the multiresistant P. falciparum strain Dd2 in the range.
The 2D structures of the molecules shown in Table1 were drawn using Marvin software (MarvinSketch, 2017), converted to 3D and optimized by HyperChem 8.03 software (HyperChem, 2007). The geometries of 2, 5-diaminobenzophenone derivatives; were first optimized by molecular mechanics, with MM+ force field (RMS = 0.001 Kcal/Å). Further, geometries were entirely re-optimized by using PM3 semi-empirical method.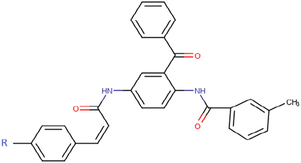
1a
-H
5a
-NH2
9a
-CH2-CH3
13a
-O-(CH2)3-(CH3)3
2a
-Cl
6a
-CH3
10a
-CH (CH3)2
3a
-NO2
7a
-CF3
11a
-C (CH3)3
4a
-Br
8a
-OCH3
12a
-O-CH2-CH3
14b
16b
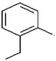
15b
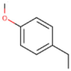
17b
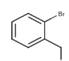
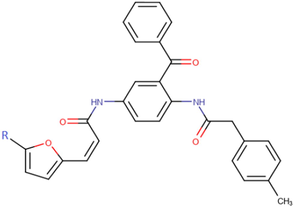
18c
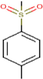
23c
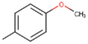
28c
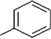
33c
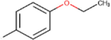
19c
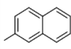
24c
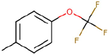
29c
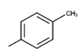
34c
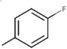
20c
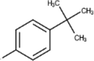
25c
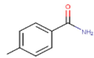
30c
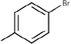
35c
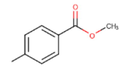
21c
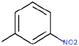
26c
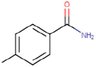
31c
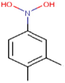
36c
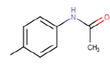
22c
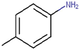
27c
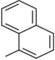
32c
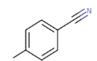
We started our investigations by choosing a suitable methodology to be used for the determination of the equilibrium structures of the 2.5-diamino-benzophenonederivatives. Our strategy consists in performing benchmark computations on the subunit of the series (Al Mogren et al., 2020; Belaidi et al., 2004; Boudergua et al., 2019; Zerroug et al., 2019).
2.2 Molecular docking simulations
Molecular docking protocols are increasingly used to predict binding affinities for a large number of ligands (Ouassaf et al., 2018; Belhassan et al., 2020). In this study, we selected the 3-chymotrypsin-like protease (3CL-protease), which is the main protease used to cleave multiple proteins into replication-linked proteins, and RdRp, the principal protein for RNA replication, as the target receptor.
The crystal structure of main protease (Mpro) from COVID-19 accessible in the Protein Data Bank (PDB) (https://www.rcsb.org/structure/6LU7) was downloaded and it is original ligand was eliminated.
Molecular docking studies are carried out using MOE (MOE Version 2007.09), developed by Chemical Computing Group, in order that to probe the interaction mechanism and binding mode of 2.5-diamino benzophenone derivatives and the FDA drug Nelfinavir, at the active site of the protein studied 6lu7. Protein energy is minimized by activating the MOE software's minimization algorithm. The final and initial energy of the proteins is calculated in kcal / mol by the MOE software using the force field MMFF94x, with a so-called conjugate gradient algorithm. All water molecules were removed and the covalently linked peptidomimetic ligand was released from Cys145. The double bond α, β of the ligand, who behaves like a Michael acceptor, was renovating.
The structure preparation module of MOE was used to correct PDB incoherence and to attribute the protonation state at pH = 7.0. The substrate/binding pockets of SARS-CoV-2 main protease are showed in Fig. 1b, having a remarkably high level of alignment (RMSD = 0.99 Å) of the key residues involved in the binding of the substrate, including the Cys145, His41, Thr45, Met49, Phe140, Asp187, Asn142, Arg188, Gln189, Met165, His172 and Glu166. It is believed that they provide the grid opening of the substrate to the active state.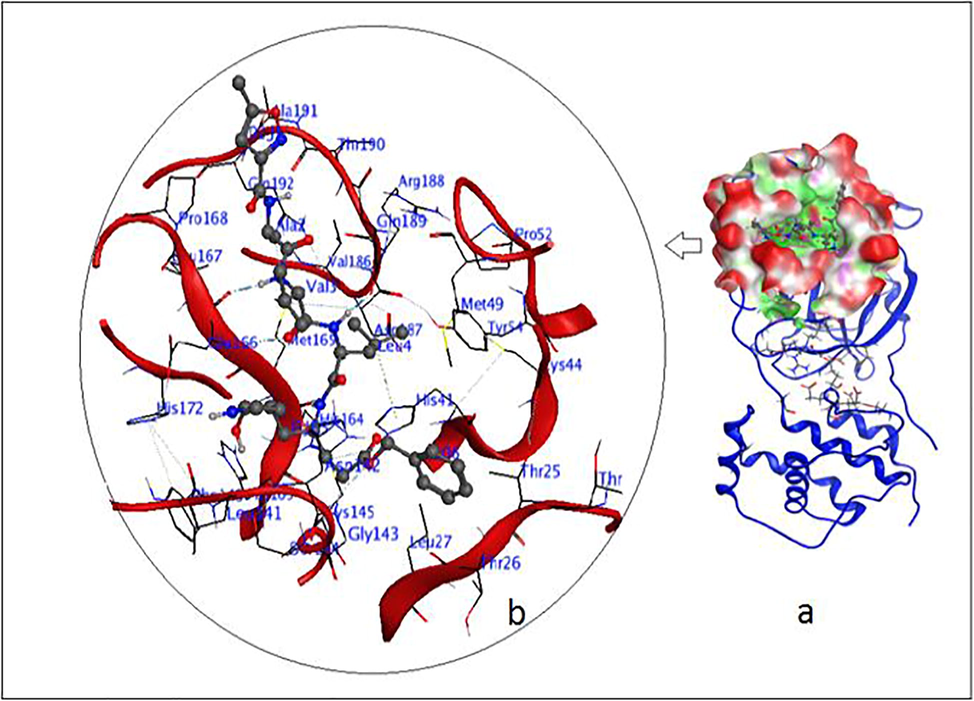
(a) Crystal structure of SARS-CoV-2virus Mpr. (b) Binding pocket with the main residues in the representation of bonds.
2.3 In silico pharmacokinetics analysis
ADME-Tox pharmacokinetics such as absorption, distribution, metabolism, excretion and toxicity of the candidate leads can be predicted using pkCSM (Pires et al., 2015).
The kCSM signatures were successfully used across five main pharmacokinetic properties classes to develop predictive regression and classification models. PK properties were calculated and checked for compliance with their standard ranges.
2.4 Molecular dynamics simulations
The YASARA Structure software (version 14.12.2) was used for all MD simulations of atoms by using of the AMBER14 force field.
The simulation cell (20 Å) filled with water, included the SARS-CoV-2 Main Protease (Mpro) protein. The experimental condition was set and maintained at the constant pressure (3 × 107 Pa) and 298 K for the entire simulation period.
We placed the Mpro protein and molecules in the center of a periodic standard cubic box and added in the counter ions, Na+ and Cl- to mimic the physiological condition at pH 7.4. Simulation was performed at constant pressure and temperature (NPT ensemble) for five nanoseconds (5 ns) with a time step of 2.5 fs. A pre-established macro script (md_run.mcr) within the YASARA software has performed all simulation steps. The MD simulation was performed using the md_run.mcr script in YASARA Structure software, and a more stable conformation came out every 100 ps.
MD simulation results were estimated for the compounds and in complex Mpro protein. The visualization was presented using YASARA Structure software (Krieger and Vriend, 2014) and Discovery Studio (Discovery Studio Modeling Environment, Release 2017, San Diego: Dassault Systèmes., 2016).
3 Results and discussions
3.1 In silico molecular docking
To know the selectivity of our compounds for SARS-CoV-2, molecular docking was carried out using MOE software. Therefore, we aim to study the molecular interactions implicated between the active binding sites of the target protein and 2,5-diaminobenzophenone derivatives and reference drug (Nelfinavir). Docking results are summarized in Table 2. The binding energy of the molecules has been shown to be negative; Nelfinavir implies that the energy required forming an interaction with the best active site of the complex with good stability. (Fig. s1)
N°
binding affinity (kcal/mol)
N°
binding affinity(kcal/mol)
N°
binding affinity (kcal/mol)
1a
−6.57
14b
−6.81
27c
−6.80
2a
−5.73
15b
−6.83
28c
−6.54
3a
−6.53
16b
−6.59
29c
−6.86
4a
−6.85
17b
−6.10
30c
−7.22
5a
−6.77
18c
−6.99
31c
−6.61
6a
−6.51
19c
−6.29
32c
−6.97
7a
−6.75
20c
−7.19
33c
−6.81
8a
−6.93
21c
−7.03
34c
−7.12
9a
−7.11
22c
−6.66
35c
−7.15
10a
−6.33
23c
−6.24
36c
−7.37
11a
−6.06
24c
−7.75
Nelfinavir
−6.83
12a
−6.29
25c
−6.71
13a
−6.67
26c
−6.57
By comparing the docked energy of all molecules studied, with is quite noteworthy that good numbers of ligands have better energy scores, showing that these compounds have a best docking pose than Nelfinavir (that the energy of interaction of nelfinavir is −6.83 kcal/mol.)
Binding analysis within the active site of SARS-COV-2 Mpro revealed that, the nelfinavir forms hydrogen bond and hydrophobic bond with amino acid residue of Thr190 and Met165 respectively.
Compounds 20c, 24c, 30c, 34c, 35c and 36c showed the best binding score with the protein 6lu7 are mentioned in Table 2.
The 2D viewing of the protein–ligand interactions of the best poses engender by the six ligands studied is illustrated in Fig. 2.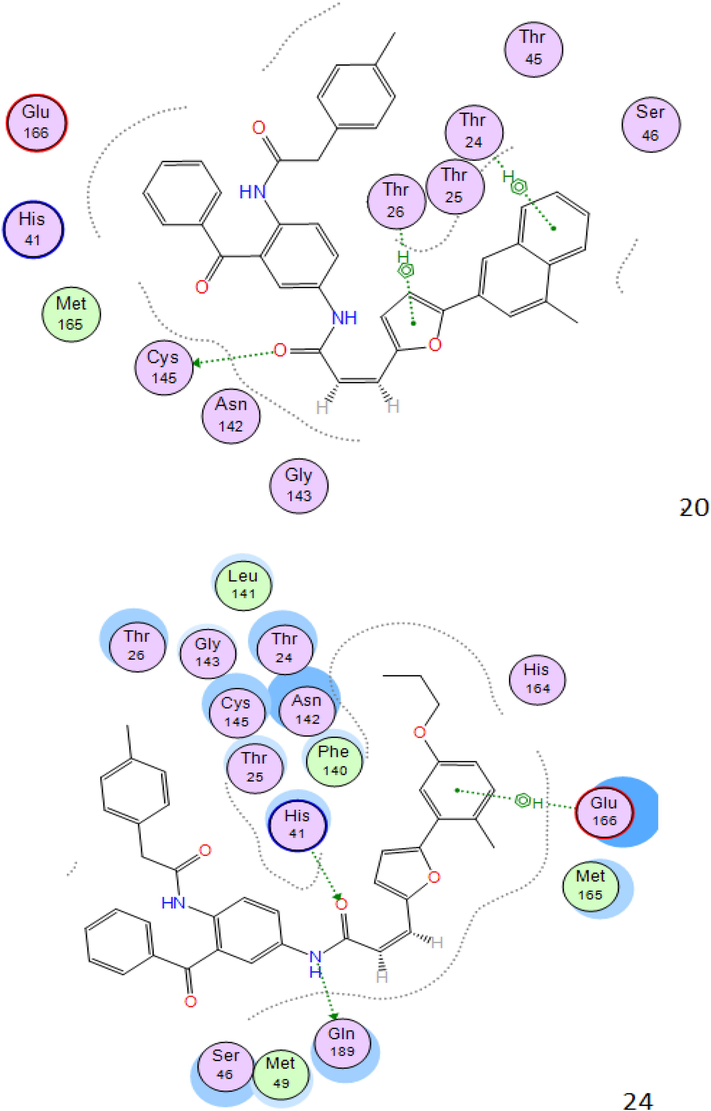
2D interactions between compounds 20c,24c,30c,34c,35c and 36c the SARS-CoV-2 main protease.
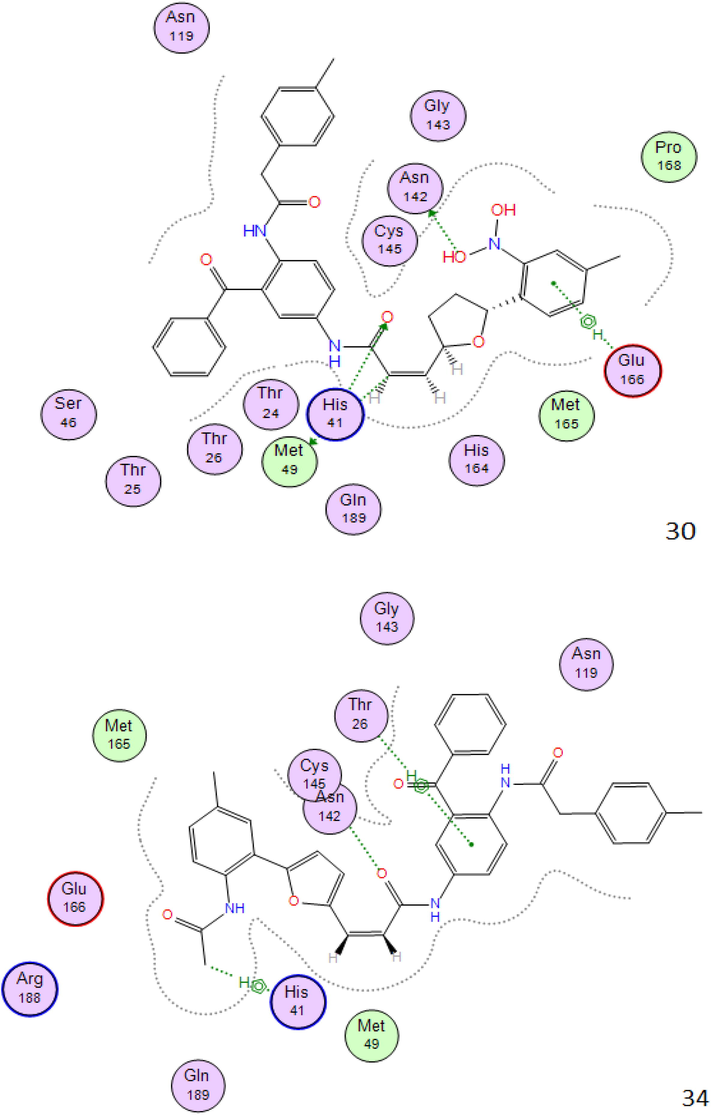
2D interactions between compounds 20c,24c,30c,34c,35c and 36c the SARS-CoV-2 main protease.
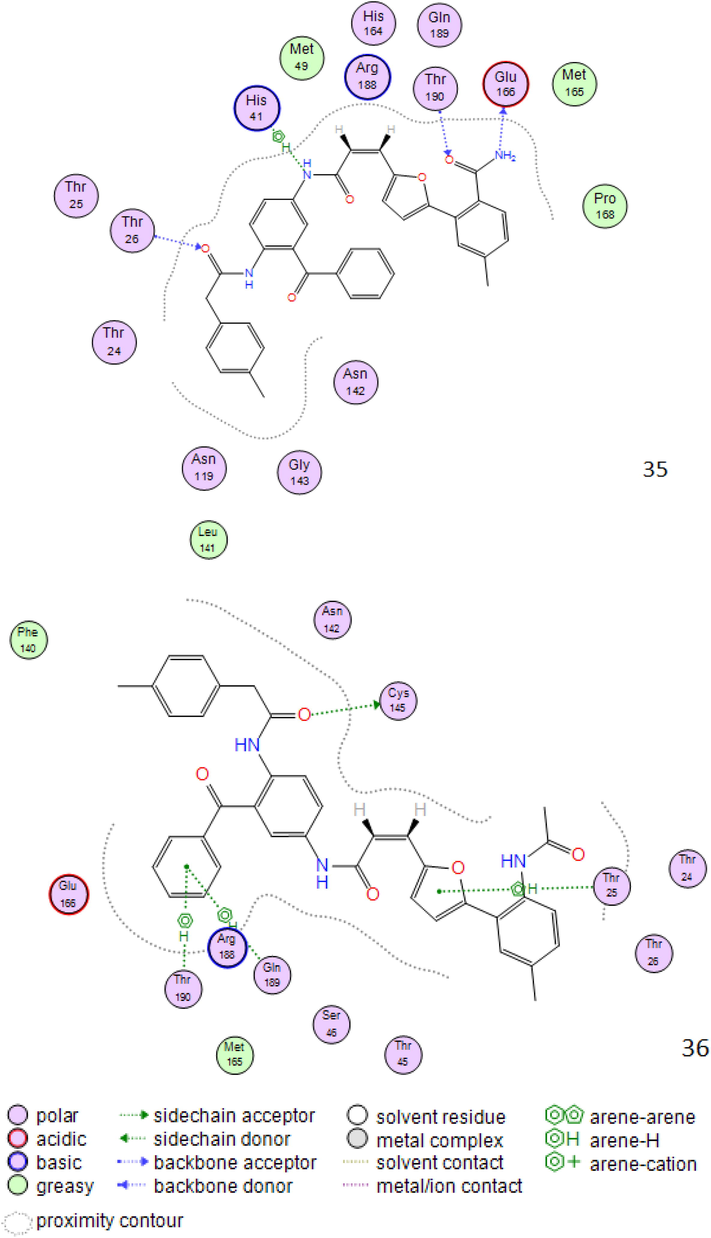
2D interactions between compounds 20c,24c,30c,34c,35c and 36c the SARS-CoV-2 main protease.
The docked conformation of the compounds has shown that the compounds interacted with the pocket-binding residues of the targeted protein through various favorable interactions, including hydrogen, polar and hydrophobic bonding.
Like the main protease enzyme of other coronaviruses, the main protease enzyme of the Novel Coronavirus (COVID-19 Mpro) has a Cys-His catalytic dyad. (Jin et al, 2020)
Residues His 41 and CYS 148 residues at the active site of the protease enzyme Middle East Respiratory Syndrome (MERS) have been implicated as important inhibitors of the enzyme's specific activity. (St John et al., 2015).
While the characteristics of the catalytic binding pocket of coronavirus major protease enzymes are retained (Yang et al., 2005), sequence alignment has shown that CYS 148 has replaced CYS 148. (Jin et al., 2020) CYS 145 in COVID-19 Mpro. In addition, a study has stated that HIS 41 and CYS 145 play a vital role and are vital for the inhibitory action of any possible and promising COVID-19 Mpro inhibitor (Jin et al., 2020).
As shown in Fig. 2 all molecules set forth the identical binding mode, significant interactions can be found between these atoms and the residues Cys145, Asn142, His41, Glu166, Thr26, which directly involved in the catalytic mechanism of this enzyme.
It should be noted that favorable interactions of hydrophobic type are seen between the benzene ring and the residues Glu166 and Thr26; we can say that the Aromatic ring was important for binding and stability of ligand-SARS-COV-2 Mpro Complex.
The structures of the molecules that have the best affinity in the binding site of SARS-CoV-2 main protease show that the present of diamine atoms (proton acceptor N atom) in the structure increase these chances for formation of H-bond interaction type.
Hydrogen bonding with the key residues inside the pocket is thought to be strong determinant for binding of ligand within the active site residues. The higher binding affinity may be assigned to the greater number of hydrogens bonds in the complex, between ligand and protein.
The distances between the residues of the active site and the top six vary between 2.08 Å and 4.48 Å, in this case it can be observed that according to Anne Imberty (Imberty et al., 2000), the interactions having distances between 2.5 Å and 3.1 Å are considered as strong (Table 3).
Compound
Interactiontype
Interacting residues of 6lu7
Distance(A°)
Strength(kcal/mol)
20c
H-donor
H-acceptor
pi-HO 9 SG Cys 145
O 9 CA Asn142
6-ring N Thr 263.27
3.48
4.48−1.0
−0.8
−1.2
24c
H-donor
H-acceptor
pi-HN 37 OE1 Gln 189
O 2 NE2 His 41
6-ring CB Glu 1663.61
2.87
4.44−0.7
−1
−0.6
30c
H-donor
H-donor H-acceptor
pi-HC 25 SD Met 49
O 77 OD1 Asn 142
O 28 NE2 His 41
6-ring CB Glu 1664.08
3.13
2.80
4.23−0.7
−1.7
−3.1
−1
34c
H-donor
H-acceptor
pi-HO 77 OD1 Asn 142
O 28 NE2 His 41
6-ring CB Glu 1663.10
2.88
4.20−1.9
−2.1
1.0
35c
H-donor
pi-H
pi-HO 74 SG Cys 145
6-ring CB Met 165
6-ring N Glu 1663.57
4.09
4.46−0.9
−0.8
−1.1
36c
H-donor H-pi
pi-HO 1 SG Cys 145
C 76 5-ring His 41
6-ring CG2 Thr 263.83
4.32
3.72−1.1
−0.9
−0.7
Nelfinavir
H-donor
pi-HO 14O Thr 190
6-ring CB Met 1652.98
4.46−2.4
−0.6
3.2 ADME-Tox predictions
In this study, we submitted the top score compounds to in silico ADMET screening, using the pkCSM software, to predict their overall absorption, distribution, metabolism, excretion, and toxicity risks.
For the discovery of oral administrative drugs, solubility is one of the major descriptors. the decimal logarithm of the molar solubility in water in log (mol/l) appear in the Table 4 (Insoluble ≤ -10 < poorly soluble < -6 < Moderately < -4 < soluble < -2 < very soluble < 0 ≤ highly soluble) From Table 4, we can see that all the compound have good solubility in the water, so no obstacle to the development and production of oral solid dosage.
compounds
Water solubility
Caco-2 permiability
Intestinal absorption
BBB permeability
CYP1A2 inhibitor
CYP2C19 inhibitor
CYP2C9 inhibitor
CYP2D6 inhibitor
CYP3A4 inhibitor
20
−3.613
0.675
93.542
0.317
Yes
No
Yes
No
No
24
−3.949
1.310
100.00
0.047
Yes
Yes
Yes
No
Yes
30
−2.892
−0.300
75.100
−0.135
Yes
No
No
No
No
34
−3.897
0.763
96.602
−1.001
Yes
Yes
Yes
No
Yes
35
−3.630
1.325
99.966
−1.000
No
Yes
Yes
No
Yes
36
−3.543
0.708
92.724
0.285
No
Yes
Yes
No
No
In general, a compound is considered to have high Caco-2 permeability if the predicted Papp log value is > 0.90 cm/s the permeability of Caco-2 of compound 35c and 24c was 1.31 10-6, 1.32 10-6 cm/s, so we could see it had a high permeability in Caco-2.
Intestinal absorption (human, % absorbed) predicted to be>80% for all the molecules except compound 30c (75.10%).
The BBB permeability of the compound 20 had a value of 0.317 log BBB and the compounds had a value in the range 0.3–1 log BBB. According to research, Pires et al, the compound would be able to cross well the blood–brain barrier, if it has a Log BB value > 0.3 and cannot be appropriately to bring if log BB < -1. Therefore, compounds 34, 35 cannot cross blood-b, but the compound would be able to cross the blood–brain barrier well. On the contrary, compound 20 can penetrate it.
Drug that inhibit or compete for CYP2D6 can cause clinical problems, this isoenzyme is highly polymorphic and is responsible for metabolizing approximately 25% of known pharmaceuticals (Ogu and Maxa, 2000). In our study, all the compounds are non-inhibitors of CYP2D6 enzymes (table 4).
ALL the compounds were AMES negative and test suggests that compound could be not mutagenic.
Drugs that block these HERG K+ channels are predicted to cause cardiac toxicity. (lee et al., 2019). The recommended range for an ideal drug should be −5 and above, as value below this level is expected to cause cardiac toxicity. The compounds 35cand 34chad a total clearance of 0.466and 0.488 log/ml/min/kg. Based on these values, they could be excreted quickly compared to other compounds.
The results of the compounds are presented in Table s1, and their high LD50 values (2.48–4.109), so therefore suggest that the compounds are fatal only at very high doses.
The most active compounds 34c and 35c were shown to possess excellent predicted absorption, distribution, metabolism and excretion parameters, so they have been chosen as models for MD to investigate their stability of those compounds in 3CLpro binding site.
3.3 Analysis of MD simulation and calculation of secondary structure content
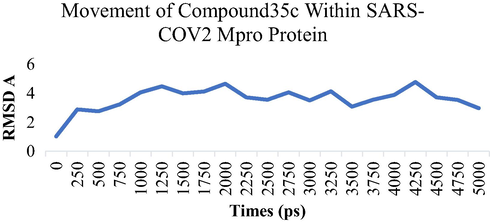
MD simulation studies were performed for five ns to study the stability of the compound 34c and compound 35cwithin SARS-COV-2 Mpro as potential Mpro protein inhibitor. The Root Means Square Displacement (RMSD) and the Root Means Square Fluctuation (RMSF) were calculated to assess the overall stability of ligands within Mpro protein complexes, C alpha, and backbone residues. Figs. 3-6 summarized the results obtained from Mpro protein backbone flexibility analysis by plotting RMSF of all residues of Mpro protein compared over full duration of simulation time.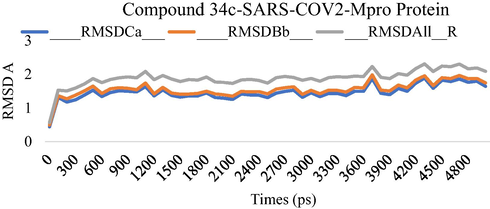
Dynamics behavior of the Compound 34c, binding to the SARS-CoV-2 Main Protease (Mpro) protein. RMSD plot as a function of 5 ns MD simulation time. Comparison of Calpha, Backbone and ovell RMSD of SARS-CoV-2 Main Protease (Mpro) protein.
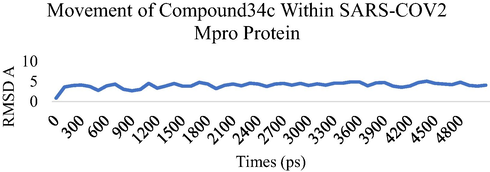
Radius of gyration, plot comparison of Compound 34c superimposed on the initial conformer at 0 ns before starting the MD simulation.
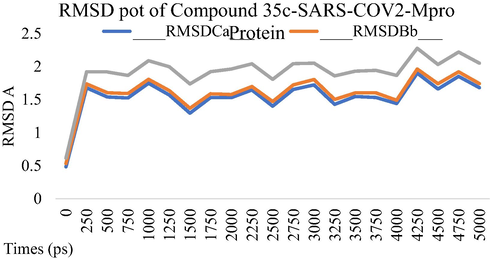
Dynamics behavior of the Compound 35c, binding to the SARS-CoV-2 Main Protease (Mpro) protein. RMSD plot as a function of 5 ns MD simulation time. Comparison of Calpha, Backbone and ovell RMSD of SARS-CoV-2 Main Protease (Mpro) protein.
Radius of gyration, plot comparison of Compound 35c superimposed on the initial conformer at 0 ns before starting the MD simulation.
4 Conclusion
COVID-19 has become a global concern, due to widespread epidemic outbreaks and uncertainty in treatment. In this study, we rely on the efficacy of hypothetical screening and the concept of reuse to identify potential new inhibitors for the main protease protein of SARS-CoV-2.
These investigations give us an interesting analysis of essential residues and ligand-receptor interactions, for the 2,5-diaminobenzophenone with SARS-CoV-2 main protease. All compounds play a key role in enzymatic catalysis, and may be considered as a potential inhibitor of Mpro. 35c and 36c compounds indicate superiority in many terms, such as interaction with both catalytic residue Cys 145 and His 41. Low free binding energy and the greater number of hydrogen bonds. Further, based on ADMET analysis, among the short-listed, two molecules with best ADME properties and least toxic compounds 35c and 36c. Were selected as models for MD simulation to illustrate the ligand-proteins stability. The ligands were stable inside the transmembrane field, which gives to their RMS deviations during the simulations. Our computational exploration indicated that after further in vitro studies these selected lead molecules 34c, 35c, could be used as effective inhibitors against the SARS-CoV-2.
Acknowledgments
We would like to express our grateful to the “Agence Universitaire de la Francophonie (AUF)” for funding research project (Reference: AUF- 463/2020. Title: Repositionnement des médicaments et le dépistage in silico de certains composés issus des ressources naturelles pour le COVID19 via les méthodes de modélisation moléculaire). We would also like to acknowledge all our colleagues at the COVID19 Project from Algeria and Morocco for their amazing support, team spirit and valuable input.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Molecular Structure, Drug Likeness and QSAR modeling of 1,2-Diazole Derivatives as Inhibitors of Enoyl-acyl carrier protein reductase. J. King Saud Univ. Sci.. 2020;32:2301-2310.
- [Google Scholar]
- Báez-Santos YM., St. John SE., Mesecar AD., 2015. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Res. 115, 21-38.
- Contribution à l’étude du contrôle stéréochimique dans les macrolides a 16 chaînons par la mécanique moléculaire. J. Soc. Alg. Chim.. 2004;14:27-39.
- [Google Scholar]
- Molecular docking analysis of N-substituted Oseltamivir derivatives with the SARS-Cov-2 main protease. Bioinformation. 2020;16:404-410.
- [Google Scholar]
- Boudergua, S., Alloui, M., Belaidi, S., Al Mogren, M. M., Abd Ellatif Ibrahim, U.A., Hochlaf, M., 2019.QSAR Modeling and Drug-Likeness Screening for Antioxidant Activity of Benzofuran Derivatives, J. Mol. Struct.1189, 307-314.
- Coronavirus Genome Structure and Replication. Curr. Top. Microbiol. Immunol.. 2005;287:1-30.
- [Google Scholar]
- Molecular Evolution of Human Coronavirus Genomes. Trend. Microbiol.. 2017;25:35-48.
- [Google Scholar]
- Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497-506.
- [Google Scholar]
- HyperChem (Molecular Modeling System) Hypercube, Inc., 1115 NW, 4th Street, Gainesville, FL 32601, USA (2007).
- An unusual carbohydrate binding site revealed by the structures of two Maackiaamurensis lectins complexed with sialic acid-containing oligosaccharides. J. Biol. Chem.. 2000;275:17541-17548.
- [Google Scholar]
- Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020
- [CrossRef] [Google Scholar]
- Targeting Zoonotic Viruses: Structure-Based Inhibition of the 3C-like Protease from Bat Coronavirus HKU4—The Likely Reservoir Host to the Human Coronavirus That Causes Middle East Respiratory Syndrome (MERS) Bioorgan. Med. Chem.. 2015;23(17):6036-6048.
- [CrossRef] [Google Scholar]
- Khaerunnisa, S., Kurniawan, H., Awaluddin, R., Suhartati, S., Soetjipto, S., 2020. Potential Inhibitor of COVID-19 Main Protease (Mpro) From Several Medicinal Plant Compounds by Molecular Docking Study. in press, https://doi.org/10.20944/preprints202003.0226.v1.
- YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics. 2014;30:2981-2982.
- [Google Scholar]
- Computational determination of hERG-related cardiotoxicity of drug candidates. BMC Bioinformatics. 2019;20
- [CrossRef] [Google Scholar]
- MarvinSketch 17.1.2, 2017. ChemAxon (http://www.chemaxon.com).
- Molecular Docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des.. 2011;7:146-157.
- [Google Scholar]
- Drug Interactions Due to Cytochrome P450. Baylor University Medical Center Proceedings; 2000. p. :421-423.
- [CrossRef]
- Molecular Docking Studies and ADMET Properties of New 1.2.3 Triazole Derivatives for Anti-Breast Cancer Activity. J. Bionanosci.. 2018;12:26-36.
- [Google Scholar]
- pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem.. 2015;58:4066-4072.
- [Google Scholar]
- Discovery of a novel lead structure for anti-malarials. Bioorg. Med. Chem.. 2001;9:785-792.
- [Google Scholar]
- Xu, Z., Peng, C., Shi, Y., Zhu, Z., Mu, K., Wang, X., Zhu, W., 2020. Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv. Doi: 10.1101/2020.01.27.921627.
- Design of Wide-Spectrum Inhibitors Targeting Coronavirus Main Proteases. PLOS Biology. 2005;3(10):e324
- [CrossRef] [Google Scholar]
- Virtual Screening in Drug-Likeness and Structure/Activity Relationship of Pyridazine Derivatives as Anti-Alzheimer Drugs. J. King Saud. Univ. Sci.. 2019;31:595-601.
- [Google Scholar]
- A Novel Coronavirus from Patients with Pneumonia in China, 2019. New England J. Med. 2020
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2021.101352.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




