

Translate this page into:
Synthesis, spectral and biological evaluation of some new heterocyclic derivatives incorporating dihydroanthracene moiety
*Corresponding author. Tel.: +60 3 8921 5412; fax: +60 3 8921 5410 jumat@ukm.my (Jumat Salimon)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Available online 20 August 2010
Peer-review under responsibility of King Saud University.
Abstract
The reaction of anthrone 1 with 4-aminoantipyrine and thiosemicarbazide afforded 4-(anthracen-9(10H)-ylideneamino)-1,5-dimethyl-2-phenyl-1,2-dihydro-3H-pyrazol-3-one 2 and anthracen-9(10H)-one thiosemicarbazone 5, respectively. Oxidation of compound 2 with potassium permanganate gave 4-(anthracen-9(10H)-ylideneamino)-1-methyl-3-oxo-2-phenyl-2,3-dihydro-1H pyrazole-5-carboxylic acid 3 which on reaction with o-phenylenediamine gave 4-(anthracen-9(10H)-ylideneamino)-5-(1H-benzimidazol-2-yl)-1-methyl-2-phenyl-1,2-dihydro-3H-pyrazol-3-one 4. Furthermore, compound 5 was condensed with different substituted phenacyl bromide to give a series of 2-(anthracen-9(10H)-ylidenehydrazono)-5-substituted-2,3-dihydro-1H-thiazole 6a–g. Compound 5 also reacted with chloroacetic acid affording 2-(anthracen-9(10H)-ylidenehydrazono)thiazolidin-4-one 7. The structures of all the products have been determined by elemental analysis and spectral studies. All compounds have been screened for their antibacterial and antifungal studies. The results are summarized in Tables 1 and 2.
Keywords
Antipyrine
Benzimidazole
Thiazole
Thiazolidin-4-ones
Antibacterial
Antifungal

1 Introduction
Research and development of potent and effective antimicrobial agents represents one of the most important advances in therapeutics, not only in the control of serious infections, but also in the prevention and treatment of some infectious complications of other therapeutic modalities such as cancer chemotherapy and surgery. Over the past decade, fungal infection became an important complication and a major cause of morbidity and mortality in immuno-compromised individuals such as those suffering from tuberculosis, cancer or AIDS and in organ transplant cases (Turan-Zitouni et al., 2005). However, in recent years, much attention has been focused on addressing the problem of multi-drug resistant (MDR) bacteria and fungi resulting from the widespread use and misuse of classical antimicrobial agents (Akbas and Berber, 2005). Such serious global health problem demands a renewed effort seeking the development of new antimicrobial agents effective against pathogenic microorganisms resistant to currently available treatments.
Antibacterial and antifungal activities of the azoles are the most widely studied and some of them are in clinical practice as antimicrobial agents. However, the azole-resistant strains led to the development of a new antimicrobial compounds. In particular pyrazole derivatives are extensively studied and used as antimicrobial agents. Pyrazole is an important class of heterocyclic compounds and many pyrazole derivatives are reported to have a broad spectrum of biological activities, such as anti-inflammatory, antifungal (Prakash et al., 2008), herbicidal (Kudo et al., 1999), antitumour, cytotoxic, molecular modelling (Vera-DiVaio et al., 2009), and antiviral (Storer et al., 1999) activities. Pyrazole derivatives also acts as antiangiogenic agents (Qiao et al., 2004), A3 adenosine receptor antagonists (Baraldi et al., 2003), neuropeptide YY5 receptor antagonists (Stamford and Wu, 2004), kinase inhibitor for treatment of type 2 diabetes, hyperlipidemia, obesity (Brown et al., 2004), and thrombopiotinmimetics (Heerding, 2004).
Antipyrine (2,3-dimethyl-1-phenyl-3-pyrazolin-5-one) was the first pyrazolone derivative used in the management of pain and inflammation, and their derivatives have attracted the attention of several research groups due to their potential activities (Jain et al., 2003). In this context, broad spectra of bioactive antipyrine derivatives have been investigated and diversities of bioactivities such as analgesic (Filho et al., 1998), anti-inflammatory (Ismail et al., 2007), antimicrobial (Mishra, 1999), and anticancer activity (Sondhi et al., 2001) have been reported. The antibacterial activity caught our attention because antimicrobial resistance developed by important pathogens has increased in the last decade (Sutcliffe, 2003). Besides, emerging and re-emerging bacterial infectious diseases still cause death and disability worldwide (Morens et al., 2004).
Benzimidazoles are remarkably effective compounds both with respect to their inhibitory activity and their favorable selectivity ratio. Extensive biochemical and pharmacological studies have confirmed that benzimidazole molecules are effective against various strains of microorganisms (Kazimierczuk et al., 2002). Benzimidazoles are regarded as a promising class of bioactive heterocyclic compounds that exhibit a range of biological activities. Specifically, this nucleus is a constituent of vitamin-B12 (Óniel et al., 2001). This ring system is present in numerous antioxidant (Ayhan-Kilcigil et al., 2007), antiparasitic (Navarrete-Vazquez et al., 2001), antihelmintics (Ravina et al., 1993), antiproliferative (Garuti et al., 2000), and anti-HIV (Rao et al., 2002) activities.
Thiazolidin-4-ones are an important group of heterocyclic compounds, having valuable biological activities in the areas of medicine. Recently, antimicrobial and antimycobacterial activities (de Aquino et al., 2008; Verma and Saraf, 2008; Küçükgüzel et al., 2006) of this framework containing compounds were explored well whereas their 2,3-disubstituted analogues have proved to be predominantly effective non-nucleoside HIV reverse transcriptase inhibitors (Barreca et al., 2001). Likewise, thiazole and their 2-substituted derivatives were also reported to exhibit diverse biological properties such as antituberculous and antimicrobial activities (Karegoudar et al., 2008). Moreover, it has been found in the drug development program for the treatment of inflammation (Suryavanshi and Pai, 2006) and HIV (Balzarini et al., 2009).
In view of the above-mentioned findings and as a continuation of our efforts (Salimon and Salih, 2010) to identify new candidates that may be of value in designing new, potent, selective and less toxic antimicrobial agents, we report herein the synthesis of some new heterocyclic derivatives starting from anthrone in order to investigate their antimicrobial activity (Fig. 1).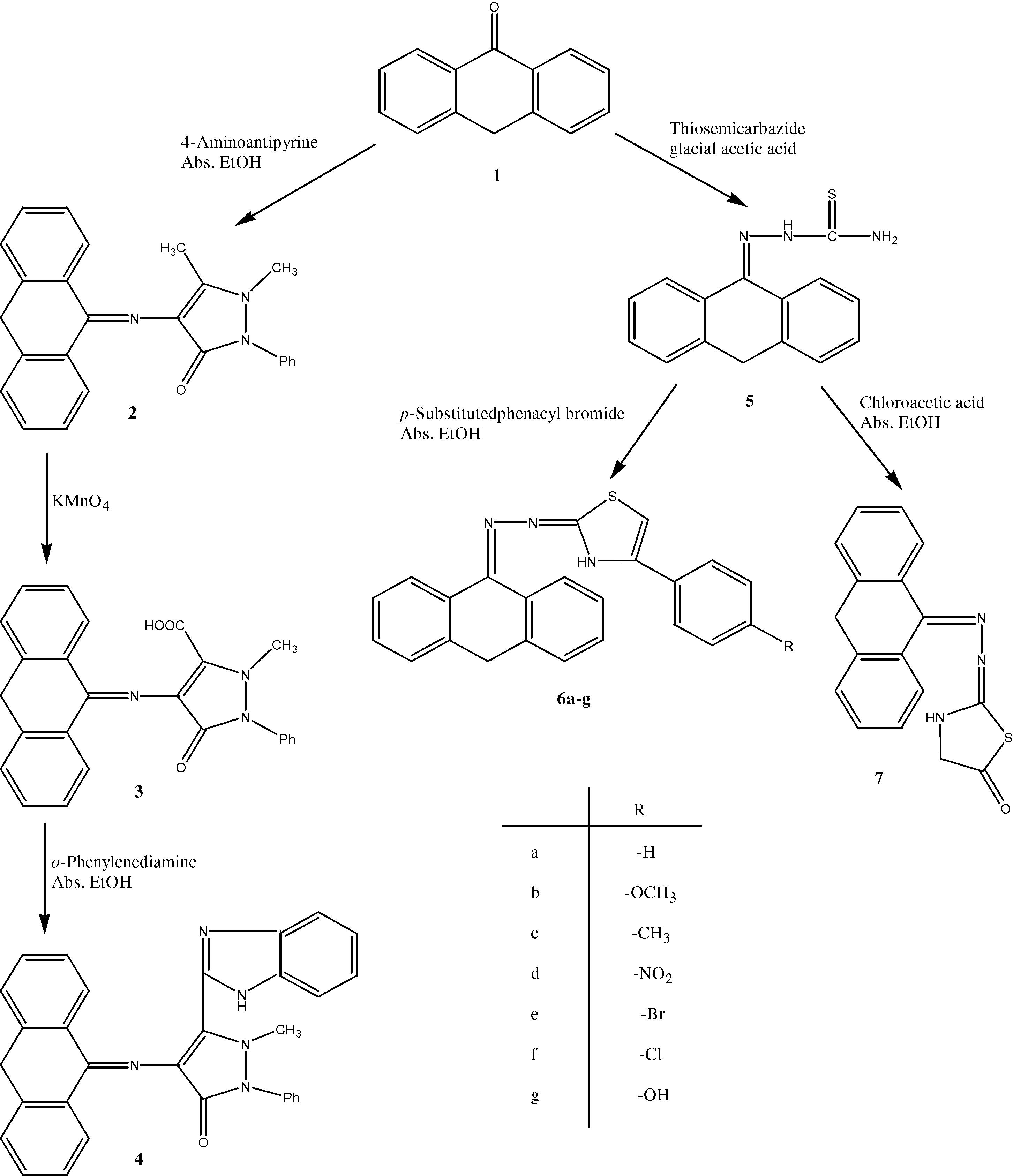
Synthetic protocol to synthesis compounds (2–7).
2 Experimental
2.1 Measurements
Melting points were determined in open glass capillaries on a Gallenkamp apparatus and are uncorrected. The percentage compositions of the elements (CHNS) for the compounds were determined using an elemental analyzer CHNS Model Fison EA 1108. The infrared spectra were recorded as potassium bromide discs using a Perkin-Elmer spectrophotometer GX. The 1H and 13C nuclear magnetic resonance spectra were recorded using the JEOL JNM-ECP 400 spectrometer in DMSO-d6 as the solvent, using TMS as an internal standard, and chemical shifts are expressed as δppm. All the reactions were followed by TLC (Silica gel, aluminum sheers 60 F254, Merck).
2.2 Synthesis of of 4-(anthracen-9(10H)-ylideneamino)-1,5-dimethyl-2-phenyl-1,2-dihydro-3H-pyrazol-3-one (2)
A mixture of anthrone 1 (10.5 g, 0.012 mol), 30 mL glacial acetic acid and 4-aminoantipyrine (8.76 g, 0.012 mol) was heated under reflux for 10 h. The reaction mixture was filtered off and recrystalized from ethanol (5.43 g, 55%); mp 67–69 °C; IR (KBr) cm−1 3089 (C–H aromatic), 2954, 2824 (C–H aliphatic), 1678 (C⚌O), 1621 (C⚌N). 1H NMR (400 MHz-DMSO-d6-ppm) δ 1.67 (s, 3H, CH3), 2.05 (s, 2H, CH2), 8.11–8.17 (d, 1H, Ar–H), 7.94–8.03 (d, 1H, Ar–H), 7.55–7.64 (d, 1H, Ar–H), 7.51–7.65 (d, 1H, Ar–H), 7.24–7.33 (d, 1H, Ar–H), 7.26–7.33 (d, 1H, Ar–H), 7.23–7.28 (t, 1H, Ar–H), 7.17–7.20 (t, 1H, Ar–H), 7.06–7.13 (t, 1H, Ar–H), 6.90–6.96 (t, 1H, Ar–H), 6.82–6.88 (t, 1H, Ar–H), 6.71–6.75 (t, 1H, Ar–H), 6.66–6.69 (t, 1H, Ar–H).13C NMR (400 MHz-DMSO-d6-ppm) δ 13.05, 13.11 (2C, 2 CH3), 14.51 (1C, CH2), 61.53 (1C, C⚌N), 131.24–135.98 (18C, aromatic carbons), 166.70 (C, C⚌O). Anal. Found (calc.) for C30H37N3O (%): C, 79.09 (79.08); H, 8.20 (8.19); N, 9.23 (9.22).
2.3 Synthesis of 4-(anthracen-9(10H)-ylideneamino)-1-methyl-3-oxo-2-phenyl-2,3-dihydro-1H pyrazole-5-carboxylic acid (3)
Compound 2 (20 g, 0.04 mol) is added to a solution of (6.32 g, 0.04 mol) of potassium permanganate and (3.32 g, 0.04 mol) sodium carbonate in (85 mL) water and the mixture is heated under reflux until the color of the permanganate has disappeared (15 h). The reaction mixture was filtered while still hot to get rid of the MnO2 precipitate. The cooled filtrate is acidified with sulphuric acid (20%), the carboxylic acid precipitate is filtered off, washed with a little cold water and crystallized from ethanol to give compound 3 as colorless crystals (18 g, 58.4%); mp 193–195 °C; IR (KBr) cm−1 3425 (O–H), 3067 (C–H aromatic), 2987, 2865 (C–H aliphatic), 1718 and 1682 (C⚌O), 1623 (C⚌N). 1H NMR (400 MHz-DMSO-d6-ppm) δ 1.69 (s, 3H, CH3), 2.04 (s, 2H, CH2), 8.12–8.18 (d, 1H, Ar–H), 7.95–8.02 (d, 1H, Ar–H), 7.54–7.65 (d, 1H, Ar–H), 7.52–7.64 (d, 1H, Ar–H), 7.25–7.32 (d, 1H, Ar–H), 7.25–7.32 (d, 1H, Ar–H), 7.24–7.27 (t, 1H, Ar–H), 7.16–7.21 (t, 1H, Ar–H), 7.05–7.12 (t, 1H, Ar–H), 6.91–6.95 (t, 1H, Ar–H), 6.83–6.89 (t, 1H, Ar–H), 6.72–6.76 (t, 1H, Ar–H), 6.65–6.68 (t, 1H, Ar–H), 11.69 (br s, 1H, O–H, D2O exchangeable). 13C NMR (400 MHz-DMSO-d6-ppm) δ 13.04 (1C, CH3), 14.50 (1C, CH2), 61.54 (1C, C⚌N), 131.23–135.97 (18C, aromatic carbons), 164.21, 171.30 (2C, 2 C⚌O). Anal. Found (calc.) for C30H35N3O3 (%): C, 74.23 (74.20); H, 7.25 (7.26); N, 8.63 (8.65).
2.4 Synthesis of 4-(anthracen-9(10H)-ylideneamino)-5-(1H-benzimidazol-2-yl)-1-methyl-2-phenyl-1,2-dihydro-3H-pyrazol-3-one (4)
To (10.8 g, 0.02 mol) of compound 3, a mixture of (2.16 g, 0.02 mol) o-phenylenediamine and a few drops of conc. hydrochloric acid in (100 mL) abs. ethanol was added. Then the mixture was heated under reflux for 24 h, the completion of the reaction was monitored by TLC. Cooled to room temperature and to the reaction mass was added 100 mL of water, stirred for 2 h, and the solid obtained was filtered and washed with water. Crystallization from ethanol gave compound 4 as colorless crystals (8.5 g, 83%); mp 120–122 °C; IR (KBr) cm−1 3356 (N–H), 3080 (C–H aromatic), 2924, 2841 (C–H aliphatic), 1683 (C⚌O), 1628 (C⚌N). 1H NMR (400 MHz-DMSO-d6-ppm) δ 1.63 (s, 3H, CH3), 2.08 (s, 2H, CH2), 8.15–8.21 (d, 1H, Ar–H), 7.92–7.97 (d, 1H, Ar–H), 7.83–7.89 (d, 1H, Ar–H), 7.73–7.78 (d, 1H, Ar–H), 7.66–7.70 (d, 1H, Ar–H), 7.54–7.61 (m, 12H, Ar–H), 8.69 (s, 1H, N–H, D2O exchangeable). 13C NMR (400 MHz-DMSO-d6-ppm) δ 12.89 (1C, CH3), 13.34 (1C, CH2), 60.47 (1C, C⚌N), 132.15–140.10 (14C, aromatic carbons), 169.23 (1C, C⚌O). Anal. Found (calc.) for C39H49N5O (%): C, 77.58 (77.57); H, 8.19 (8.18); N, 11.58 (11.60).
2.5 Synthesis of anthracen-9(10H)-one thiosemicarbazone (5)
Thiosemicarbazide (4.6 g, 0.05 mol) was added to (13 g, 0.05 mol) of anthrone 1 dissolved in glacial acetic acid (100 mL), the reaction mixture was refluxed for 10 h and the completion of the reaction was monitored by TLC. Cooled to room temperature and the reaction mass poured into 250 mL ice water, the solid obtained was filtered, washed with water, and recrystallized from methanol yielding 5 (11.3 g, 81.2%); mp 210–212 °C; IR (KBr) cm−1 3445 and 3350 (NH2), 3063 (C–H aromatic), 1620 (C⚌N). 1H NMR (400 MHz-DMSO-d6-ppm) δ 1.89 (s, 2H, CH2), 8.10–8.13 (d, 1H, Ar–H), 7.98–8.04 (d, 1H, Ar–H), 7.85–7.89 (d, 1H, Ar–H), 7.72–7.75 (d, 1H, Ar–H), 7.65–7.69 (t, 1H, Ar–H), 7.55–7.60 (t, 1H, Ar–H), 7.46–7.50 (t, 1H, Ar–H), 7.38–7.42 (t, 1H, Ar–H), 9.38 (s, 2H, NH2, D2O exchangeable). 13C NMR (400 MHz-DMSO-d6-ppm) δ 11.05 (1C, CH2), 62.17 (1C, C⚌N), 130.41–136.25 (12C, aromatic carbons). Anal. Found (calc.) for C19H25N3S (%): C, 69.67 (69.68); H, 7.65 (7.69); N, 12.80 (12.83); S, 9.80 (9.79).
2.6 General synthesis procedure for compound (6a–g)
A mixture of compound 5 (3 g, 0.007 mol) and different p-substituted phenacyl bromide (1.39 g, 0.007 mol) in (50 mL) abs. ethanol was refluxed for 24 h, the completion of the reaction was monitored by TLC. It was then cooled to room temperature and poured into ice-cold water, stirring for 30 min. The solid was filtered, washed with water, and then crystallized from ethanol
2.6.1 Synthesis of 2-(anthracen-9(10H)-ylidenehydrazono)-5-phenyl-2,3-dihydro-1H-thiazole (6a)
(2.5 g, 81.2%); mp 220–222 °C; IR (KBr) cm−1 3273 (N–H), 3075 (C–H aromatic), 1626 (C⚌N). 1H NMR (400 MHz-DMSO-d6-ppm) δ 1.56 (s, 2H, CH2), 8.13–8.16 (d, 1H, Ar–H), 8.05–8.09 (d, 1H, Ar–H), 7.90–7.95 (d, 1H, Ar–H), 7.83–7.86 (d, 1H, Ar–H), 7.75–7.79 (t, 1H, Ar–H), 7.66–7.71 (t, 1H, Ar–H), 7.59–7.62 (t, 1H, Ar–H), 7.52–7.55 (t, 1H, Ar–H), 7.43–7.49 (m, 5H, Ar–H), 8.18 (s, 1H, proton of thiazole ring), 9.21 (s, 1H, N–H, D2O exchangeable). 13C NMR (400 MHz-DMSO-d6-ppm) δ 10.99 (1C, CH2), 60.78, 61.63 (2C, 2C⚌N), 130.98–137.09 (18C, aromatic carbons). Anal. Found (calc.) for C28H33N3S (%): C, 75.79 (75.80); H, 7.52 (7.50); N, 9.45 (9.47); S, 7.24 (7.23).
2.6.2 Synthesis of 2-(anthracen-9(10H)-ylidenehydrazono)-5-methoxyphenyl-2,3-dihydro-1H-thiazole (6b)
(0.75 g, 55%); mp 249–250 °C; IR (KBr) cm−1 3272 (N–H), 3072 (C–H aromatic), 1625 (C⚌N). 1H NMR (400 MHz-DMSO-d6-ppm) δ 1.60 (s, 2H, CH2), 1.89 (s, 3H, OCH3), 8.08–8.11 (d, 1H, Ar–H), 7.97–8.03 (d, 1H, Ar–H), 7.88–7.91 (d, 1H, Ar–H), 7.80–7.83 (d, 1H, Ar–H), 7.71–7.74 (t, 1H, Ar–H), 7.57–7.60 (t, 1H, Ar–H), 7.50–7.53 (t, 1H, Ar–H), 7.43–7.46 (t, 1H, Ar–H), 7.35–7.38 (m, 5H, Ar–H), 8.17 (s, 1H, proton of thiazole ring), 9.20 (s, 1H, NH, D2O exchangeable). 13C NMR (400 MHz-DMSO-d6-ppm) δ 11.08 (1C, CH2), 14.69 (1C, OCH3), 61.57, 63.24 (2C, 2C⚌N), 129.51138.79 (18C, aromatic carbons). Anal. Found (calc.) for C29H35N3OS (%): C, 73.52 (73.53); H, 7.44 (7.45); N, 8.86 (8.87); S, 6.76 (6.77).
2.6.3 Synthesis of 2-(anthracen-9(10H)-ylidenehydrazono)-5-methylpheny-2,3-dihydro-1H-thiazole (6c)
(0.8 g, 64%); mp 245–248 °C; IR (KBr) cm−1 3275 (N–H), 3078 (C–H aromatic), 1626 (C⚌N).1H NMR (400 MHz-DMSO-d6-ppm) δ 1.23 (s, 3H, CH3), 1.57 (s, 2H, CH2), 8.10–8.13 (d, 1H, Ar–H), 8.03–8.06 (d, 1H, Ar–H), 7.92–7.95 (d, 1H, Ar–H), 7.83–7.86 (d, 1H, Ar–H), 7.76–7.80 (t, 1H, Ar–H), 7.69–7.73 (t, 1H, Ar–H), 7.60–7.64 (t, 1H, Ar–H), 7.52–7.55 (t, 1H, Ar–H), 7.44–7.47 (m, 5H, Ar–H), 8.16 (s, 1H, proton of thiazole ring), 9.22 (s, 1H, N–H, D2O exchangeable). 13C NMR (400 MHz-DMSO-d6-ppm) δ 8.07 (1C, CH3), 12.15 (1C, CH2), 62.39, 63.70 (2C, 2C⚌N), 131.34–140.27 (18C, aromatic carbons). Anal. Found (calc.) for C29H35N3S (%): C, 76.11 (76.10); H, 7.70 (7.71); N, 9.19 (9.18); S, 7.03 (7.01).
2.6.4 Synthesis of 2-(anthracen-9(10H)-ylidenehydrazono)-5-nitropheny-2,3-dihydro-1H-thiazole (6d)
(0.78 g, 52%); mp 240–242 °C; IR (KBr) cm−1 3236 (N–H), 3075 (C–H aromatic), 1624 (C⚌N), 1545 and 1316 (NO2). 1H NMR (400 MHz-DMSO-d6-ppm) δ 1.55 (s, 2H, CH2), 8.01–8.05 (d, 1H, Ar–H), 7.93–7.96 (d, 1H, Ar–H), 7.84–7.86 (d, 1H, Ar–H), 7.77–7.81 (d, 1H, Ar–H), 7.70–7.74 (t, 1H, Ar–H), 7.64–7.66 (t, 1H, Ar–H), 7.58–7.61 (t, 1H, Ar–H), 7.50–7.53 (t, 1H, Ar–H), 7.46–7.49 (m, 5H, Ar–H), 8.14 (s, 1H, proton of thiazole ring), 9.25 (s, 1H, N–H, D2O exchangeable). 13C NMR (400 MHz-DMSO-d6-ppm) δ 11.68 (1C, CH2), 61.30, 63.57 (2C, 2C⚌N), 130.67–139.13 (18C, aromatic carbons). Anal. Found (calc.) for C28H32N4O2S (%): C, 68.81 (68.82); H, 6.59 (6.60); N, 11.46 (11.47); S, 6.57 (6.56).
2.6.5 Synthesis of 2-(anthracen-9(10H)-ylidenehydrazono)-5-bromopheny-2,3-dihydro-1H-thiazole (6e)
(0.65 g, 50%); mp 255–257 °C; IR (KBr) cm−1 3274 (N–H), 3078 (C–H aromatic), 1628 (C⚌N). 1H NMR (400 MHz-DMSO-d6-ppm) δ 1.57 (s, 2H, CH2), 8.11–8.15 (d, 1H, Ar–H), 7.90–7.93 (d, 1H, Ar–H), 7.81–7.84 (d, 1H, Ar–H), 7.72–7.75 (d, 1H, Ar–H), 7.65–7.68 (t, 1H, Ar–H), 7.60–7.63 (t, 1H, Ar–H), 7.53–7.56 (t, 1H, Ar–H), 7.43–7.45 (t, 1H, Ar–H), 7.37–7.40 (m, 5H, Ar–H), 8.17 (s, 1H, proton of thiazole ring), 9.15 (s, 1H, N–H, D2O exchangeable). 13C NMR (400 MHz-DMSO-d6-ppm) δ 10.90 (1C, CH2), 60.57, 61.89 (2C, 2C⚌N), 128.93–130.88 (18C, aromatic carbons). Anal. Found (calc.) for C28H32BrN3S (%): C, 64.37 (64.36); H, 6.16 (6.17); N, 8.05 (8.04); S, 6.15 (6.14).
2.6.6 Synthesis of 2-(anthracen-9(10H)-ylidenehydrazono)-5-chloropheny-2,3-dihydro-1H-thiazole (6f)
(0.80 g, 54%); mp 267–269 °C; IR (KBr) cm−1 3270 (N–H), 3080(C–H aromatic), 1630 (C⚌N). 1H NMR (400 MHz-DMSO-d6-ppm) δ 1.61 (s, 2H, CH2), 8.08–8.11 (d, 1H, Ar–H), 7.99–8.02 (d, 1H, Ar–H), 7.90–7.93 (d, 1H, Ar–H), 7.85–7.87 (d, 1H, Ar–H), 7.78–7.81 (t, 1H, Ar–H), 7.71–7.73 (t, 1H, Ar–H), 7.65–7.68 (t, 1H, Ar–H), 7.58–7.61 (t, 1H, Ar–H), 7.52–7.55 (m, 5H, Ar–H), 8.15 (s, 1H, proton of thiazole ring), 9.11 (s, 1H, N–H, D2O exchangeable). 13C NMR (400 MHz-DMSO-d6-ppm) δ 12.01 (1C, CH2), 60.76, 61.43 (2C, 2C⚌N), 129.20–131.54 (18C, aromatic carbons). Anal. Found (calc.) for C28H32ClN3S (%): C, 70.33 (70.34); H, 6.76 (6.75); N, 8.80 (8.79); S, 6.72 (6.71).
2.6.7 Synthesis of 2-(anthracen-9(10H)-ylidenehydrazono)-5-hydroxypheny-2,3-dihydro-1H-thiazole (6g)
(0.78 g, 52.7%); mp 280–282 °C; IR (KBr) cm−1 3272 (N–H), 3079 (C–H aromatic), 1627 (C⚌N). 1H NMR (400 MHz-DMSO-d6-ppm) δ 1.60 (s, 2H, CH2), 8.17–8.15 (d, 1H, Ar–H), 8.04–8.07 (d, 1H, Ar–H), 7.94–7.97 (d, 1H, Ar–H), 7.87–7.90 (d, 1H, Ar–H), 7.80–7.83 (t, 1H, Ar–H), 7.73–7.75 (t, 1H, Ar–H), 7.66–7.69 (t, 1H, Ar–H), 7.59–7.62 (t, 1H, Ar–H), 7.52–7.56 (m, 5H, Ar–H), 8.19 (s, 1H, proton of thiazole ring), 9.20 (s, 1H, N–H, D2O exchangeable), 12.31 (s, 1H, O–H, D2O exchangeable). 13C NMR (400 MHz-DMSO-d6-ppm) δ 11.13 (1C, CH2), 60.98, 61.36 (2C, 2C⚌N), 129.48–131.05 (18C, aromatic carbons). Anal. Found (calc.) for C28H33N3OS (%): C, 73.18 (73.16); H, 7.23 (7.24); N, 9.15 (9.14); S, 6.97 (6.98).
2.7 Synthesis of 2-(anthracen-9(10H)-ylidenehydrazono)thiazolidin-4-one (7)
To a mixture of compound 5 (5 g, 0.015 mol) and potassium hydroxide (0.84 g, 0.015 mol) in (10 mL) abs. ethanol, chloroacetic acid (1.42 g, 0.015 mol) was added gradually. The reaction mixture was refluxed for 24 h, the completion of the reaction was monitored by TLC. The cooled solution was diluted with 100 mL of ice-cold water and the precipitate obtained was filtered to yield 2.5 g of the crude product. Crystallization from ethanol gave compound 7 as colorless crystals (2.1 g, 44.6%); mp 270–273 °C dec; IR (KBr) cm−1 3321 (N–H), 3090 (C–H aromatic), 1657 (C⚌O), 1625 (C⚌N). 1H NMR (400 MHz-DMSO-d6-ppm) δ 1.67 (s, 2H, protons of anthracene ring), 2.04 (s, 2H, protons of thiazolidinone ring), 8.00–8.03 (d, 1H, Ar–H), 7.94–7.96 (d, 1H, Ar–H), 7.88–7.91 (d, 1H, Ar–H), 7.80–7.83 (d, 1H, Ar–H), 7.75–7.77 (t, 1H, Ar–H), 7.69–7.71 (t, 1H, Ar–H), 7.62–7.65 (t, 1H, Ar–H), 7.54–7.57 (t, 1H, Ar–H), 8.89 (s, 1H, N–H, D2O exchangeable). 13C NMR (400 MHz-DMSO-d6-ppm) δ 10.79 (1C, CH2 of anthracene ring), 14.21 (1C, CH2 of thiazolidinone ring), 61.88 (1C, C⚌N), 63.46 (1C, C⚌N), 132.04–139.15 (12C, aromatic carbons), 170.78 (1C, C⚌O). Anal. Found (calc.) for C22H29N3OS (%): C, 68.88 (68.89); H, 7.63 (7.62); N, 10.95 (10.96); S, 8.37 (8.36).
3 Results and discussion
3.1 Chemistry and characterization
The synthetic procedure adopted to obtain the target compounds is depicted in Fig. 1. The starting compound anthrone 1 was obtained from Aldrich Company and used directly without further purification. Thus, refluxing of compound 1 with 4-aminoantipyrine in boiling glacial acetic acid afforded the Schiff base derivative 2. The oxidation of 2 with KMnO4 was studied with the aim of formation of benzimidazole derivatives. Thus, it was oxidized using KMnO4 to give compound 3, and then cyclocondensation of compound 3 with o-phenelynediamine in boiling ethanol containing a catalytic amount of HCl gave the benzimidazole derivative 4. Structures of these products are based on analytical and spectral data. The IR spectrum of compound 2 showed four characteristic absorption bands at 2987, 2865, 1685 and 1624 cm−1 due to methyl, C⚌O and C⚌N groups. While the characteristic IR absorption bands of compound 3 appeared at 1718, 1682 and 1623 cm−1 due to C⚌O and C⚌N groups. On the other hand, IR spectrum of compound 4 showed bands at 3356 and 1683 cm−1 due to N–H and C⚌O absorption bands. The 1H NMR spectrum of compound 2 revealed the appearance of two singlets at δ 1.56 and 1.67 ppm assigned to two methyl groups. The spectrum of compound 3 revealed one D2O-exchangable singlet at δ 11.69 ppm assigned to one O–H proton, in addition to one singlet at δ 1.69 ppm due to one methyl proton. Furthermore, the 1H NMR spectrum of compound 4 showed one D2O-exchangable singlet at δ 7.64 ppm due to one N–H proton. The 13C NMR spectra of compounds 2, 3 and 4 showed signals at 167.43, 164.21 and 171.30, 169.23, respectively, assigned to C⚌O groups.
Recently, we have reported the reaction of anthrone with l-histidine which represents a new, simple and efficient synthetic route for the synthesis of imidazole derivatives (Salimon and Salih, 2010). Therefore, it was interesting to study the reaction of 1 with thiosemicarbazide to yield compound 5, then cyclocondensation of 5 with p-substituted phenacyl bromide and chloroacetic acid in boiling ethanol furnished the thiazole derivatives, compounds 6a–g and 7, respectively. The analytical and spectral data are in agreement with the proposed structures. The IR spectrum of compound 5 showed two absorption bands at 3445 and 3350 cm−1 due to NH2 group beside one C⚌N absorption bands at 1620 cm−1. It's 1H NMR revealed one D2O-exchangable singlet at δ 7.56 ppm due to NH2 protons. Furthermore, its 13C NMR spectrum showed multiplet signals at 130.41–136.25 ppm assigned to aromatic protons. On the other hand, the IR spectra of compounds 6a–g showed characteristic absorption bands at about 3275, 3080 and 1625 cm−1 due to N–H, aromatic protons and C⚌N group. The 1H NMR spectrum showed one D2O-exchangable singlet at δ about 9.22 due to one thiazole N–H proton (Sarojini et al., 2010), in addition to two singlets at δ 1.57 assignable for methylene protons of the anthracene. 13C NMR revealed signals at δ 60.34 and 130.19–134.78 ppm due to C⚌N group and aromatic carbons. Elemental analysis, IR, 1H NMR and 13C NMR are in agreement with the proposed structures
Also, the structure of compound 7 was established on the basis of its elemental analysis and spectral data. Its IR spectrum displayed an absorption band at 1657 cm−1 due to C⚌O group. The 1H NMR spectrum showed one D2O-exchangable singlet at δ 8.89 ppm due to N–H proton; beside this a singlet signal appeared at δ 2.02 ppm corresponding to the thiazolidinone methylene group (Bondock et al., 2007). The C⚌O signal of the thiazolidinone ring appeared at δ 170.78 ppm in 13C NMR spectrum.
3.2 Biological activity
3.2.1 Antibacterial activity
The newly synthesized compounds were screened for their antibacterial activity against Escherichia coli (ATTC-25922), Staphylococcus aureus (ATTC-25923), Pseudomonas aeruginosa (ATTC-27853), and Bacillius subtilis (recultured) bacterial stains by the disk-diffusion method (Cruickshank et al., 1975; Collins, 1976). Disks measuring 6.25 mm in diameter were punched from Whatman no. 1 filter paper. Batches of 100 disks were dispensed to each screw-capped bottle and sterilized by dry heat at 140 °C for an hour. The test compounds were prepared with different concentrations using DMSO. One milliliter containing 100 times the amount of chemical in each disk was added to each bottle, which contained 100 discs. Disks of each concentration were placed in triplicate in nutrient agar medium seeded with fresh bacteria separately. The incubation was carried out at 37 °C for 24 h. Streptomycin was used as a standard drug at a concentration of 10 μg/mL. Solvent and growth controls were kept and the zones of inhibition were noted. The results of such studies are given in Table 1. Zone of inhibition in millimeters.
Compound
Escherichia coli (ATTC-25922)
Staphylococcus aureus (ATTC-25923)
Pseudomonas aeruginosa (ATTC-27853)
Bacillius subtilis (recultured)
1
–
–
–
–
2
12
12
13
11
3
14
13
17
15
4
26
26
22
23
5
15
18
8
–
6a
20
18
17
18
6b
–
–
13
13
6c
–
13
14
14
6d
28
25
24
26
6e
20
18
17
18
6f
19
19
20
21
6g
18
15
12
12
7
24
22
20
20
Standard (Streptomycine)
20
21
24
24
The above data showed that compound 2-(anthracen-9(10H)-ylidenehydrazono)-5-nitrophenyl-2,3-dihydro-1H-thiazole 6d was the most potent compound, exhibited very good activity against the four organisms. The compounds 4-(anthracen-9(10H)-ylideneamino)-5-(1H-benzimidazol-2-yl)-1-methyl-2-phenyl-1,2-dihydro-3H-pyrazol-3-one 4 and 2-(anthracen-9(10H)-ylidenehydrazono)thiazolidin-4-one 7 exhibited very good activity against E. coli and S. aureus. The remaining compounds were found to have a slight or moderate activity against the tested organisms and some of the compounds were found to be inactive (indicated – sign).
3.2.2 Antifungal activity
Newly prepared compounds were screened for their antifungal activity against Aspergillus flavus (NICM No. 524), Aspergillus fumigatus (NCIM No. 902), Candida albicans (NCIM No. 300), Penicillium marneffei (recultured), and Trichophyton mentagrophytes (recultured) in DMSO by the serial plate dilution method (Khan, 1997; Varma, 1998). Sabouraud's agar media were prepared by dissolving peptone (1 g), d-glucose (4 g), and agar (2 g) in distilled water (100 mL) and adjusting the pH to 5.7. Normal saline was used to make a suspension of the spore of fungal strain for lawning. A loopful of a particular fungal strain was transferred to 3 mL saline to get a suspension of the corresponding species. Agar media (20 mL) were poured into each petri dish. Excess suspension was decanted and the plates were dried by placing in an incubator at 37 °C for 1 h. Using agar punch, holes were made into each agar and labeled. A control was also prepared in triplicate and maintained at 37 °C for 3–4 days. Antifungal activity was determined by measuring the diameter of the inhibition zone. Activity of each compound was compared with that of flucanazole as the standard.
The antifungal data showed that the newly prepared compounds have moderate to good activity against the above-mentioned organisms. The compound 4-(anthracen-9(10H)-ylideneamino)-1,5-dimethyl-2-phenyl-1,2-dihydro-3H-pyrazol-3-one 2 exhibited very good activity against A. flavus, A. fumigatus, and T. Mentagrophytes. Compound 2-(anthracen-9(10H)-ylidenehydrazono)-5-nitrophenyl-2,3-dihydro-1H-thiazole 6d showed very good activity toward A. flavus and A. fumigatus. The remaining compounds were found to have slight or moderate activity against the tested organisms and some compounds were found to be inactive (indicated – sign) (Table 2). Zone of inhibition in millimeters.
Compound
Aspergillus flavus (NICM No. 524)
Aspergillus fumigatus (NCIM No. 902)
Penicillium marneffei (recultured)
Candida Albicans (NCIM No. 300)
Trichophyton Mentagrophytes (recultured)
1
–
–
10
8
–
2
25
22
19
18
24
3
15
16
21
17
16
4
20
15
18
14
16
5
–
17
–
–
–
6a
19
21
14
16
18
6b
20
18
17
16
19
6c
16
18
15
18
19
6d
25
24
20
17
19
6e
12
10
13
18
–
6f
15
18
23
16
17
6g
16
12
15
20
14
7
10
12
21
17
16
Standard (Flucanazole)
21
18
21
20
19
4 Conclusion
The main aim of the present study is to synthesize and investigate the antimicrobial activity of new heterocyclic derivatives containing pyrazole, benzimidazole, thiazole and thiazolidin-4-one moieties with the hope of discovering new structures serving as potential broad spectrum antimicrobial agents. The antibacterial and antifungal data revealed that the compounds 2–7 showed good to moderate antimicrobial activity. Basically introduction of pyrazole moiety in the structure of compound 2 has increased the antifungal activity compared to the other. On the other hand, the presence of a pyrazole ring together with a benzimidazole ring in compound 4 has a positive influence on antibacterial activity. Further, incorporation of –NO2 group in the phenyl ring, compound 6d, increased lipophilicity as well as a remarkably increased the antibacterial and antifungal activity.
Acknowledgments
The authors acknowledge the Universiti Kebangsaan Malaysia for funding (Codes “UKM-GUP-NBT-08-27-113” and “UKM-OUP-NBT-29-150/2010”) and the direct contributions of the support staff from the School of Chemical Sciences and Food Technology and the faculty of the Science and Technology department at the Universiti Kebangsaan Malaysia. Special thanks granted to SRF, IIE.
References
- Eur. J. Med. Chem.. 2005;40:401-405. References are cited therein
- Arch. Pharm.. 2007;34:607-611.
- Eur. J. Med. Chem.. 2009;44:303-311.
- Bioorg. Med. Chem.. 2003;11:4161-4169.
- Bioorg. Med. Chem. Lett.. 2001;11:1793-1796.
- Eur. J. Med. Chem.. 2007;42:948-954.
- Brown, M.L., Cheung, M., Dickerson, S.H., Drewy, D.H., Lackey, K.E., Peat, A.J., Thomson, S.A., Veal, J.M., Wilson, J.L., 2004. PCT Int. Appl. WO 9596 (Chem. Abstr. 140, 128436y).
- Microbiological Methods (2nd ed.). London: Butterworth; 1976.
- Cruickshank, R., Duguid, J.P., Marion, B.P., Swain, R.H., 1975. In Medicinal Microbiology, 12th ed., vol. II, Churchil Livingstone, London, pp. 196–202.
- Bioorg. Med. Chem.. 2008;16:446-456.
- Il Farmaco. 1998;53:55-57.
- Bioorg. Med. Chem. Lett.. 2000;10:2193-2195.
- Heerding, D.A., 2004. PCT Int. Appl. WO 03 103686 (Chem. Abstr. 140, 42170v).
- Arch. Pharm. Life Sci.. 2007;340:476-482.
- Synth. Commun.. 2003;33:563-577.
- Eur. J. Med. Chem.. 2008;43:261-267.
- Acta Biochim. Pol.. 2002;49:185-195.
- Khan, Z.K., 1997. In vitro and vivo screening techniques for bioactivity screening and evaluation, Proceeding Int. Workshop UNIDO-CDRI, pp. 210–211.
- Eur. J. Med. Chem.. 2006;41:353-359.
- Chem. Pharm. Bull.. 1999;47:857-868.
- J. Indian Chem. Soc.. 1999;76:35-39.
- Nature. 2004;430:242-249.
- Bioorg. Med. Chem.. 2001;11:187-190.
- Óniel, M.J., Smith, M., Heckelman, P.E., 2001. The Merck Index, 13th ed. Merck & Co. Inc., New Jersey, P-1785, Monograph Number, p. 10074.
- Eur. J. Med. Chem.. 2008;43:435-440.
- Qiao, J.X., Pinto, D.J., Orwat, M.J., Han, W., Friedrich, S.R., 2004. PCT Int. Appl. WO 03 99, 276 (Chem. Abstr. 140, 16722g).
- Arzneim. Forsch.. 1993;43:684-694.
- Il Farmaco. 2002;57:819-823.
- Int. J. Pharm. Tech. Res.. 2010;2:205-208.
- Sarojini, B.K., Krishna, B.G., DarshanRaj, C.G., Bharath, B.R., Manjunath, H., 2010. Eur. J. Med. Chem., doi: 10.1016/j.ejmech.2010.03.039.
- Indian J. Chem. B. 2001;40:113-119.
- Stamford, A.W., Wu, Y., 2004. PCT Int. Appl. WO 5262 (Chem. Abstr. 140, 11141p).
- Nucleosides Nucleotides Nucleic Acids. 1999;18:203-216.
- Indian J Chem.. 2006;45B:1227-1230.
- Bioorg. Med. Chem. Lett.. 2003;13:4159-4161.
- Turan-Zitouni, G., Kaplancikli, Z.A., Yildiz, M.T., Chevallet, P., Kaya, D., 2005. Eur. J. Med. Chem. 40, 607–613. References are cited therein.
- Varma, R.S., 1998. Antifungal Agents: Past, Present and Future Prospects, National Academy of Chemistry and Biology, India, Lucknow.
- Bioorg. Med. Chem.. 2009;17:295-302.
- Eur. J. Med. Chem.. 2008;43:897-905.




