

Translate this page into:
Spectroscopic exploration in aggregation with in silico molecular modeling and in vitro assay on antioxidant 7-chloro-2-4-fluorophenyl-3-hydroxychromen-4-one
⁎Corresponding author. benaezhil@yahoo.com (V. Bena Jothy)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
1 Introduction
Compound containing benzopyran moiety is an important pharmacophore that has talent in interacting with a variety of cellular targets possessing different pharmacological activities. Benzopyran moiety played an essential role in chemistry of pharmaceuticals along with chemistry of coordination that attracted a significant attention to researchers. Benzopyran nucleus bearing structural skeletons such as chromene, 2H-chromene and 4H-chromene points to wide range of biological activities such as antitumor, antihepatotoxic, antioxidant, anti-inflammatory, diuretic, anticoagulant, antispasmolytic, estrogenic, antiviral, antifungal, antimicrobial, anti-helminthic, hypothermal, vasodilatory, anti-HIV, anti-tubercular, herbicidal, anticonvulsant and analgesic activity. Chromene is a heterocyclic scaffold of fused benzene and γ-pyrone ring, which constitutes basic backbone of various types of polyphenol, widely found in alkaloids, tocopherols, flavonoids, and anthocyanins. These compounds are biologically active natural products and promising synthetic compounds in the field of medicinal, agrochemical, cosmetics and pigment industries. Diverse applications in the fields of medicine, agriculture and related pharmacology of chromene derivatives have been developed during last several years. Drug molecules bearing chromene unit have been investigated for their cytotoxic (anticancer), neuroprotective, human immunodeficiency virus-inhibitory, antimicrobial, antifungal and antioxidant activity (Laskar and Brahmachari, 2014; Ren and Yew, 2011). Fluorine, the most electronegative atom of all the compounds has become a prevalent part in anti-inflammatory drug category for the effect it exerts in molecules and are designed to treat rheumatoid arthritis (Ramalingam et al., 2012). Fluorine substituents play an important role in increasing the fat solubility of molecules thereby increasing their bioavailability in pharmaceutical field.
Polycyclic organic compound, 7-chloro-2-(4-fluorophenyl)-3-hydroxychromen-4-one (CFPHC) is a derivative of chromene with highly electronegative chlorine and fluorine atoms in γ-pyrone ring system ensuring good electron charge-transfer properties. Spectroscopic techniques such as Fourier-transform Infrared (FT-IR) spectroscopy, Fourier-transform Raman (FT-Raman) spectroscopy, ultraviolet–visible (UV–vis) spectroscopy, nuclear magnetic resonance (NMR) spectroscopy and thermogram-differential thermal analysis (TG-DTA) along with density functional theory (DFT) were used to investigate the structural and thermodynamical properties of CFPHC. Experimental and computational investigations for determining mentioned properties of CFPHC have not been reported in literature so far. The present research work accentuate to in-depth information about unit cell dimensions, geometrical parameters, electronic properties, and physical properties at the molecular level along with thermal behaviour about sample with an inert reference as a function of increasing temperature undergoing identical thermal cycles. Investigations on Highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy gap, molecular electrostatic potential (MEP) surface and natural population analysis (NPA) have been done in optimized molecular structure by density functional theory. In silico molecular modeling using AutoDock 4.2 has been implemented for conformational complementarity between ligand and receptor. It also aims to focus the chromophore characteristics including the free hydroxyl groups, carbonyl group, electronegative atoms such as chlorine, fluorine, C⚌C double bond in relation to their antioxidant activity, which strongly supported its biological and pharmacological activity via antioxidant and anti-inflammatory activities of the title compound. Antimicrobial activity (in vitro) of the compound was evaluated against four pathogenic bacterial strains such as Escherichia coli, Bacillus subtilis, Staphylococcus aureus and Klebsiella pneumoniae along with two fungal strains Aspergillus niger and Candida albicans.
2 Experimental details
Bruker AXS Kappa Apex2 CCD single crystal X-ray diffractometer was used to find the crystal structure of CFPHC. FT-IR spectrum of CFPHC was recorded using Perkin Elmer spectrophotometer in the region 4000–400 cm−1 with the spectral resolution of 1 cm−1 and FT-Raman spectrum of CFPHC was recorded in the region 4000–50 cm−1 using Bruker RFS 27 spectrophotometer using 1064 nm excitation from an Nd:YAG (neodymium-doped yttrium aluminium garnet) laser source with the spectral resolution of 2 cm−1. UV–vis spectrum of the compound was examined in the range 200–800 nm based on ASTME 169-04 using Varian, CARY 100 BIO UV–Visible spectrophotometer in ethanol solution. 1H, 13C NMR spectra of CFPHC were recorded with the aid of Bruker Avance III 500 MHz multi nuclei solution NMR spectrometer while Inverse Quad Probe head 5 mm ‘QXI’ with 2H lock was used to record 19F NMR spectrum. Thermal analytical study of CFPHC was carried out using a SDT Q600 V20.9 Build 20 thermal analyzer in an inert nitrogen atmosphere.
Radical scavenging activity of the title compound against stable 2,2-diphenyl-1-picrylhydrazyl (DPPH•) was determined by the slightly modified method of Brand-Williams et al. (Brand-Williams et al., 1995). DPPH• reacts with an antioxidant compound, which can donate hydrogen, and reduce DPPH• and change in color (from deep violet to light yellow) was measured at 517 nm on a UV–visible spectrophotometer. Solution of DPPH• in methanol 60 µM was prepared fresh before UV–vis measurements. 3.9 mL of this solution was mixed with 100 µL of test solution at various concentrations. Sample was kept in the dark for 15 min at room temperature, decrease in absorbance was measured and the experiment was carried out in triplicate.
Antibacterial assay was studied using Agar well diffusion method recommended by Smânia et al. (1999). Bacterial inoculum was spread uniformly using sterile cotton swab on a sterile Petri dish Mueller Hinton Agar allocated with four wells. Two wells were filled with 2 mg as well as 4 mg of title compound an others with positive control Gentamycin and negative control methanol. Systems were incubated for 24 h at 36 ± 1 °C, under aerobic conditions. After incubation, clear zone observed and inhibitions of bacterial growth zones were measured. Antifungal assay was studied with Fluconozole as positive control and methanol as negative control. A sterile swab was used to evenly distribute fungal culture over Potato Dextrose Agar (PDA) agar medium allocated with four wells. The plates were allowed to dry for 15 min before use in the test and 2 mg and 4 mg of the sample were added into two wells and others with positive and negative controls. The plates were incubated at 26 °C for 72 h after which they were examined for inhibition zones. Anti-inflammatory activity was studied using inhibition of protein denaturation technique according to procedure recommended by Mizushima and Kobayashi (1968). Reaction mixture consists of test sample and 1% aqueous solution of bovine albumin fraction and the pH of the reaction mixture was adjusted using small amount of 1normal-hydrochloride. Sample was incubated at 37 °C for 20 min and then heated to 51 °C for 20 min, after cooling the samples the turbidity was measured at 660 nm and experiment was performed in triplicate.
3 Computational details
ORTEP 3 (Johnson and Burnett, 1996; Farrugia, 1999) and Mercury 3.9 (Macrae et al., 2008) are an excellent visualization tools for crystallographic structural analysis. Structural refinement details and crystal data of grown crystal of CFPHC were obtained using ‘publcif’ (Westrip, 2010). Unit cell parameters of CFPHC were determined by least-square technique using many reflections and the structure solved was further refined by full-matrix least squares method using SHELXL97 (Westrip, 2010). Geometry optimization and vibrational wavenumber were performed using Becke-3-Lee-Yang-Parr (B3LYP) gradient correlation functional with 6-311++G(d,p) the basis set (Hariharan and Pople, 1973; Krishnan et al., 1980; Becke, 1993; Becke, 1988; Lee et al., 1988) using Gaussian 09W program package developed by Frisch and co-workers (Frisch et al., 2009). Scaling factor (0.9673) (Scott and Radom, 1996; Foresman and Frisch, 1996) is used for the corresponding basis set in order to correct the theoretical errors, such as basis set deficiencies caused by neglecting anharmonicity while potential energy distribution (PED) for the normal modes were computed using MOLVIB 7.0 program (Sundius, 2002; Sundius, 1990). To improve the agreement between predicted and observed wavenumbers, computed harmonic wavenumbers were scaled according to the scaled quantum mechanical force field (SQMFF) procedure (Pulay et al., 1983). Deviation from the experiments after scaling was found to be less than 9 cm−1 with a few exceptions. Descriptions of predicted wavenumbers during the scaling process were followed by potential energy distribution matrix and Cartesian representation of the force constants were transferred to a non-redundant set of symmetry coordinates, chosen in accordance with the recommendations of Pulay et al. (1979). Natural bond orbital (NBO) (Glendening et al., 1998) calculation was performed using NBO 3.1 program as implemented in the Gaussian 09 W package which was performed in gas phase at DFT-B3LYP/6-311++G(d,p) (Kohn and Sham, 1965; Parr and Yang, 1994) level of theory. HOMO and LUMO energies were computed using time-dependent density functional theory (TD-DFT) calculation that was carried out in ethanol solvent to predict electronic excitation energies of CFPHC. NMR serves as a great resource in determining the structure of an organic compound by revealing the hydrogen and carbon skeleton and Gauge-Invariant atomic orbital (GIAO) (Wolinski et al., 1990) calculation performed for collecting datas. Perl script called thermo.pl extracted the essential data from a Gaussian output file and compute thermodynamic functions at several temperatures (Irikura, 2002).
Molecular docking study was performed with the aid of AutoDock 4.2, a computer-aided software (Morris et al., 1998, 2009), for predicting the interaction of ligands with bio-macromolecular targets and provides reproducible results to estimate the free energy of binding of ligands to receptors. Ligand was created from the structure optimization of the ground state of title compound that were performed using B3LYP/6-311++G(d,p)level of basis set. Parameters like binding energy, inhibition constant and hydrogen bond interactions were determined. Three-dimensional (3D) X-ray crystal structures of different antibacterial protein receptors in PDB format retrieved from research collaboratory for structural bioinformatics (RCSB) protein data bank (Rose et al., 2015; Berman et al., 2000) used for docking poses. PyMOL 1.2r1 molecular graphics system used to visualize and analyze the binding poses regarding protein-ligand interactions (PyMOL, Schrödinger, LLC).
4 Results and discussion
4.1 Recrystallization process
7-Chloro-2-(4-fluorophenyl)-3-hydroxychromen-4-one was purchased (powder) with a stated purity of greater than 94% from OTAVA Chemicals, Canada. Methanol was used as solvent for recrystallization process in order to reproduce crystal sample. Solution saturates over a period of 26 days and small needle shape grown crystals with crystal size of 0.25 × 0.25 × 0.20 mm were used for further studies.
4.2 X-ray crystallography analysis
X-ray crystal data of the compound was collected at 293 K and intensity data collected shows h from −9 to 9, for k from −10 to 10 and for l from −14 to 14 in the range of 2.7–29.2° totaling 2639 unique reflections. After so many cycles of refinement, R factor dropped down to 0.059. Table S1 (Supplementary information) illustrates the complete crystallographic data, data collection and structural refinement details of the grown crystal CFPHC that exists in triclinic system with P space group, exhibiting lattice and unit cell parameters α = 101.87(1)°, β = 103.14 (1)°, γ = 95.09° and a = 7.19 (1) Å, b = 7.86(2) Å, c = 11.36 (2) Å, respectively. Number of molecules per unit cell (Z) and the volume (V) are 2 and 606.23(2) Å3, respectively. Final anisotropic full-matrix least-squares refinement on F2 with 182 variables and values of largest peak in final difference electron density synthesis are 0.29 e/Å−3, largest hole was −0.36 e/Å−3 and calculated density was 1.592 mg/m3. Crystallographic information file of CFPHC with reference number CCDC 1530535 were deposited in Cambridge crystallographic data centre.
4.2.1 Halogen and hydrogen bonding interaction analysis
Charge-transfer interaction from phenyl rings to chlorophenyl ring and fluorophenyl ring reduces bond lengths confirming halogen-bonding interactions (C–H⋯Cl and C–H⋯F) from crystal structure having bond angles at 128° and 145° having symmetry x + 1, y, z + 1 and −x + 1, −y, −z + 1, correspondingly. Carbonyl group oxygen atom binds to O⋯H (2.29 Å) confirms O–H⋯O bonding interactions through symmetry neighbouring oxygen molecule (−x + 3, −y + 1, −z + 1) in benzopyran moiety and the geometrical bonding interactions predicted by X-ray diffraction shown in Table S2.
4.3 Optimized structural geometry analysis
Bond length of C2–O11 is 1.23 Å, which has fine agreement with reported C–O bond length (1.2 Å) of related compounds with small variations (Robinet et al., 2001). Owing to hyperconjugation, the bond length of hydroxyl group in pyran ring reduces also by negative inductive effect, delocalization of lone pairs happens by releasing electrons, reduced the corresponding bond angle C3–O12–H13 = 104°. Bond lengths associated with electron donating oxygen atom (C–O) reduces from the expected value 1.43 Å (Sawant and Nirwan, 2012) due to electron delocalized ring system. C–H bond parameters vary due to the electron cloud surrounding each atom repelling each other leading to destabilization along with steric hindrance. Bond angles connecting C8 alters due to the negative inductive effect of chlorine atom. Hydrogen and fluorine forms a polar covalent bond by sharing electrons thereby electrons being attracted to one atom more strongly than other, which results in partial positive and negative charges. Separation of partial charges creates a dipole and electrons are more pulled towards fluorine forming C20–H25⋯F26 and C22–H27⋯F26 halogen bonding interactions. Simulated geometric parameters show good correlations with the crystallographic bond parameters while slight deviations arise because theoretical calculations was done for single isolated stable molecule in gas phase and experimental values predicted from crystalline solid phase and are summarized, given in Table S3. Fig. 1(a) shows ORTEP diagram while (b) represents optimized molecular structure of CFPHC and (c) crystal pack diagram of CFPHC.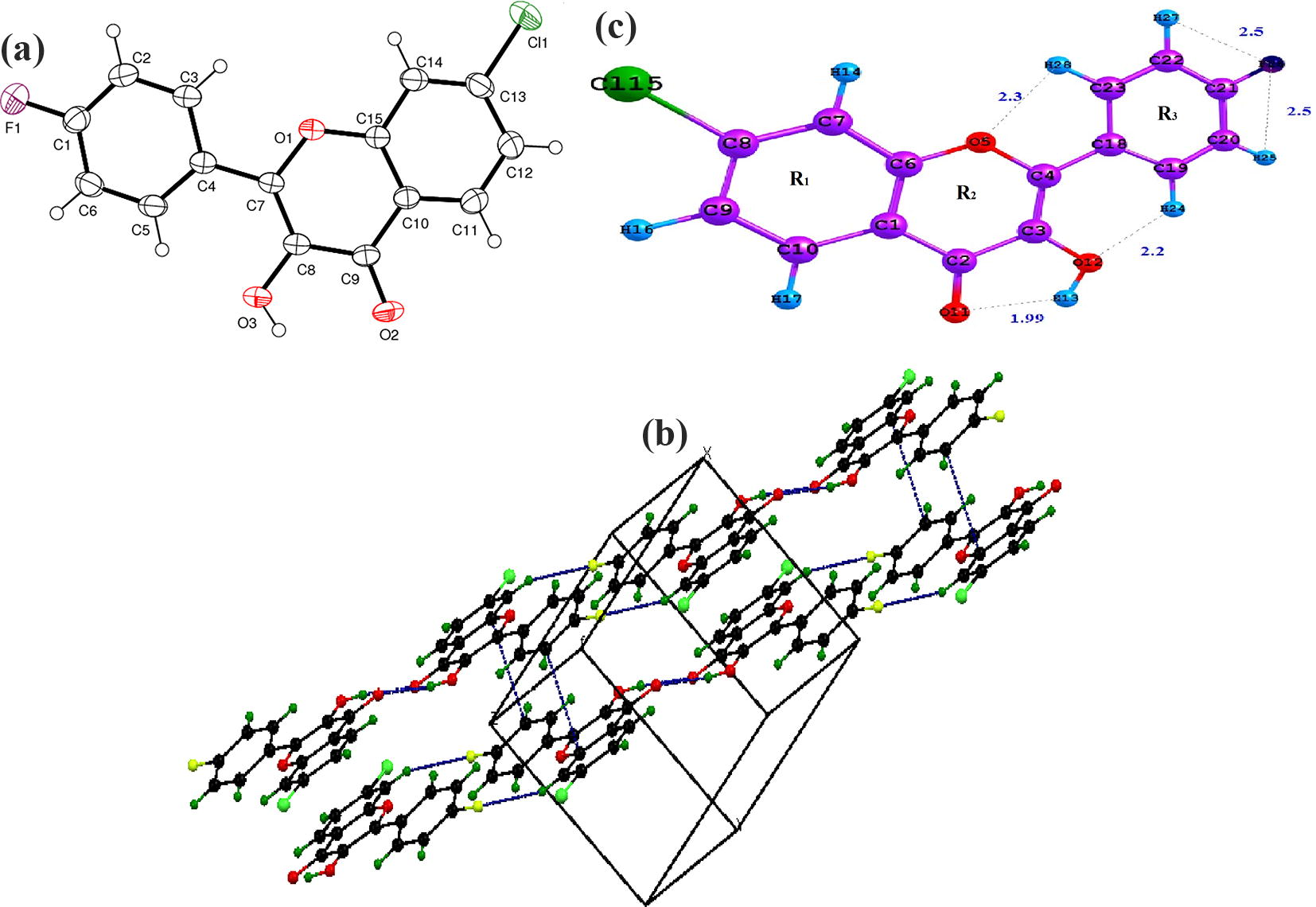
(a) ORTEP diagram (b) Crystallographic view (c) Optimized geometrical structure of CFPHC.
4.4 Spectral assignments via experimental and computational view
CFPHC has 28 atoms and according to group theory analysis, 78 normal modes of vibrations are distributed as: Γ3N-6 = 53 À (in-plane) + 25 Ȁ (out-of-plane); harmonic wavenumbers are adjusted by the scaling factor and detailed vibrational assignments of CFPHC carried out with the aid of normal co-ordinate analysis illustrates in Table 1. Theoretical IR and Raman spectra of CFPHC are shown in Fig. 2(a,b). Slight differences concerning experimental and calculated vibrational modes observed perhaps due to hydrogen bonding leading to strong perturbation of wavenumbers and intensities of various PED modes. vs-very strong; s-strong; m-medium; w-weak; vw-very weak; ν-stretching; β-in-plane bending; γ-out-of plane bending; puck-puckering; τ-torsion; τ′-out-of-plane torsion; τAS-asymmetric torsion; τ′AS-out-of-plane asymmetric torsion; SD-symmetric deformation; AD-asymmetric deformation; ADO-out-of-plane asymmetric deformation; TD-trigonal deformation; (1)-Phenyl ring 1; (2)-Phenyl ring 2; (3)-Phenyl ring 3.
Experimental Wavenumbers (cm−1)
Scaled Wavenumbers (cm−1)
Assignment with (PED ≥ 10%)
νIR
νRaman
νCalc.
3486 s
3486 w
3487
νOH(1)(100)
3068 w
3067
νCH(3)(99)
1629 vs
1626 vs
1630
νCO(2)(58), νCC(1)(25)
1604 vs
1605 vs
1601
νCC(1)(37), νCO(2)(17), νCC(2)(13)
1393 s
1393 s
1394
νCC(2)(77), νCC(1)(17)
1308 m
1310
νC(1)(20), νCO(2)(16), νCC(3)(13), βOH(1)(12)
1351 s
βCH(2)(40), νCC(2)(38), νCC(1)(12)
1298 s
1297 s
1298
βOH(1)(34), νCC(3)(48), νCC(2)(16)
1265 s
βCH(3)(78), νCC(3)(10)
1250 s
βCH(2)(36), νCC(1)(22)
1230 s
βCH(2)(24), βCH(3)(15), νCF(3)(13)
1205 s
1204 s
1201
νCC(1)(39), νCC(2)(32), νCC(3)(34)
1166 s
βCH(3)(52), νCF(3)(19), νCC(3)(18)
1174 m
νCC(3)(34), νCF(3)(17), νCC(1)(13)
1107 s
1104 s
1102
γCH(3)(70), Rpuck(3)(17)
913 m
915 w
912
RTD(3)(47), RADO(3)(21)
842 s
840
νCC(1)(15), νCCl(1)(12)
772 w
772
τCCCH(3)(92), RTD(3)(36),RAD(3)(23)
691 w
698
RADO(2)(27), βCO(1)(22), βCC(3)(12)
610 w
616 w
612
βCO(1)(65)
465 w
468
βCF(3)(41), RADO(3)(25), βR(3)(15)
419 w
410
τ′AS (2)(36), Rpuck(2)(18)
371 vw
374
βCCl(2) (27), RADO(2)(25)
351 vw
351
Rpuck(1)(42), RADO(1)(28), Rpuck(3)(14), βCF(3)(15)
278 vw
275
Rpuck(1)(29), τ′AS (1)(26), τAS (2)(11)
198 vw
βCCl(2)(39), βCC(3)(22)
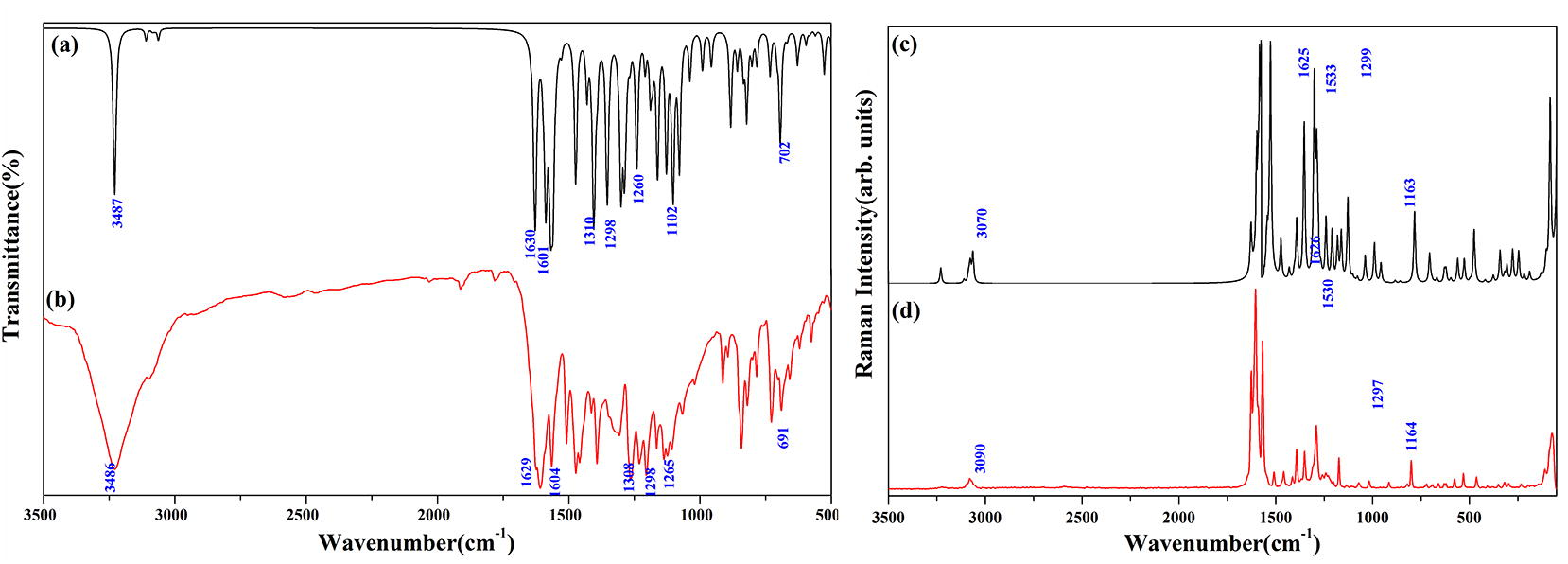
(a) IR Spectrum (b) Raman Spectrum of CFPHC.
4.4.1 Hydroxyl vibrations
Hydroxyl stretching vibration generally occurs in the region 3700–3500 cm−1 (Kurt et al., 2011) however it red shifts to wavenumber region 3550–3200 cm−1 due to intramolecular hydrogen bonding with the carbonyl group of γ-pyrone ring. Strong broad IR peak at 3486 cm−1 assigned to O–H stretching mode vibration calculated wavenumber at 3487 cm−1. In-plane bending modes generally appear as strong peak in the region 1330–1420 cm−1 which are viewed as a strong IR peak at 1298 cm−1 and as a strong Raman peak at 1297 cm−1 coupled with C–C stretching vibrations.
4.4.2 Carbonyl vibrations
An electron-attracting group with negative inductive effect (−I), positive Mesomeric effect (+M) and resonance lowers C2⚌O11 stretching vibrations which usually occur in the region 1715–1680 cm−1 (Bellamy, 1975) in ketones. The peaks observed as very strong IR absorption peak at 1629 cm−1 and at 1626 cm−1 in Raman. Red shifting of this mode is supported by DFT calculations, which locate this mode at 1630 cm−1 with weak hydrogen bonding as well as resonance stabilized delocalization effect. Calculated hyperconjugative interactions LP1(O11)/LP2(O11) → σ∗O12-H13 also supports O–H⋯O intramolecular interactions (Table S6). C–C⚌O bending mode at 610 cm−1 (IR), 616 cm−1 (Raman) for chromenes results the wavenumber of this mode at 612 cm−1. Spectral appearance of peak with weak intensity at 691 cm−1 in IR and at 419 cm−1 in Raman are attributed to deformation (scissoring) peak δ(C⚌O) which usually occur in IR region 625 ± 70 cm−1 and Raman region 540 ± 80 cm−1 (Arjunan and Mohan, 2009). Migani et al. (2005) assigned skeletal νC–O mode with large amplitude on C⚌C bond at 1299 cm−1 and IR medium peak at 1308 cm−1 with PED (16%) at 1310 cm−1 assigned to CFPHC coupled with C–C and OH bending vibrations.
4.4.3 Hydrocarbon vibrations
C–H stretching modes usually appear with the strong Raman intensity and are highly polarized (Sundaraganesan et al., 2008) and give rise to multiple peak in the region 3100–3000 cm−1. C–H stretching vibrations occur as a weak Raman peak at 3068 cm−1 and predicted DFT mode at 3067 cm−1 with PED (99%). In-plane C–H bending vibrations appear in the range 1400–1000 cm−1 and the observed IR peaks are at 1230 (strong), 1265 (strong) and Raman peaks at 1250 (strong), 1351 (strong) cm−1. γ(C–H) mode of pyrone ring appear as a broad and strong peak absorbing near 1107 cm−1 (IR) and 1104 cm−1 (Raman) and 1102 cm−1 at DFT mode. γ-Pyrone ring exhibit a medium IR peak at 772 cm−1, which is attributed to asymmetric deformation and trigonal deformation modes.
4.4.4 C–F vibrations
Fluorine atom at para-position of the skeletal ring 3, absorbs strongly over a wide range between 1360–1000 cm−1 (Arivazhagan and Jeyavijayan, 2011) due to C–F stretching modes. Corresponding C–F stretching vibration is observed as strong infrared peak at 1166 cm−1 and medium Raman peak at 1174 cm−1. Mono fluorinated benzene shows high wavenumber at 478 cm−1 (Varsányi and Szöke, 1969) and Raman intensity peak at 465 cm−1 with 41% PED is attributed as C–F in-plane bending vibration mode. C–F out-of-plane bending vibration is assigned as depolarized Raman lines at 351 cm−1 and remains more or less unaltered computational mode with 14% PED.
4.4.5 C–Cl vibrations
Stretching vibration of carbon–halogen bond in ring 1 generate a strong IR peak at 842 cm−1 occurring around the region 850–550 cm−1 having fine agreement with the literature (Silverstein et al., 1991). Most chloro compounds have absorptions around 390–165 cm−1 due to out-of-plane bending modes and in CFPHC, in-plane as well as out-of-plane bending C–C–Cl modes are observed at 198 cm−1 and 371 cm−1 with very weak Raman intensity.
4.4.6 Phenyl ring vibrations
Spectral region of 1430–2000 cm−1 is assigned as υ(C⚌C) vibration mode by (Barroso-Bogeat et al., 2014) and very strong IR peaks at 1205, 1393 cm−1 and Raman peaks at 1204, 1393 cm−1 are visualized and DFT mode predicts this at 1201 and 1394 cm−1. Trigonal deformation of ring is calculated at 912 cm−1 and is assigned to weak Raman peak at 915 cm−1 and IR medium peak at 913 cm−1. Low Raman intensity peak at 278 cm−1 is assigned as asymmetric torsion of the ring with DFT at 275 cm−1.
4.5 UV–vis spectral analysis
UV–vis spectroscopy studies the changes in electronic energy levels within the molecule arising due to transfer of electrons from π- or non-bonding orbitals and provides the knowledge about π-electron systems and conjugated non-bonding electron systems. Ultraviolet spectrum of 4-chromene showed absorption peaks around 260 nm and 330 nm when measured in ethanol (Ellis, 2009). C⚌C bond prevents conjugation in γ-pyrone carbonyl group thereby reducing involvement of flurophenyl ring to UV–vis spectrum. Electron-donating groups such as –CH3, –Cl, –Br, –OH, –OCH3, –NH2 etc. increases secondary peak absorption. The presence of conjugate double bond and functional group –OH also influence the conjugated systems, decreases the energy difference between HOMO and LUMO and the radiations of longer wavelength are absorbed. Consequently a peak of very low intensity in 300–330 nm range (peak I) have been assigned in chromenes. Increased electron drift from electron-donating hydroxyl group and chlorine atom in γ-pyrone ring through π-bond of benzene ring shows shoulder peak at 309 nm and bathochromic (red) shift to 347 nm occurs due to the solvent stabilization of excited or ground electronic states showing π→π∗ transition with 56% minor contribution from HOMO-2 → LUMO excitation. UV–vis spectrum of CFPHC shows n→π∗ transition around carbonyl chromophore undergoing hypsochromic (blue) shift showing sharp intensity peak at 253 nm in γ-pyrone moiety for ethanol solvent stabilized by hydrogen bonding having polar excited state. Table S4 shows UV absorption corresponding to experimental peaks (Fig. 3) along with calculated excitation energies; absorbance and oscillator strength (f) for title molecule for ethanol solvent. Maximum absorption wavelengths obtained, viz. 350, 301, and 265 nm, are in good agreement with experimental values of 347, 309, and 253 nm, attributed to the excitation with oscillator strengths 0.53, 0.17, 0.12, respectively.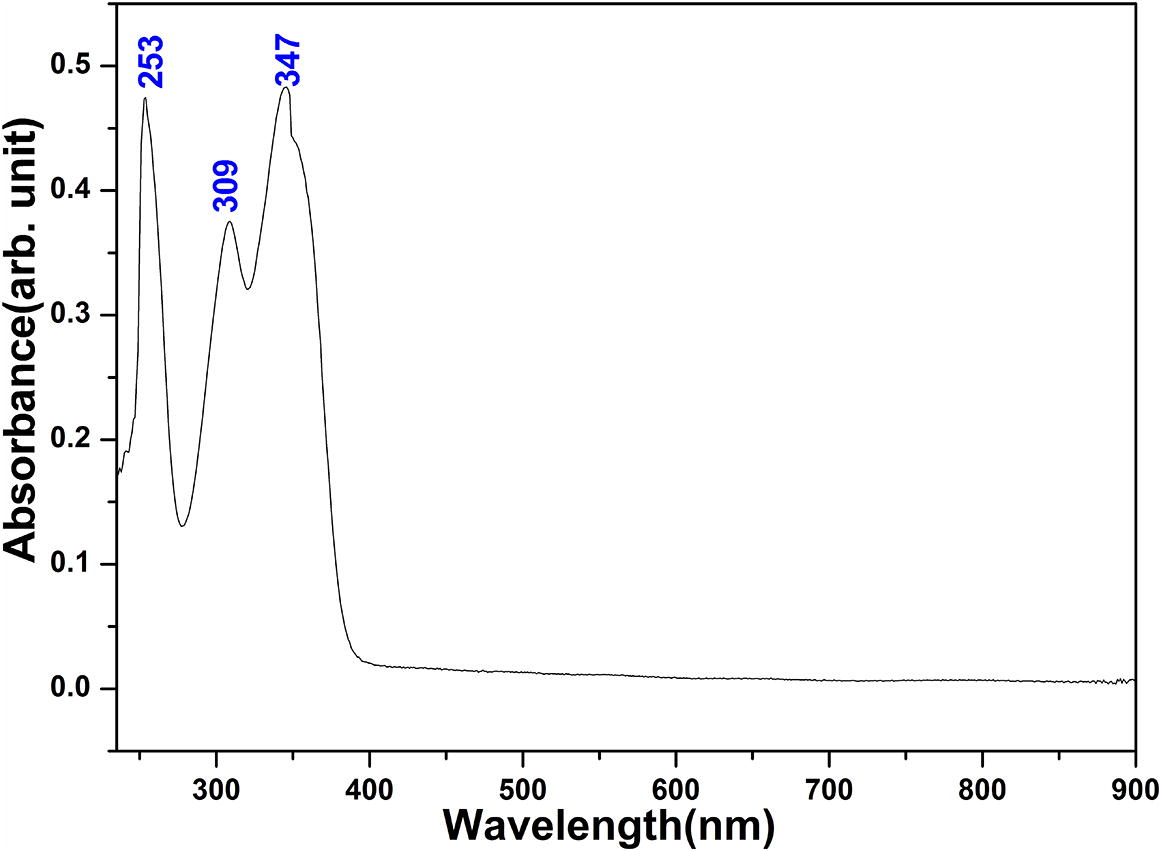
UV–vis spectrum of CFPHC.
4.5.1 HOMO and LUMO analysis
Compounds having high EHOMO are good electron donors while those having low ELUMO are good electron acceptors therefore a significant degree of intramolecular charge transfer (ICT) from electron donor groups to electron acceptor groups transpire through a π-conjugated pathway (Xavier et al., 2011). Frontier orbital analysis of CFPHC shows that EHOMO value is calculated as −6.43 eV, displaying more electron accepting capability while ELUMO value is −2.63 eV. HOMO-LUMO orbital energy gap can provide useful information about the charge transfer interaction, chemical reactivity and biological activities such as antibacterial, antioxidant as well as DNA binding (Fleming, 1976; Mu et al., 2015; Rahmounia et al., 2014). By comparing the DFT computations of fluorine and chlorine substituted compounds (Nilesh and Anil, 2017), molecular orbital energy gap of CFPHC is found to be 3.80 eV and the corresponding wavelength fall in the UV region. Frontier orbital energy values such as HOMO − 1 = −7.36 eV; LUMO + 1 = −1.47 eV; HOMO − 2 = −7.69 eV; LUMO + 2 = −1.18 eV are also calculated. In CFPHC, HOMO and HOMO-1 are delocalized and predominantly located on hydroxyl groups, chlorophenyl ring and carbonyl group showing major contribution of 93% HOMO → LUMO transition, which entails an electron density transfer from pyran ring through oxygen atoms to chlorophenyl ring, while HOMO − 2 are confined with 22% major influence on chlorophenyl ring. As the number of conjugated double bonds is increased, the gap between highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) is progressively lowered. LUMO populates on all atoms except hydrogen atoms while LUMO + 1 mainly populates on hydroxyl group oxygen atom and carbon atoms and LUMO + 2 on in both chloro- and fluro-phenyl ring carbon atoms. Molecular orbital plots for ground state of CFPHC molecule in gas phase including the HOMO, LUMO, LUMO + 1, LUMO + 2 are shown in Fig. 4.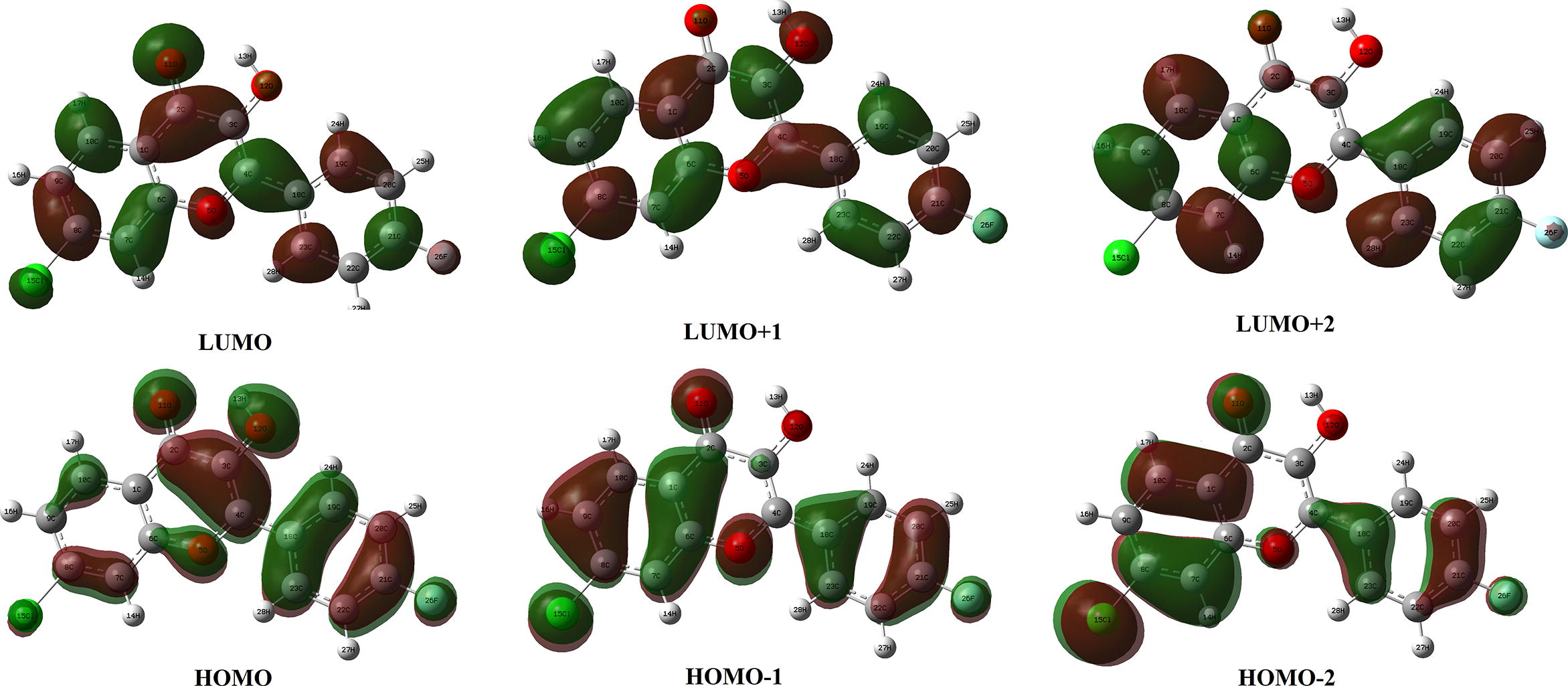
HOMO-LUMO plot of CFPHC.
4.6 Natural population analysis
Population analysis assign the atomic population for each atom in a molecule (Reed et al., 1985) and the summary of natural population charges from NBO output is listed in Table S5. Natural population distribution chart of CFPHC is as shown in Fig. 5. Charge distribution of optimized monomer CFPHC produces a total dipole moment of 1.61 Debye and points out that all hydrogen atoms possess a positive charge and among them H13 of hydroxyl group possess highest positive charge due to intramolecular bonding interaction sites. Oxygen draws the electrons in the bonds and shares with the hydrogen atoms towards it while effect of electronegative halogen atom in the π system is weak, because the lone pair of electrons are so tightly held, and they are strong σ acceptors (Fleming, 2010). All oxygen atoms acquire negative charge while hydroxyl oxygen atom (O12) having highest negative charge and oxygen atom of carbonyl group also possess high value of negative charge due to O–H⋯O intramolecular hydrogen bonding. Since oxygen and chlorine atoms are highly electronegative, the effective charges on oxygen and hydrogen caused by the chlorine drawing electrons from the carbon are high, thereby reducing the charge in chlorine atom Cl15.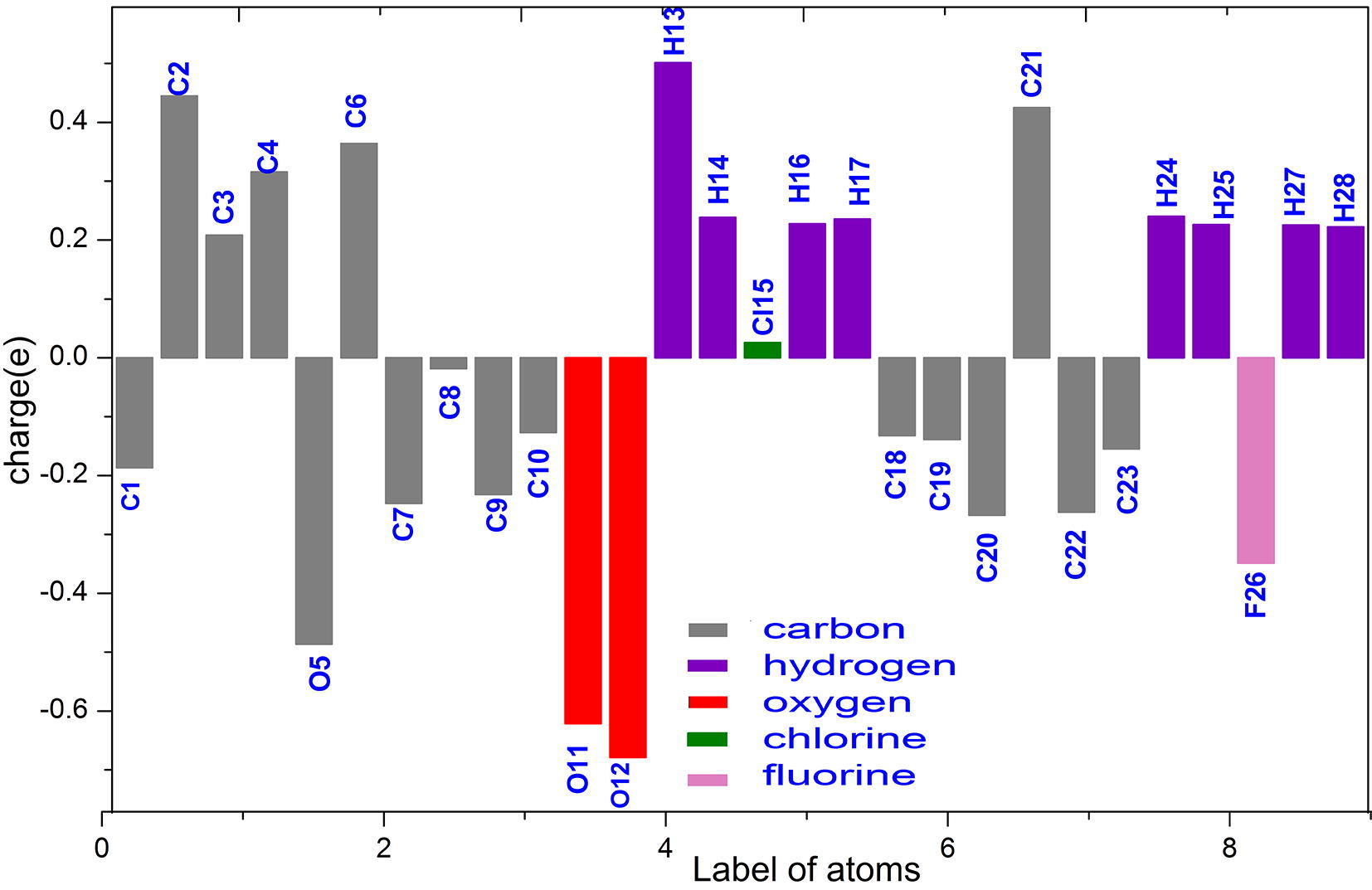
Natural Population Charges distribution chart of CFPHC.
4.7 Molecular electrostatic potential surface analysis
Molecular electrostatic potential surface (MESP) analysis serves to explain hydrogen bonding, reactivity and structure-activity relationship of CFPHC especially its bioactivity. Electrostatic potential at different points on the electron density surface is shown in Fig. S1 and the reported values are in Table S5. In CFPHC, γ-pyrone ring carbon atoms are blue in color characterized by a relative absence of electrons. Negative (red) potential (−22.37a.u) mostly projects above the carbonyl oxygen atom and hydroxyl group contributing hydrogen bonding interactions resulting in concentrated electron density across it. F26 with electric potential (−26.57a.u) shows red color exposing its greater electronegativity difference between atoms in the bond. Cl15 has a highest electric potential (−64.37a.u) showing slight yellow color and due to the greater electronegativity difference between atoms in a bond, makes it the more polar bond.
4.8 Natural bond orbital analysis
NBO analysis summarizes hybridization of atomic lone pairs and of the atoms involved in bond orbitals. Mixing of the donor and acceptor orbitals treated with second-order perturbation theory expounds the intramolecular interactions, rehybridization and delocalization of electron density within the molecule and second order perturbation analysis of Fock matrix using NBO basis of CFPHC is as shown in Table S6. Interaction energies from electron donating groups lone pair LP(O) to the anti-bonding acceptor σ∗(C–C) of phenyl rings showing high stabilization energies due to resonance inside the ring as well as hyperconjugation between the electron donating groups and phenyl ring. Lone pair interactions such as LP2O11→σ∗(C2–C3) is 20.83 kcal/mol; LP2O12→σ∗(C2–C3) is 16.45 kcal/mol; LP2O11→σ∗(C1–C2) is 18.40 kcal/mol; LP2O12→σ∗(C3–C4) is 23.95 kcal/mol increases electron density (ED) in C–C antibonding orbital thereby weakening the respective bonds.
Corresponding stabilization energies associated with dihedral angles of γ-pyrone ring and phenyl ring show high values as 991.69 kcal/mol and 55.55 kcal/mol resulting charge transfer from γ-pyrone ring to flurophenyl ring. Red shift of O–H stretching wavenumber confirms O–H⋯O hydrogen bonding with stabilization energies 0.59 and 4.59 kcal/mol due to the lone pair (LP) interactions, LP1(O11)/LP2(O11)→σ∗O12–H13. Intramolecular charge transfer from σC4–O5→σ∗(C23–H28) with stabilization energy 16.71 kcal/mol exposes C–H⋯O hydrogen bonding with increase in electron density (ED) in C–H antibonding orbital weakening the respective bonds and the corresponding interactions are LP1(O5)→σ∗(C23–H28) is 43.06 kcal/mol and LP1(O12)→σ∗(C19–H24) is 0.82 kcal/mol for CFPHC. Interaction involving carbon-fluorine bond C–H⋯F having highest stabilization energies are obtained by overlapping orbital atoms LP1(F26)→σ∗(C22–H27), LP1(F26)→σ∗(C20–H25) having very high stabilization energies 7828.52 kcal/mol and 926.52 kcal/mol, correspondingly. Hydroxyl group and carbonyl group increases the number of double bonds in chromene moiety due to the presence of highly electronegative oxygen atom, stabilizing molecular π-system and distressing π-electronic system at junction σC4–C18→σ∗C18–C19 of neighboring flurophenyl ring partly destroying its aromaticity showing less stabilization energy 1.94 kcal/mol.
4.9 NMR analysis
Nuclear magnetic resonance spectroscopy is a powerful analytical technique used to characterize organic molecules by identifying carbon-hydrogen frameworks within the molecules using 1H, 13C and 19F NMR analyses. NMR solvent deuterated dimethyl sulfoxide (DMSO)-d6 is liable to dissolve compounds of reasonably varying polarity.
4.9.1 1H NMR spectral analysis
1H NMR spectrum absorption region for common organic solvents in DMSO-d6 is around δ 6.0–7.9 ppm (Jacobsen, 2016). Chemical shift for protons attached to the ring C appears in the range δ = 7.79 ppm and the experimental values to some extent fall in the same range coinciding with the theoretical data. Among all protons in the title compound, H24 acquires high chemical shift value δ = 9.82 ppm confirming intramolecular hydrogen bonding and displays a very low-field resonance (Kalinowski et al., 1988). Increasing electronegativity of chlorine deshields nearby hydrogen atoms H14 and H16 resulting in a downfield shift of δ = 8.03 and 7.62 ppm, correspondingly.
4.9.2 13C NMR spectral analysis
Chemical shift is substantially determined by the electron population surrounding the nucleus and shielding it from the applied field. 13C chemical shifts are used to probe the total electron population and are extremely sensitive to substitution and molecular geometry. Spectrum appears between low field carbonyl carbons and high field methyl carbons in the range 122–132 ppm (Jacobsen, 2016). C3 attached with highly electronegative hydroxyl group and adjacent carbonyl group results high peak at δ = 139.52 ppm. sp2 hybridized C1 due to resonance effect of adjacent carbonyl group C2⚌O11 absorbs downfield having chemical shift δ = 127.1 ppm (Kalinowski et al., 1988). Meanwhile C2 of carbonyl group absorbs far downfield exhibiting highest chemical shift of δ = 172.89 ppm. Attachment of electronegative atom to carbon bearing proton causes a shift of δ = 164.13 ppm (Jacobsen, 2016) and hydrogen bonding [C22–H27⋯F26, C20–H25⋯F26] decreases the electron density around the proton causing deshielding to move the proton absorption to lower field with lower chemical shift values around δ = 7.6 ppm.
4.9.3 19F NMR spectral analysis
Fluorine NMR is used as an analytical tool for drug analysis and serves as a powerful tool for qualitative and quantitative analyses of biological systems. Fluorine attached with benzene also couples with the nuclear protons and positive (+) values indicate downfield shifts, lower-shielding, or higher wavenumber and negative values indicate up field shifts, higher-shielding, or lower wavenumber. Replacement of hydrogen by isotope fluorine in para position of phenyl ring gives rise to measurable shift and negative value −110.00 ppm shifts to lower wavenumber. As a result of van der Waals forces, deshielding influence of carbon (C–F) attached restricts the motion of electrons on the fluorine and thus makes fluorine nucleus respond to magnetic field as if electron density were lowered. Solvent induced shifts relative to DMSO-d6 for both experimental and calculated 1H, 13C and 19F NMR chemical shifts of the title molecule are shown in Table 2. Fig. S2 (a,b,c) illustrate the experimental 1H, 13C and 19F NMR spectra of CFPHC.
13C
1H
19F
Atoms
Expt.
Calc.
Atoms
Expt.
Calc.
Atoms
Expt.
Calc.
C1
125.50
121.81
H13
7.54
7.45
F26
−110.00
−109.92
C2
172.89
172.91
H14
8.03
8.09
C3
139.52
141.08
H16
7.62
7.77
C4
145.05
146.62
H17
8.58
8.56
C6
162.15
158.62
H24
9.82
9.35
C7
120.57
120.42
H25
7.61
7.60
C8
155.05
151.65
H27
7.67
7.64
C9
128.07
127.99
H28
8.74
8.69
C10
129.85
128.75
C18
130.57
130.54
C19
138.49
134.76
C20
116.19
118.04
C21
164.13
170.64
C22
118.07
118.48
C23
130.61
131.04
4.10 Thermo gravimetric and differential thermal analysis
When sample CFPHC was heated beyond 340 °C, single stage decomposition curve shows two stages of decomposition. First weight loss occurs in the temperature range 98 °C amounting to 2.40% and the second one between 98.70 and 317.50 °C which amounts to the rest of the sample 91%. Presence of multiple weight loss steps indicates the presence of chromophore present in the sample and endothermic dip observed at 98.7 °C is due to their decomposition. DTA curve shows an endothermic dip at 196.20 °C which is due to the melting point of the compound. Complete decomposition at temperature 317.50 °C reflects in DTA thermograms. TG and DTA thermograms of CFPHC are shown in Fig. S3.
Thermodynamic functions such as heat capacity (
), entropy (
) and enthalpy (Δ
) changes for CFPHC molecule were computed from theoretical harmonic wavenumbers in gas phase and tabulated in the Table 3. Thermodynamic parameters calculated shows rise in temperature ranging from 100 to 1000 K due to the fact that, the molecular vibrational intensity increases with increase in temperature (Bevan and Boerio-goates, 2000). Correlation equations connecting heat capacity, entropy, enthalpy changes and temperatures are been fitted by quadratic formulas and the corresponding polynomial curve fitting factors (R2) for these thermodynamic properties are 0.99, 0.99 and 0.99, respectively. Thermochemical field can use these parameters to compute the other thermodynamic energies and estimate directions of chemical reactions according to relationships of thermodynamic functions and using second law of thermodynamics. The corresponding fitting equations ((1)–(3)) are as follows and the correlation graphics are shown in Fig. S4.
Temperature T (K)
Specific Heat Capacity (
) (cal/mol.k)
Entropy (
) (cal/mol.k)
Enthalpy (Δ
) (kcal/mol)
100
32.76
91.66
2.02
200
55.58
121.45
6.44
298.15
77.41
147.73
12.98
300
77.81
148.21
13.12
400
98.20
173.46
21.94
500
115.24
197.27
32.65
600
128.92
219.54
44.88
700
139.87
240.27
58.34
800
148.75
259.54
72.79
900
156.04
277.50
88.04
1000
162.10
294.27
103.96
4.11 In silico study
Molecular docking, the In silico analysis is a computational structure-based design method for assessing ligand molecule to select those that are likely to bind a protein with highest affinity (Shimada, 2006) estimating the bioactivity of the ligand. Hydrogen bonding and hydrophobic interactions play an important role in stabilizing a ligand energetically at the interface of a protein structure. Ligand binding with receptor protein involves the breakage of hydrogen bonds with the release of water molecules and the formation of new hydrogen bonds between the protein and ligand. Ligand CFPHC created from the optimized ground state structure (gas phase) was successively docked with seven reported protein receptors, having PDB id: 2GG5 (Evdokimov et al., 2007), PDB id: 2KCE (Rutenber and Stroud, 1996) as Escherichia coli PDB proteins along with PDB id: 1GAL (Hecht et al., 1993), [organism: Aspergillus niger], PDB id: 4L2J (Moreno et al., 2014), PDB id: IJZQ (Nakama et al., 2001), PDB id: 2MHV (Fizil et al., 2015), (antifungal proteins) and PDB id: 2K35 (Jung et al., 2008) (antimicrobial protein), respectively. Fig. 6 shows ligand-targeted complexes with hydrogen bonding interactions and binding pockets of protein-ligand interactions was visualized using PyMOL1.2r1. Yellow colored dotted lines shows hydrogen bonding of protein-ligand interactions while green and blue color indicates the hydrophobic interactions. Binding affinity of a ligand to its target protein represented in Table 4.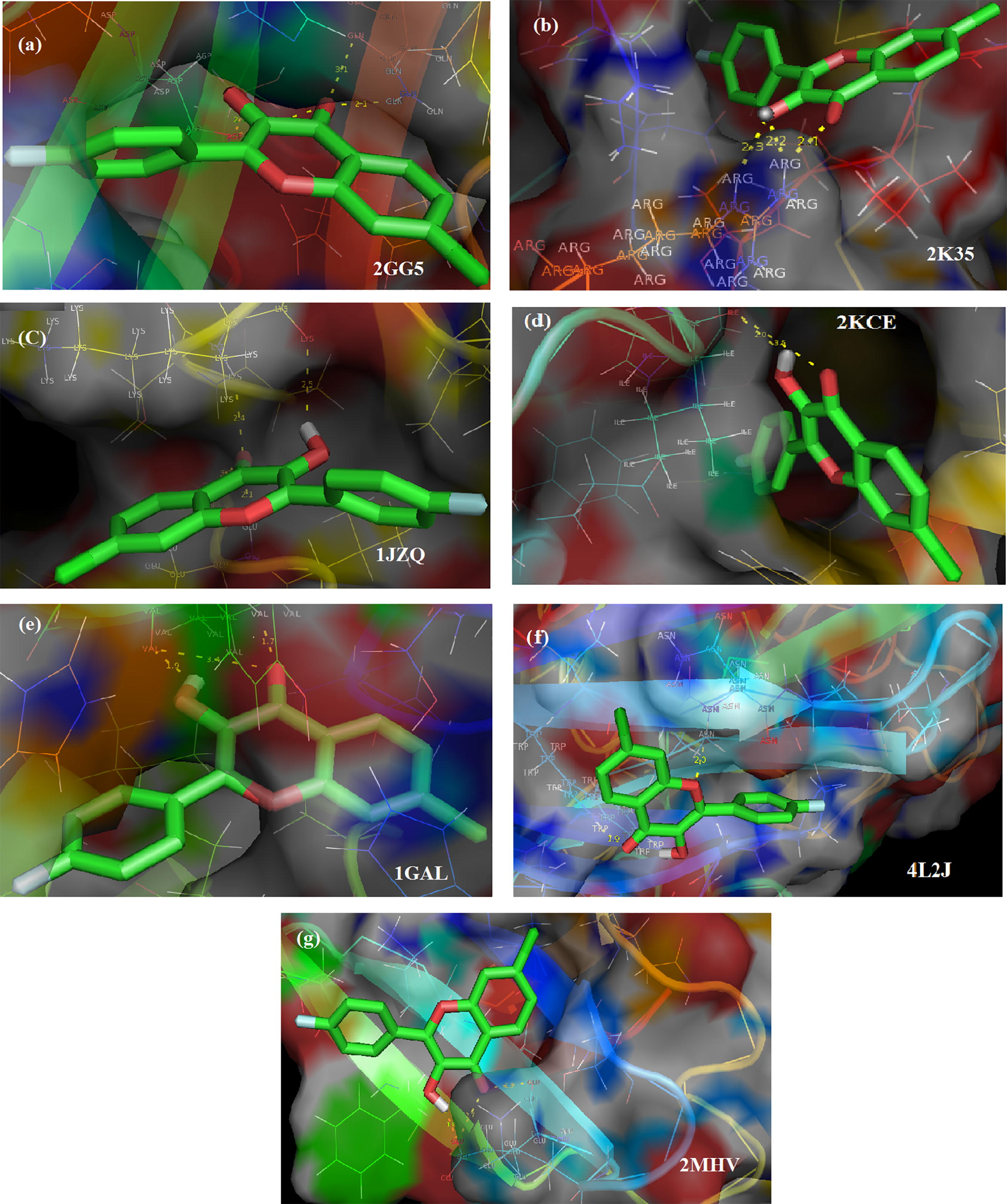
Ligand CFPHC interactions with target proteins.
Protein PDB ID
Bonded Residues
H-Bond Distance (Å)
Estimated Inhibition Constant (Ki) (μM)
Reference RMSD (Å)
Binding energy (ΔG)(kcal/mol)
2GG5
Asp 97
3.3
23.36
21.84
−6.99
2.1
Gln 233
2.1
3.1
2KCE
Ile 79
2.0
18.79
39.35
−6.45
3.5
1GAL
Ile 6
1.9
868.8
78.94
−8.27
3.4
1.7
4L2J
Asn 35
2.0
48.58
20.24
−5.88
Trp 32
1.9
1JZQ
Glu 499
2.1
52.18
67.83
−5.84
3.2
Lys 482
2.5
2.4
2MHV
Glu 13
2.8
94.58
20.15
−5.49
2.7
1.8
2K35
Arg 47
2.3
34.52
8.15
−6.09
2.2
2.1
Residue Asp 97 of 2GG5 attached to carbonyl group at 3.3 Å and hydroxyl group at 2.1 Å with average inhibition constant (Ki) = 23.36 µM while Gln 233 interact with ligand at 3.1 and 2.1 Å with oxygen atom of carbonyl group in chromene moiety having binding energy −6.99 kcal/mol. Interactions regarding the carbonyl and hydroxyl group of ligand CFPHC with the residue Ile 79 of 2KCE, respectively at 2.0 Å and 3.5 Å possess binding energy (ΔG) = −6.45 kcal/mol with very low inhibition constant of 18.79 µM. Hydrogen atom of hydroxyl group possess binding energy of −8.27 kcal/mol and Root-mean-square-deviation (RMSD) 78.94 Å that holds Val 250 residue of 1GAL at 1.9 Å while carbonyl oxygen interact with this residue forming two hydrogen bonds with high inhibition constant 868.8 µM at 3.4 Å and 1.7 Å, respectively. Oxygen atoms in both carbonyl and hydroxyl chromophore show the hydrogen bonding interaction with residue Gln-89 having ΔG = −5.66 kcal/mol and Ki = 70.61 µM] for 4L2J protein at bond distances of 2.0 Å and 2.3 Å. In addition, Trp 32 attached with carbonyl group at 1.9 Å with RMSD of 20.24 Å. Lys 482 residue of antifungal protein 1JZQ forms a hydrogen bond with hydrogen atom of hydroxyl group at 2.5 Å and carbonyl group at 2.4 Å having Ki = 52.18 µM and ΔG = −5.84 kcal/mol. Likewise carbonyl group forms two hydrogen bonds [2.1 and 3.2 Å] with residue Glu 499 having an inhibition constant of 52.18 µM at RMSD 67.83 Å. Amino acid protein Glu13 of antifungal protein 2MHV interacts with hydroxyl hydrogen atom at 1.8 Å with binding energy −5.49 kcal/mol also at 2.7 Å and 2.8 Å with carbonyl oxygen atom with RMSD of 20.15 Å with very high inhibition constant of 94.58 µM. Arg 37 of 2K35 antimicrobial protein interacts with both carbonyl group and hydroxyl group forming three hydrogen bond at 2.1 Å, 2.3 Å and 2.2 Å, respectively with an inhibition constant of 34.52 µM having binding energy −6.09 kcal/mol.
4.12 In vitro assays
4.12.1 DPPH• radical scavenging assay for antioxidant activity
Title compound showed 14, 35, 64 and 82% of scavenging activity at various concentration (12.5, 25, 50 and 100 μg/mL), respectively. 50% scavenging activity (inhibition constant (IC50) was calculated as 35 μg/mL. While comparing with standard antioxidant agent ascorbic acid (IC50 = 23 μg) test compound showed significant antioxidant activity. Antioxidant activity of CFPHC is shown in Table 5a and is represented as bar diagram shown in Fig. S5 (a).
CFPHC (μg)
% inhibition
T1(12.5)
14
T2(25)
35
T3(50)
64
T4(100)
82
Ascorbic acid (μg)
% inhibition
T1(5µg)
11.43
T2 (10 µg)
26.29
T3 (20 µg)
34.79
T4 (40 µg)
71.55
4.12.2 Agar well-diffusion method for antimicrobial activity
The test compound CFPHC showed significant antibacterial activity against bacterial strains Bacillus subtilis and Escherichia coli, but only average antibacterial activity has shown against gram-positive bacteria Staphylococcus aureus and gram-negative bacteria Klebsiella pneumoniae. Table 5b shows Bacillus subtilis with zones of inhibition (12 and 15 mm) and Escherichia coli (12 and 14 mm) while positive control gentamycin also shows significant inhibition zone.
Biological activity
Organism
Zone of inhibition (mm)
Positive Control
Concentration
2 mg
4 mg
Antibacterial
Gentamycin
Escherichia coli
40
12
14
Staphylococcus aureus
42
11
12
Klebsiella pneumoniae
40
11
12
Bacillus subtilis
45
12
15
Antifungal
Fluconazole
Aspergillus niger
35
14
11
Candida albicans
32
–
–
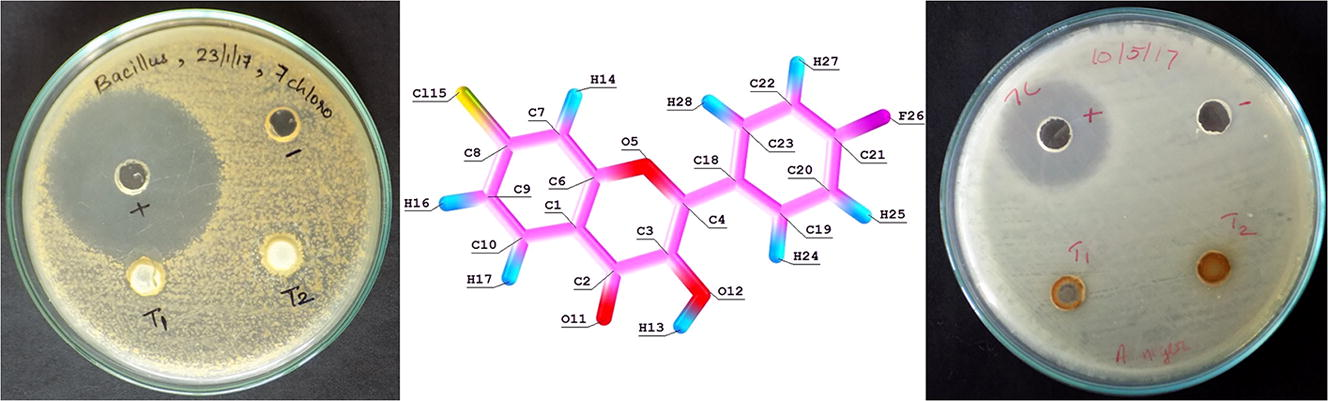
Average antifungal activity has shown against Aspergillus niger with zone of inhibition (14 mm and 11 mm) and positive control fluconazole shows inhibition zone while no zone of inhibition is seen in the case of Candida albicans. Photographs of antimicrobial activity of the compound is shown in Fig. S6.
4.12.3 Protein denaturation inhibition assay for anti-inflammatory activity
Protein denaturation inhibition assay was performed to analyze the anti-inflammatory activity of CFPHC. Assay was performed using bovine serum albumin and Aspirin as a standard. When compared with percentage of albumin denaturation inhibition of standard drug Aspirin 68% at 100 μg and CFPHC showed moderate protein denaturation inhibition activity around 65% at 500 μg and is presented in Table 5c. Bar diagrams show the comparison of Aspirin and title compound as shown as Fig. S5(b).
Concentration (μg)
% inhibition
100
35%
200
47.50%
300
55%
400
62.50%
500
65%
Aspirin (100 μg)
68%
5 Conclusion
Single crystal of CFPHC exists in the triclinic system with P space group. Calculated structural geometry parameters are in good correlation with the XRD results. Vibrational analysis clearly exposes red shifting of O–H stretching wavenumber confirms the existence of O–H⋯O intramolecular hydrogen bonding interaction. Downshielding of H24 is due to the formation of improper C–H⋯O hydrogen bonding determined by NMR spectral analysis This interaction also confirmed by LP1(O12)→σ∗(C19–H24) by NBO analysis. HOMO–LUMO energy gap explains the charge transfer within the molecule. Molecular electrostatic potential helps to identify the nucleophilic and electrophilic regions of CFPHC. Molecular docking results indicated that the compound is an effective antifungal agent capable of interacting with the target proteins (1GAL and 2MHV). DPPH Radical Scavenging Assay explore excellent antioxidant activity. Significant antimicrobial and anti-inflammatory activities confirmed by in vitro assay against various microorganisms. Therefore, the title compound can be considered as a better pharmacological agent and is suitable for antifungal treatment.
References
- Molecular structure and vibrational spectroscopic studies of 1-niro-4-(trifluoromethoxy) benzene by density functional method. Indian J. Pure Appl. Phys.. 2011;49:516-522.
- [Google Scholar]
- Fourier transform infrared and FT-Raman spectra, assignment, ab initio, DFT and normal co-ordinate analysis of 2-chloro-4-methylaniline and 2-chloro-6-methylaniline. Spectrochim. Acta A. 2009;72:436-444.
- [Google Scholar]
- FT-IR analysis of pyrone and chromene structures in activated carbon. Energy Fuels. 2014;28:4096-4103.
- [Google Scholar]
- Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys.. 1993;98:5648-5652.
- [Google Scholar]
- Density-functional exchange energy approximation with correct asymptotic behaviour. Phys. Rev. A. 1988;38:3098-3100.
- [Google Scholar]
- The Infrared Spectra of Complex Molecules. London: Chapmann & Hall; 1975.
- Calculations from Statistical Thermodynamics. New York: Academic Press; 2000.
- Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol.. 1995;28:25-30.
- [Google Scholar]
- The Chemistry of Heterocyclic Compounds, Chromenes, Chromanones, and Chromones. New York: John Wiley & Sons; 2009.
- Serendipitous discovery of novel bacterial methionine aminopeptidase inhibitors. Protein Sci.. 2007;66:538-546.
- [Google Scholar]
- WinGX suite for small molecule single-crystal crystallography. Appl. Cryst.. 1999;32:837-838.
- [Google Scholar]
- “Invisible” conformers of an antifungal disulfide protein revealed by constrained cold and heat unfolding, CEST-NMR experiments, and molecular dynamics calculations. Chem. Eur. J.. 2015;21:5136-5144.
- [Google Scholar]
- Molecular Orbitals and Organic Chemical Reactions. United Kingdom: John Wiley and Sons Ltd; 2010.
- Frontier Orbitals and Organic Chemical Reactions. New York: John Wiley and Sons; 1976.
- Exploring Chemistry with Electronic Structure Methods. Pittsburgh: Gaussian; 1996.
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B.,Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Jr., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R. L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O¨, Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J., 2009. Gaussian 09, Revision A. 02, Gaussian Inc.: Wallingford, CT.
- Glendening, E.D., Reed, A.E., Carpenter, J.E., Weinhold, F., 1998. “NBO 3.1”, Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin, Madison, WI.
- The influence of polarization functions on molecular orbital hydrogenation energies. Theoret. Chim. Acta. 1973;28:213-222.
- [Google Scholar]
- Crystal structure of glucose oxidase from Aspergillus niger refined at 2.3 Å resolution. J. Mol. Biol.. 1993;229:153-172.
- [Google Scholar]
- Irikura, K.K., 2002. THERMO.PL, National Institute of Standards and Technology.
- NMR Data Interpretation Explained: Understanding 1D and 2D NMR Spectra of Organic Compounds and Natural Products. New York: Wiley; 2016.
- ORTEP3. Report ORNL-6895. Tennessee, USA: Oak Ridge National Laboratory; 1996.
- Hydramacin-1, structure and antibacterial activity of a protein from the basal metazoan hydra. J. Biol. Chem.. 2008;284:1896-1905.
- [Google Scholar]
- Carbon-13 NMR Spectroscopy. Chichester: John Wiley & Sons; 1988.
- Self-consistent equations including exchange and correlation effects. Phys. Rev. A. 1965;140:1133-1138.
- [Google Scholar]
- Self-consistent molecular orbital methods XX. A basis set for correlated wave functions. J. Chem. Phys.. 1980;72:650-654.
- [Google Scholar]
- Molecular structure, vibrational, UV and NBO analysis of 4-chloro-7-nitrobenzofurazan by DFT calculations. Spectrochim. Acta A. 2011;79:1162-1170.
- [Google Scholar]
- Access to biologically relevant diverse chromene heterocycles via multicomponent reactions (MCRs): recent advances. J. Org. Biomol. Chem.. 2014;2:1-50.
- [Google Scholar]
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785-789.
- [Google Scholar]
- Mercury CSD 2.0 – new features for the visualization and investigation of crystal structures. J. Appl. Cryst.. 2008;41:466-470.
- [Google Scholar]
- The ring-opening reaction of chromenes: a photochemical mode-dependent transformation. J. Phys. Chem. A. 2005;109:8684-8692.
- [Google Scholar]
- Interaction of anti-inflammatory drugs with serum proteins, especially with some biologically active proteins. J. Pharm. Pharmacol.. 1968;20:169-173.
- [Google Scholar]
- Moreno, F.B.M.B., de Oliveira, R.S.B., de Azevedo Moreira, R., Lobo, M.D.P., de Freitas, C.D.T., Ramos, M.V., Grangeiro,T.B., Monteiro-Moreira, A.C.O., 2014. Crystal structure of osmotin, an antifungal laticifer protein. RCSB PDB. To be published <http://www.rcsb.org/pdb/explore.do?structureId=4l2j> accessed February 15, 2017.
- AutoDock4 and Auto Dock Tools 4: automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30:2785-2791.
- [Google Scholar]
- Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem.. 1998;19:1639-1662.
- [Google Scholar]
- Synthesis, crystal structure and DFT studies of 8-chloro-3-((3-chlorobenzyl)thio)-[1,2,4] triazolo[4,3-a] pyridine. Crystals. 2015;5:491-500.
- [Google Scholar]
- Structural basis for the recognition of isoleucyl-adenylate and an antibiotic, mupirocin by isoleucyl-tRNA synthetase. J. Biol. Chem.. 2001;276:47387-47393.
- [Google Scholar]
- Effect of electronegativity on structural, spectrophotometric and thermo-chemical properties of fluorine and chlorine substituted isoxazoles by DFT method. Cogent Chem.. 2017;3:1296342.
- [Google Scholar]
- Density-Functional Theory of Atoms and Molecules. London: Oxford University Press; 1994.
- Combination of theoretical ab Initio and experimental information to obtain reliable harmonic force constants. Scaled quantum mechanical (SQM) force fields for glyoxal, acrolein, butadiene, formaldehyde, and ethylene. J. Am. Chem. Soc.. 1983;105:7037-7047.
- [Google Scholar]
- Molecular geometries, force constants, and dipole moment derivatives. J. Am. Chem. Soc.. 1979;101:2550-2560.
- [Google Scholar]
- J. Mol. Struct.. 2014;1075:254-263.
- Spectroscopic analysis (FT-IR/FT-Raman) and molecular structure investigation on m-fluoronitrobenzene using hybrid computational calculations. Spectrochim. Acta A. 2012;94:318-330.
- [Google Scholar]
- Expeditious assembly of a 2-amino-4H-chromene skeleton by using an enantioselective Mannich intramolecular ring cyclization-tautomerization cascade sequence. Chem. Eur. J.. 2011;17:7781-7785.
- [Google Scholar]
- Molecular mechanics: the cross-conjugated carbonyl group in heterocyclic compounds. 3. Parameterisation (MM2) of the adjacent C=C bond: evaluation tests. J. Mol. Model.. 2001;7:43-53.
- [Google Scholar]
- The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucl. Acids Res.. 2015;43:345-356. <www.rcsb.org>
- [Google Scholar]
- Binding of the anticancer drug ZD1694 to E. coli thymidylate synthase: assessing specificity and affinity. Structure. 1996;4:1317-1324.
- [Google Scholar]
- Synthesis, characterization and DFT studies of 6.8-dichloro-2-(4-chlorophenyl)-4H-chromene-4-one. Indian J. Pure Appl. Phys.. 2012;50:308-313.
- [Google Scholar]
- Harmonic vibrational frequencies: an evaluation of Hartree-Fock, Moller-Plesset, quadric configuration interaction, density functional theory and semiempirical scale factors. J. Phys. Chem.. 1996;100:16502-16513.
- [Google Scholar]
- The challenges of making useful protein-ligand free energy predictions for drug discovery. In: Ekins S., ed. Computer Applications in Pharmaceutical Research and Development. Hoboken: John Wiley and Sons; 2006. p. :321-351.
- [Google Scholar]
- Spectrometric Identification of Organic Compounds. New York: John Wiley & Sons; 1991.
- Antibacterial activity of Steroidal compounds isolated from Ganodermaapplanatum (Pers.) Pat. (Aphyllophoromycetideae) fruit body. Int. J. Med. Mushrooms. 1999;1:325-330.
- [Google Scholar]
- Molecular structure and vibrational spectra of 3-chloro-4-fluorobenzonitrile by ab initio HF and density functional method. Spectrochim. Acta A. 2008;7:1134-1139.
- [Google Scholar]
- MOLVIB-a flexible program for force field calculations. J. Mol. Struct.. 1990;218:321-326.
- [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.2r1, 2009. Schrödinger, LLC, <https://www.pymol.org>.
- Vibrational Spectra of Benzene Derivatives. New York: Academic Press; 1969.
- publCIF: software for editing, validating and formatting crystallographic information files. J. Appl. Crystallogr.. 2010;43:920-925.
- [Google Scholar]
- Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc.. 1990;112:8251-8260.
- [Google Scholar]
- Spectrochim. Acta A. 2011;78:319-326.
Appendix A
Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.jksus.2017.12.016.
Appendix A
Supplementary data




