

Translate this page into:
1,4,9,9-tetramethyloctahydro-4,7-(epoxymethano)azulen-5(1H)-one, a natural product as a potential inhibitor of COVID-19: Extraction, crystal structure, and virtual screening approach
⁎Corresponding authors. yns.elbakri@gmail.com (Youness El Bakri), sosaid@alfaisal.edu (Souraya Goumri Said)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
In the present work, we describe the extraction of a natural product namely 1,4,9,9-tetramethyloctahydro-4,7-(epoxymethano)azulen-5(1H)-one, and its structure was confirmed by single crystal X-ray diffraction analysis. The conformations of the 5-, 6-, and 7-membered rings in the title compound, C15H24O2, have been probed by a Cremer-Pople puckering analysis. C—H···O hydrogen bonds generate chains in the crystal that stretch along the c-axis direction. The Hirshfeld surface analysis method was used to stabilize the crystal packing of the natural compound. Accompanied by experimental studies, quantum chemical calculations were also performed to compare the structural elucidation and the results of these geometrical parameters exhibited excellent agreement. The compound was also docked with several drug targets of the SARS-CoV-2 virus and found to show the best binding with the main protease enzyme, having a binding energy of −12.31 kcal/mol and interacting with His41 and Cys145 residues. The dynamic stability deciphered the complex to be stable with an average RMSD of 3.8 Å. The compound dynamics with the enzyme showed the compound conformation to be highly stable. The intermolecular binding free energy determined the compound-main protease enzyme to show high interaction energy of < 40 kcal/mol. Together, these studies demonstrate the compound to be a lead structure against SARS-CoV-2.
Keywords
Natural product
SARS-CoV-2
COVID-19
DFT study
Molecular Docking
Molecular dynamics

1 Introduction
Infection with the novel coronavirus SARS-CoV-2, which is currently causing a global epidemic with over two million deaths so far, causes coronavirus disease (COVID-19). Though a variety of vaccinations against the virus have recently been licensed, availability remains a major difficulty, and adoption by the general public has become a contentious issue.
The COVID-19 epidemic has had a major influence on countries all around the globe. In the last year and a half, this subject has piqued the interest of international scientists and researchers, prompting them to investigate and develop potential remedies in the areas of prevention and cure (Salehi et al., 2020; Yuki et al., 2020). The focus of research in this area has been on developing an appropriate vaccine and discovering a cure for this condition. Chemical treatments have traditionally been the most popular treatment for viral infections (De Clercq and Li, 2016; Kumar et al., 2022). However, in recent years, the use of conventional synthetic organic compound-based allopathic drugs for therapy has been criticized, mostly due to the various negative effects of such treatments. Multiple pharmaceuticals are frequently provided by doctors to counteract the negative effects of allopathic treatments. The impact on the human body as a whole becomes complicated. Another issue is that long-term use of allopathic medicines causes the human body to acquire resistance to them, necessitating higher doses of these drugs, which are not regarded as good for the body (Attena, 2016; Bennadi, 2013; Gawde et al., 2013).
Chemical extracts from natural plants, also known as “herbal medicines,” have been discovered to be beneficial for a variety of ailments over the years. This includes anticancer, antifungal, antiviral, antimicrobial, antimalarial, and anti-HIV agents (CA et al., 1996; Verma and Singh, 2008). Chemical treatments have always been popular because of their immediate action against the ailment they are meant to treat (Moon and Alam, 2020). Herbal medications also have been chastised for their sluggish action on the human body (Abdalla et al., 2021; Boufissiou et al., 2022). Additionally, herbal medications have a big advantage in terms of their benign nature, which means they are far less likely to harm humans.
In the last several months, several papers have appeared in the literature demonstrating a promising technique to treat the COVID-19 epidemic (Bailly and Vergoten, 2020; Cai et al., 2020; Chen et al., 2020; Sahraei et al., 2020; Touret and de Lamballerie, 2020) However, they all suggest using chemical treatments to cure the pandemic or a potential vaccination to heal the epidemic.
Herein, we used several computational methods to demonstrate the binding affinity of the studied compound against several targets of SARS-CoV-2. The binding conformation and affinity were determined using molecular docking studies, while the dynamics of the docked complex were investigated using molecular dynamics simulations. Further, cross-validation of the docking results was obtained using MMGBSA and MMPBSA binding free energy methods. In recent years, these computational methods have been widely used to identify possible therapeutic compounds against a variety of infections.
2 Material and methods
2.1 Extraction and isolation
The air-dried aerial parts of A. lanatum (1.2 Kg) were powdered and extracted with a mixture of CH2Cl2:MeOH (1:1) (10 L) at room temperature. The extract was concentrated in vacuo, obtaining a residue of 94 g. The residues were re-fractionated by column chromatography on silica gel and eluted with n-hexane-Et2O (1:1) fractions 1-A and 1-B. Fraction 1-A was further purified on a Sephadex LH-20 column eluted with n-hexane-CH2Cl2-MeOH (4:7:0.5) to give impure compound 1 (Mahmoud and El-Sayed, 2019), which was further purified by flash chromatography (eluent: n-hexane-CH2Cl2 (1:2)) to yield pure 1 as a colorless crystal (Fig. 1a).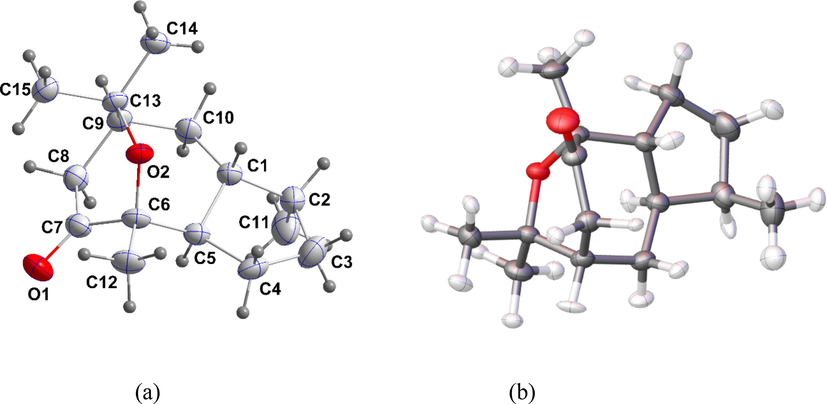
The labeling strategy for the title compound and 50% probability ellipsoids of both (a) IAM and (b) HAR models.
2.2 X-ray diffraction studies
For the X-ray crystallographic study, a colorless block-like specimen of C15H24O2 with approximate dimensions of 0.29 × 0.23 × 0.16 mm3 was used. The data for X-ray intensity were collected using a Bruker D8 VENTUREPHOTON 100 CMOS system outfitted with an 'INCOATEC IμS micro-focus source' (Cu, λ = 1.54178 Å) and a mirror monochromator. During data collection, the crystal was kept at 150.15 K utilizing Olex2 (Dolomanov et al., 2009), and the structure was solved with the SHELXT (Sheldrick, 2015) structure solution program utilizing Intrinsic Phasing and refined with the olex2. Refine (Bourhis et al., 2015) refinement package utilizing Gauss-Newton minimization. Selected crystal structure data are listed in Table S1.
2.3 Hirshfeld atom refinement (HAR) and Hirshfeld surface analysis (HS)
From X-ray diffraction data, Hirshfeld atom refinement is one of the interesting methods for determining structural parameters for hydrogen atoms. Therefore, HAR refinement was performed for the titled natural compound, which only contains C–H bonds, using the NoSpherA2 module (Kleemiss et al., 2021) with the help of ORCA 4.2.1 (Neese, 2012) in the Olex 1.5 software (Dolomanov et al., 2009) by the PBE method and the def2-TZVPP basis set. Then, the HS and the associated 2D fingerprint plots of the natural derivative allowed us to understand the additional information about crystal packing and the nature of interactions that were executed using CrystalExplorer21.5 (Spackman et al., 2021). The dnorm and shaped index maps were plotted using different color ranging, such as dnorm: from −0.11 (red) to 1.36 (blue) Å and shaped index: from −0.99 (concave) to 0.99 (convex) Å. The visualization of the supramolecular architecture of the molecule in the solid space is shown in energy frameworks, which were calculated for a reference molecule with a 3.8 Å radius of clusters using the accurate method of CE-B3LYP level and 6-311G(d,p) basis set. Furthermore, energy frameworks, which are graphical representations of the strength of crystal packing in different directions at the solid-state level, assist us in calculating the interaction energies with the cylinders.
2.4 DFT studies
The atomic coordinates were calculated from the crystallographic data for the calculation of the Hirshfeld surface analysis and the DFT calculation. The molecular structure of the natural compound was optimized with the B3LYP method and a 6-311G** basis set using the Gaussian 09 software package (Frisch et al., 2004). To understand the delocalization of bonding orbitals and dipole electric field polarizabilities, the NBO and NLO analyses were carried out during the geometry optimization at the same level of theory. The electrostatic potential map, HOMO, and LUMO of the natural compound were drawn.
The graphic illustration of an iso-surface with HOMO and LUMO values of 0.2 was employed. The Gauss View (Dennington et al., 2009) and 3plot (Stash and Tsirelson, 2014) programs were used to visualize all of these investigations.
2.5 Molecular docking studies
In search of novel inhibitory compounds against COVID-19, a molecular docking of the synthesized compound was carried out against multiple drug targets of the ARS-CoV-2 virus. The targets selected were the main protease enzyme (PDB ID: 7kx5), RNA-dependent RNA polymerase (RdRp)(PDB ID: 7,bv2), and Nucleocapsid(PDB ID: 6wzo). The structures were then imported to UCSF Chimera v1.15 (Pettersen et al., 2004) where co-crystallized associated molecules were removed and the energy minimized. During the process, missing hydrogen atoms were added and steepest descent (steps, 1000) and conjugate gradient (steps, 1000) algorithms were applied to remove steric clashes if any were present in the enzyme structure. The synthesized compound was sketched via ChemDraw 12.0 (Milne, 2010), followed by protonation and energy minimization in UCSF Chimera v1.15. The active site information of targeted enzymes was extracted from published literature. Molecular docking studies were performed using AutoDockVina in PyRx v0.8 (Dallakyan and Olson, 2015). Both enzyme and compound were converted into.pdbqt format. The grid box in each docking case was placed around the active pocket of the enzyme with dimensions of 25 Å along the XYZ planes. The number of docked compound poses generated with the enzymes is 20 and is assigned a binding energy value of kcal/mol. The best-docked conformation of the compound that revealed acceptable rmsd_refine values and was similar to the binding mode of the native co-crystallized inhibitor was selected for further study. Only the docked complex of the compound with the target enzyme showing the best intermolecular affinity was visualized for chemical interactions and docked pose investigation. This was achieved using UCSF Chimera v1.15 and (Pettersen et al., 2004) Discovery Studio Visualizer 2021 (Studio, 2008).
2.6 Molecular dynamics simulation
The best affinity docked model of the compound was set as starting coordinates for a long run of 200 ns of molecular dynamics simulation using AMBER20 simulation software (Lee et al., 2020). The enzyme-ligand complex was solvated in transferable intermolecular potential with a three-point (TIP3P) water model, keeping the marginal distance between the water box edge and enzyme at 12 Å. Enzyme and compound parameters were produced by FF14Sb (Maier et al., 2015) and GAFF (Sprenger et al., 2015), respectively. The enzyme residues were then treated for standard ionization state at pH 7, and the complex was neutralized by adding a sufficient number of counterions. The simulation procedure was conducted in three stages: complex geometry optimization using the steepest descent algorithm for 100 ps; system equilibration for 1000 ps under a constant number of particles, volume, and temperature (NVT) ensemble; system equilibration constant Number of particles, Pressure, and Temperature (NPT) ensemble at a pressure of 1 atm. Afterward, simulation was performed for 200 ns using constant pressure (NPT ensemble). The Particle Mesh Ewald algorithm was used for estimating long-range interactions (Essmann et al., 1995). The simulation trajectories were evaluated for structural data like root mean square deviation (RMSD) using the CPPTRAJ module of AMBER (Roe and Cheatham, 2013). The binding free energy between the protein and compound was determined using the MMPBSA.py (Miller et al., 2012) module. The module was set to pick simulation frames from trajectories at regular intervals. In total, 1000 frames were selected.
3 Results and discussion
3.1 X-ray diffraction analysis
Cremer-Pople puckering analyses of the 5-, 6- and 7-membered rings (Cremer &Pople, 1975) (Cremer and Pople, 1975) yielded the following sets of parameters. For the C1···C5 ring, Q(2) = 0.432(3)Å, φ(2) = 348.3(3)°, and the conformation is considered to be a twist on the C1···C5 axis. For the O2/C6···C9/C13 ring, Q = 0.714(3)Å, θ = 94.20(16)° and φ = 82.36(19). For the O2/C6/C5/C1/C10/C9/C13 ring, Q(2) = 1.168(2)Å, Q(3) = 0.040(2)Å, φ(2) = 219.36(12)° and φ(3) = 229(4)° with a total puckering amplitude of 1.169(2)Å. Finally, for the C1/C5···C10 ring, Q(2) = 0.726(3)Å, Q(3) = 0.642(3)Å, φ(2) = 160.0(2)° and φ(3) = 356.5(2)° and its total puckering amplitude is 0.969(3)Å. In the crystal, C15—H15C···O1 hydrogen bonds form chains extending along the c-axis direction (Table S2 and Fig. 2).
(a) Side view of portions of three chains seen along the a-axis direction with C—H···O hydrogen bonds shown as dashed lines; (b) End view of three chains seen along the c-axis direction with C—H···O hydrogen bonds shown as dashed lines.
3.2 Hirshfeld atom refinement and Hirshfeld surface analysis
Fig. 1b shows the anisotropic refinement of hydrogen atoms and their ADPs are in the supporting (Table S3). Also, Fig. 3 and supporting tables (Table S4) display the geometrical parameters of the molecule as obtained from the IAM and HAR models. A comparison of the geometrical parameters of both models allows us to understand the best model. Their bond lengths, bond angles, and torsion angles reveal the precise geometrical discrepancies. The C–H bond distance of the methyl group in the IAM model is ∼ 0.98 Å whereas it is extended in the HAR model, the value is ∼ 1.056 Å. Similarly, the C–H bond distance of CH and CH2 groups is also slightly increased from ∼ 0.99 Å to 1.11 Å. From the inspection, the bond length of carbon-hydrogen bonds obtained from the HAR model is in good agreement with neutron bond lengths (Steiner and Saenger, 1993) and a third decimal variation has been observed in the non-hydrogen bonds. Therefore, we can conclude that the HAR model reproduces the hydrogen bond lengths from neutron data. The deformation density map (Fig. 4) shows the charge accumulation in the keto group oxygen, which confirms the lone-pair region.
Shows C–H bond lengths of the molecule (a) IAM and (b) HAR model.
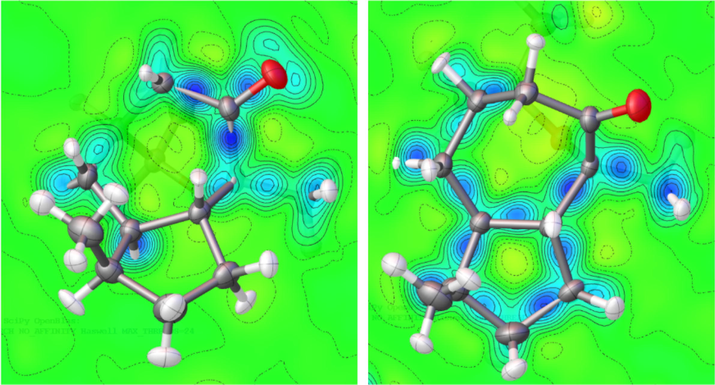
Deformation density map of the molecule, drawn at 0.1 eÅ−3.
The dnorm and shaped index maps of the molecule are shown in Fig. 5. The visualization of the molecular surface for the selected parts is permitted to be transparent, in which case the red spots explain the hydrogen bonding, such as C-H‧‧‧O contacts, and white as well as blue areas explain the weaker and longer contacts. The two-dimensional fingerprint plots show the discrete intervals of di and de, in which the molecule forms only O‧‧‧H and H‧‧‧H types of interactions that are displayed in the different shapes, like wings and cam-lobe shapes, where the values are 14.5 and 85.5 % respectively. These two types of interactions are the major driving forces in the solid-state packing, which is widely scattered in the plot. Notably, the molecule is found to have a small C‧‧‧H contribution and there are no C‧‧‧C, C‧‧‧O, and O‧‧‧O contributions.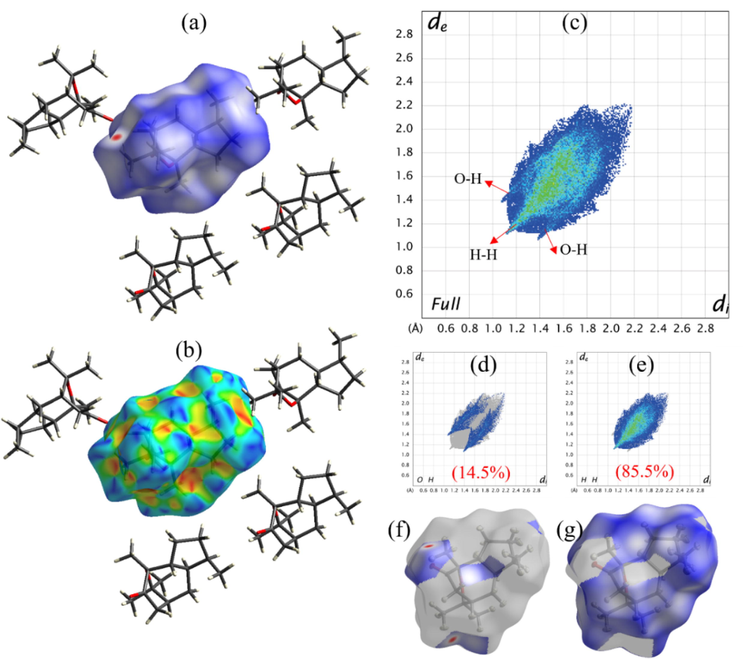
HS study of the product; (a, f-g) dnorm, (b) shaped index, and (c-e) fingerprint 2D plots with their contribution in percentages.
The pairwise interaction energies of the reference molecule with a 3.8 Å cluster of adjacent molecules were calculated. Here the total energy is the contribution of electrostatic, dispersion, polarization, and repulsion energies (Fig. 6 and Table S5). According to the table, the most energetically stable dimers are formed in the symmetry of -x, y + 1/2, and -z + 1/2 with a distance of 5.86 Å and they carry higher electrostatic, dispersion, and total energies than the other symmetry-related compounds. The different colored (red, green, and blue) cylinders are represented by their energy components of electrostatic, dispersion, and total energies, and the directionality of the energy frameworks is found to be very similar in all three energy patterns.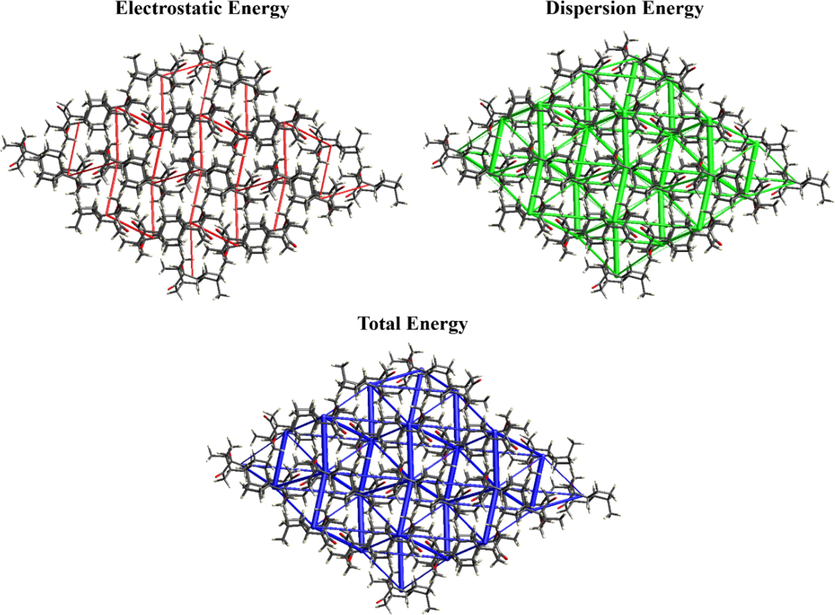
Energy frameworks of the molecule.
3.3 DFT studies
The comparison of geometrical parameters of both the IAM and HAR models was discussed in the previous section (2.3); here, The IAM model was compared to the selected structural information acquired from the optimized molecule. The geometrical parameters of hydrogen and non-hydrogen atoms calculated by the OPT and HAR models are found to be almost similar, whereas the IAM model is slightly shorter than the other two models. The effect of the base set is responsible for this difference. (Table S4).
3.3.1 Molecular electrostatic potential
The MEP map depicts the electron-rich and electron-deficient portions of the molecule in general; it also helps predict the electrophilic and nucleophilic sites in the molecule. The electrostatic potential and total electron density calculations were performed to draw the two different views of the MEP map for the molecule (Fig. 7), in which the vicinity of the keto group region shows a dark red surface in both maps, which indicates electron-rich and electrophilic attacks, whereas other regions exhibited a blue surface, which was found to be electron-poor and nucleophilic regions. Further, the minima and maxima of the electrostatic potential map provide the role and ability of non-covalent interactions, which ranged from 55.28 to −47.36 kcal/mol. The most prominent negative values are observed around the keto (O2) group (-47.36 kcal/mol) and the most positive regions are around aromatic H-atoms (55.28 kcal/mol). In the solid state, this keto group forms a C-H‧‧‧O type of interaction and can interact with the active site of protein or DNA.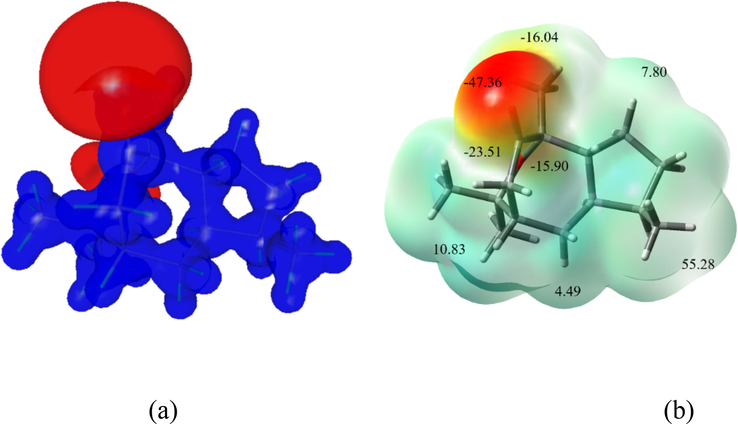
(a) ESP maps of the natural molecule in the two different views; (a) 3D electrostatic potential map and (b) minima and maxima of ESP with total electron density map.
3.3.2 Molecular orbital analysis
According to the scientific reports, molecular reactivity such as bandgap energy, ionization potential, electron affinity, electronegativity, electrophilicity, and global hardness have been globally calculated from the DFT calculation (Table S6). Therefore, the DFT calculation allows us to explore the most important information on reactivity as well as site selectivity. As we know, HOMO energy is closely tied to ionization energy, whereas LUMO energy is directly associated with electron affinity. The stability of the molecule can be confirmed from the bandgap energy, the value is 5.65 eV. Koopman's theorem is used to calculate molecular orbital energies, which allows closed-shell molecules to duplicate the molecule's reactivity.
Based on the global reactivity characteristics, the molecule's calculated ionization potential, electron affinity, and electronegativity are 5.65, 0.91, and 3.74 eV. The chemical hardness is also another important molecular property that explains the resistance of the atoms in the molecule to either polarize or deform their electron density clouds, the value is 2.83 eV. Fig. 8 depicts the HOMO and LUMO energy levels, with blue and red color surface distributions indicating positive and negative locations in the molecular orbital wave function, respectively. The keto and methyl groups in the molecule have a high concentration of HOMO and LUMO.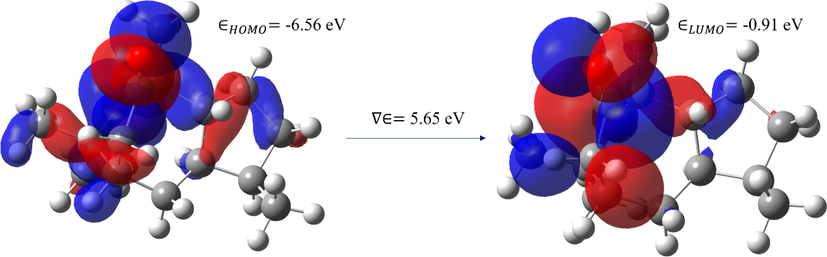
The HOMO and LUMO map of the molecule.
3.3.3 Nonlinear optical properties (NLO)
Because of the rapid increase of optical, storage, and electronic devices, organic compounds with NLO properties are being studied in various research fields like material science, medicine, and molecular and solid-state physics. In general, the organic materials exhibited intermolecular charge transfer with excellent NLO response due to π-electrons with strong electric polarization, charge distribution from donors to acceptor, and low dielectric coefficient. Also, the presence of delocalized electrons allows one to enhance the NLO property. Importantly, a clear understanding of NLO properties such as polarizability (liner effect) and hyperpolarizability (nonlinear effect) of the materials is vital to designing a material. As a result, the DFT/B3LYP 6-311G** technique computes various components of the 3 × 3 × 3 matrix from the molecule's dipole moment (µ).The complete equations for determining the dipole moment (µ), polarizability (αtotal), and first-order hyperpolarizability (βtotal) of the molecule, which is responsible for the non-linear optical features of the molecule, are shown in many studies, the values are shown in Table S7. The polarizability and first-order hyperpolarizability values are converted to electrostatic units (esu) (α: 1a.u = 0.1482 × 10-24esu and β: 1a.u = 8.6393 × 10-33esu). The dipole moment (µ), the polarizability (αtotal) and first-order hyperpolarizability (βtotal) of the molecule are 3.241 Debye, −10.75 × 10-24esu, and 332.44 × 10-33esu respectively. These values are significantly higher than urea, indicating that the title molecule is an excellent candidate for NLO applications. Due to a lack of hydroxyl groups in the molecule, these values are slightly lower than those published earlier for a natural chemical (Ourhzif et al., 2021).
3.3.4 Naturel bond orbital study (NBO)
The NBO analysis is a potential method to explain the charge transfer and conjugated interactions between the donor and acceptor moieties. Also, it explains the electron distribution in different subshells of their atomic orbitals of the titled molecule. At the B3LYP level, using the 6-311G** basis set, the precise interactions between lone-pair-filled orbitals (bonding) and Rydberg empty orbitals (anti-bonding) of the molecule were computed. Generally, to study the electron transmission from donor orbital to acceptor orbital, a large stabilization energy E(2) value allows validation of the interaction between electrons of the donor and electrons of the acceptors. Also, the stabilization energies are connected with electron delocalization between donor and acceptor. The calculated NBO analysis of the titled compound shows weak hydrogen bonding between oxygen atoms and C–H bonds, the stabilization energy of the interactions is found to be greater than 0.5 kcal/mol (Table S8). Moreover, the stabilization energy of oxygen (O1) atoms with carbon (C6 and C7) is 21.39 and 19.87 kcal/mol, respectively. This high stabilization energy elongates the bond with the electron density of the antibonding orbitals.
3.4 Molecular docking study
Molecular docking is a very useful method for determining a ligand's optimum binding mode in terms of binding to a receptor molecule. Among the receptors used, the compound showed the best binding with the SARS-CoV-2 main protease enzyme having a binding affinity score of −12.31 kcal/mol. The binding energy of the compound with SARS-CoV-2 Nucleocapsid and RdRp enzyme is −10.11 kcal/mol and 9.21 kcal/mol, respectively. The compound was found to interact with the active pocket of the main protease enzyme and form strong hydrophilic and hydrophobic contact with critical residues such as His41 and Cys145. The docked conformation and interactions are given in Fig. 9.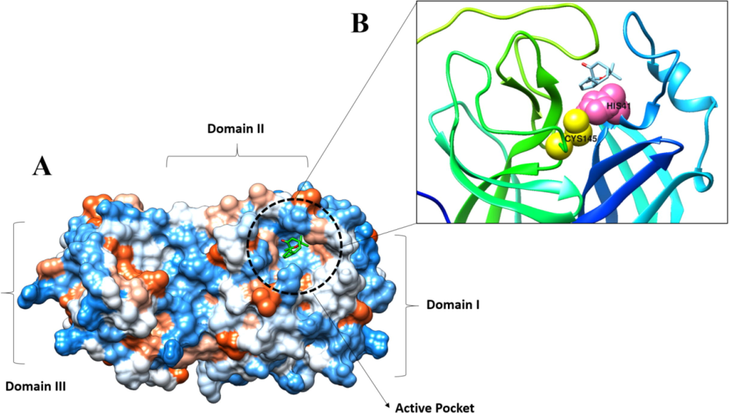
A. Docked compound at SARS-CoV-2 main protease enzyme active pocket. B. Key active pocket residues interacting with the compound.
3.5 Molecular dynamics
The dynamics simulation data revealed the formation of a stable docked complex and very few structural deviations are observed in the compound binding conformation. Both receptor and compound RMSD are provided in Fig. 10. The receptor dynamics were seen to be stable in the first 80 ns, followed by continuous structural changes until 160 ns. Toward the end of the simulation, the receptor achieved very stable behavior. The loops of the receptor were revealed to be responsible for middle phase unstable dynamics. However, these changes do not affect the compounds binding with the enzyme. On the other hand, the compound binding conformation remained in a very stable state throughout the length of simulation time T.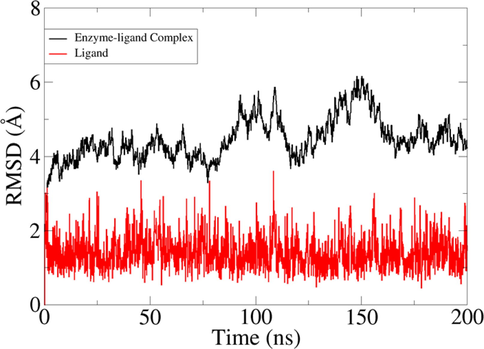
RMSD analysis of docked complex and ligand molecules in given simulation time.
Furthermore, complex binding free energies were estimated. It was found that the electrostatic (-14.36 kcal/mol) and Van der Waals energies (-42.41 kcal/mol) are the most favorable in complex formation. It seems that hydrophobic regions of the compound play a major role in binding with the SARS-CoV-2 main protease enzyme. Both the energies when added together revealed significant gas-phase energy of −56.77 kcal/mol. The solvation energy of the complex is 8.01 kcal/mol and 12.12 kcal/mol in MM/GBSA and MM/PBSA, respectively. The total binding free energy of the complex is −48.76 kcal/mol in MM/GBSA and −44.65 kcal/mol in MM/PBSA, respectively (Table S9).
4 Conclusions
A new natural product was extracted and characterized by a single X-ray diffraction technique. According to HS research of the crystal structure, the most significant contributors to crystal packing are just O⋅⋅⋅H and H⋅⋅⋅H contributions. The calculation of electrostatic, dispersion, and total energy frameworks confirms the stabilization by the dispersion energy contribution. The DFT of the optimized structure was compared with the experimental molecular structures of both the IAM and HAR models to understand the properties like dipole moment, frontier orbital energy gap, natural bond orbital calculation, and molecular electrostatic potential map. The study was further stretched to include molecular docking analysis of the compounds against different SARS-CoV-2 enzymes. It was revealed that the main protease enzyme is the main target for the compound and was reported to show the best binding conformation and interactions with the hotspot residues of the enzyme.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- In silico studies on phytochemicals to combat the emerging COVID-19 infection. J. Saudi Chem. Soc.. 2021;25:101367
- [Google Scholar]
- Limitations of western medicine and models of integration between medical systems. J. Altern. Complement. Med.. 2016;22:343-348.
- [Google Scholar]
- Glycyrrhizin: An alternative drug for the treatment of COVID-19 infection and the associated respiratory syndrome? Pharmacol. Ther.. 2020;214:107618
- [Google Scholar]
- In-silico investigation of phenolic compounds from leaves of Phillyrea angustifolia L. as a potential inhibitor against the SARS-CoV-2 main protease (Mpro PDB ID: 5R83) using a virtual screening method. J. Saudi Chem. Soc.. 2022;26:101473
- [Google Scholar]
- The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment–Olex2 dissected. Acta Crystallogr. Sect. A Found. Adv.. 2015;71:59-75.
- [Google Scholar]
- CA, Ÿ.N., Anderson, L.A., Phillipson, J.D., 1996. Herbal medicines. A Guid. Heal. Prof. Pharm. Press. London 25.
- Experimental treatment with favipiravir for COVID-19: an open-label control study. Engineering. 2020;6:1192-1198.
- [Google Scholar]
- Chen, J., Xia, L., Liu, L., Xu, Q., Ling, Y., Huang, D., Huang, W., Song, S., Xu, S., Shen, Y., 2020. Antiviral activity and safety of darunavir/cobicistat for the treatment of COVID-19, in: Open Forum Infectious Diseases. Oxford University Press US, p. ofaa241.
- Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev.. 2016;29:695-747.
- [Google Scholar]
- Dennington, R., Keith, T., Millam, J., 2009. GaussView, version 5.
- OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr.. 2009;42:339-341.
- [Google Scholar]
- Knowledge, attitude, and practices toward ayurvedic medicine use among allopathic resident doctors: a cross-sectional study at a tertiary care hospital in India. Perspect. Clin. Res.. 2013;4:175.
- [Google Scholar]
- Accurate crystal structures and chemical properties from NoSpherA2. Chem. Sci.. 2021;12:1675-1692.
- [Google Scholar]
- Reaction of 7α-bromo-6-nitrocholest-5-enes with hydrazine: Formation of steroidal pyrazolines and molecular docking against SARS-CoV-2 omicron protease. Steroids. 2022;188:109120
- [Google Scholar]
- Alchemical binding free energy calculations in AMBER20: Advances and best practices for drug discovery. J. Chem. Inf. Model.. 2020;60:5595-5623.
- [Google Scholar]
- The anti-proliferative activity of anisosciadone: A new guaiane sesquiterpene from Anisosciadium lanatum. Anti-Cancer Agents Med. Chem. (Formerly Curr. Med. Chem. Agents). 2019;19:1114-1119.
- [Google Scholar]
- ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput.. 2015;11:3696-3713.
- [Google Scholar]
- MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput.. 2012;8:3314-3321.
- [CrossRef] [Google Scholar]
- Aluminum chloride-functionalized silica gel synthesis as a catalyst for the preparation of biologically active oxazolidinethiones: Antioxidant and molecular docking studies. J. Saudi Chem. Soc.. 2020;24:906-914.
- [Google Scholar]
- Crystal structure, Hirshfeld surface analysis and DFT studies of Euphorbioside monohydrate a major bisnorsesquiterpene isolated from Euphorbia resinifera latex. J. Mol. Struct.. 2021;1241:130511
- [Google Scholar]
- UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem.. 2004;25:1605-1612.
- [Google Scholar]
- PTRAJ and CPPTRAJ: Software for processing and analysis of molecular. Trajectory Data: Dyn; 2013. p. :9.
- Aminoquinolines against coronavirus disease 2019 (COVID-19): chloroquine or hydroxychloroquine. Int J Antimicrob Agents. 2020;55:105945
- [Google Scholar]
- Coronavirus disease 2019 (COVID-19): a systematic review of imaging findings in 919 patients. Ajr Am J Roentgenol. 2020;215:87-93.
- [Google Scholar]
- CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr.. 2021;54
- [Google Scholar]
- The general AMBER force field (GAFF) can accurately predict thermodynamic and transport properties of many ionic liquids. J. Phys. Chem. B. 2015;119:5882-5895.
- [Google Scholar]
- Developing WinXPRO: a software for determination of the multipole-model-based properties of crystals. J. Appl. Crystallogr.. 2014;47:2086-2089.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2023.102628.
Appendix A
Supplementary data
The following are the Supplementary data to this article:




