

Translate this page into:
Synthesis and anti-tubercular evaluation of some novel (E)-5-(4-(benzylidene amino) phenyl)-1,3,4-oxadiazole-2-thiol derivatives
⁎Corresponding authors at: Department of Mathematics, College of Applied Sciences, AlMaarefa University, Ad Diriyah 13713, Saudi Arabia (F. Yasmin); Department of Pharmacy Practice, College of Pharmacy, AlMaarefa University, Dariyah, 13713 Riyadh, Saudi Arabia (S.M.B. Asdaq); Department of Pharmaceutical Chemistry, Krupanidhi College of Pharmacy, Bengaluru 560 035, India (P. Das). faryasmin79@gmail.com (Farhana Yasmin)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Background
Tuberculosis remains a ninth global health cause affecting millions of people. The susceptibility and resistance caused by first and second-line drugs have not changed for decades. There is a need to develop novel drugs with better pharmacological profiles.
Methods
In this study, a series of (E)-5-(4-(benzylidene amino) phenyl)-1,3,4-oxadiazole-2-thiol derivatives were synthesized, docked, and ADMET studies were performed. Based on binding affinity, the compounds were evaluated for their ability to inhibit the M. tuberculosis H37Rv strain.
Results
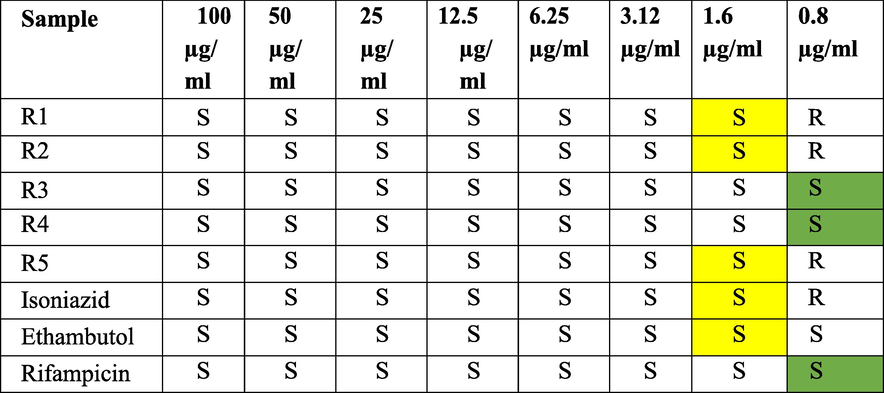
The compounds showed binding energy between −8.2 and −10.0 Kcal/mol. Molecular simulations benefited the representation of the actual biological conditions with a significant outcome. The compound 5-(4-{(E)-[(2-nitrophenyl) methylidene] amino} phenyl)-1,3,4-oxadiazole-2-thiol (R4) showed the best binding −10.0 Kcal/mol, MIC of 0.8 µg/ml, the IC50 value of 49.01 and the Selectivity Index of 61.33. The synthesized compounds were evaluated for anti-mycobacterial activity against M. tuberculosis (H37Rv) using MABA assay and compared with the standards; R3 and R4 were sensitive at 0.8 µg/ml.
Conclusion
Among the designed compounds 5-(4-{(E)-[(2-nitrophenyl)methylidene]amino}phenyl)-1,3,4-oxadiazole-2-thiol showed the best activity with higher IC50 values. As a result, molecular hit can be good lead for further development for tuberculosis treatment.
Keywords
1,3,4-Oxadiazole
Docking
Simulation
M. tuberculosis
ADMET studies

1 Introduction
About 10.6 million people suffer from tuberculosis (TB), and 1.6 million died in 2021. According to WHO, 21.4 lakhs of cases were reported in India in 2021 (Bagcchi, 2023). TB is associated with HIV in 11% of people (Galemore, 2016). Mycobacterium transmits by direct ingestion of tiny aerosol droplets containing M. tuberculosis. The causative agent Mycobacterium bovis was primarily found in soil and affected cattle; later, this species infected humans and evolved into Mycobacterium tuberculosis. The first-line treatment leads to recovery in about 90% of patients but long-term causes multi-drug resistance to Mycobacterium. Patientsmust take second and third-line anti-TB drugs (Kavitha et al., 2014). During long-term chemotherapy, first-line drugs are denied. As a result, only 50% of patients with MDRTB have been successfully treated with the drugs available. The global number of official TB-related deaths (1.3 million) was almost double that of HIV/AIDS (0.68 million), and TB mortality was impacted by the Covid-19 pandemic more than HIV /AIDS. One of the key contributors to TB deaths and poor treatment outcomes is drug resistance, and this has become a global challenge. The challenge occurs in the treatment of various forms of multi-drug resistance (MDR-TB), total drug resistance (TDR-TB), and Extensively-drug resistance (XDR-TB). About 470,000 people fall ill, and about 180,000 die annually due to MDR-TB (https://www.who.int/teams/global-tuberculosis-programme/tb-reports).
Recently, azo-based heterocycles have been broadly studied that are responsible for a wide range of pharmacological activities. Oxadiazoles are five-membered aromatic heterocyclic and have the potential as good bio-isosteric replacements for carbonyl functional groups. Molecules having oxadiazole in their backbone have demonstrated an array of pharmacological activities, including widely studied for various pharmacological activities (Karol et al., 2020) comprising anti-inflammatory (Gobec et al., 2015), analgesic (Leite et al., 1999), anesthetic (Kayukova et al., 2011), anthelmintic, anti-allergic (Guda et al., 2013), anti-Alzheimer (Wang et al., 2021), anti-microbial, anti-tumor (Kumar et al., 2011), anti-epileptic (Lankauet al., 2007), anti-depressant (Ergün et al., 2010), anti-fungal, anti-retroviral, anti-parasitic (Filho et al., 2016), anti-platelet and anti-thrombotic (Bethge et al., 2005), anti-tubercular (Makane et al., 2019a,b), anti-tussive (Harsányi et al., 1966), and anti-viral. Chemo-informatics approaches have been promising over the past few years in finding pharmacologically active compounds across a broad range of therapeutic areas. The presence of toxophoric —N⚌C—O— linkage in the 1,3,4-oxadiazole ring is responsible for their potent pharmacological activities. Amongst substituted 1,3,4-oxadiazoles, 2,5-disubstituted-1,3,4-oxadiazole derivatives are stable, especially aryl substituted 1,3,4-oxadiazoles are more stable than the corresponding alkyl derivatives. In the present study, we report the anti-tubercular activity of these aryl-substituted 1,3,4-oxadiazoles against the H37Rv strain of Mtb, and in silico analyses were implemented to reveal the molecular level of interaction with the ligand.
2 Materials and methods
2.1 Computational methods
2.1.1 Docking
Using a standard protocol (Bala et al., 2014), a protein (PDB ID: 2NSD) and designed molecules were docked into their active sites. ChemDraw (ChemDraw 16.0) was used to draw the chemical structures of the ligands with proper 2D orientations, and ChemBio3D was used to minimize the energy of each molecule. The protein Enoyl Co-A reductase (PDB ID: 2NSD) structure was prepared by eliminating the co-crystallized ligand, H2O molecules, and cofactors, which were eliminated, as reported in the literature (Garrett et al., 1998). The target protein file was prepared to leave the associated residues with the protein. The graphical user interface was used to create the grid box for docking. The grid box was made with a grid point spacing of 0.667 Å, with 23, 23, and 23 points in the x, y, and z directions, respectively. A grid box was observed (51 Å, 49 Å, and 35 Å).
2.1.2 Analysis
The results were visually analyzed using Pymol (2.5.3), and the 2D- interactions were seen using Discovery Studios (Stryjska et al., 2021).
2.1.3 In-silico drug-likeness properties of the compounds:
The drug-like characteristics of molecules were envisaged based on a previous idea by Lipinski. Molecules were converted to their default simplified molecular-input line-entry format (SMILE). Swiss ADME estimated the in-silico pharmacokinetics of isolated substances, including the no. of H-bond donors, H-bond acceptors, rotatable bonds, and TPSA (El-Ganiny et al., 2022). OSIRIS Properties were used to forecast the toxicity and toxicological outcomes of the designed substances. OSIRIS Properties were utilized to determine whether substances had drug potential (Guan et al., 2018). The greater the drug score, the more likely the substance would be considered a drug candidate (Zoppi et al., 2020).
2.1.4 Molecular dynamic simulations
Molecular dynamics has been performed to study the function and dynamics of proteins and ligands. Molecular docking has limitations, unlike biological procedures in which proteins and ligands are dissolved in water. A simulation cycle was run for 20 ns (ns) for the protein to understand further the non-bonding interaction between the ligand and the protein and the lead stability. The system was submerged in a simple point charge (SPC) solvent model to complete the dynamics. It was necessary to maintain the orthorhombic box shape of the boundary condition during the system development process. A buffer containing 0.15 M NaCl was added to the OPLS3e force field in the System Builder tool for neutralization. Additionally, minimization was performed using a minimization tool. During the experiment, the MD method kept temperature (K) and pressure (bar) at 300 K and 1.01325 bar, respectively. The reports were obtained using Desmond's Simulation Interaction Diagram tool (SID) (Le et al., 2019).
2.2 Chemicals and reagents
The p-nitro benzhydrazide, powdered stannous and various aromatic aldehyde were purchased from CDH. The purity of the compound was obtained by precoated TLC plates (Merck 60F 254). IR spectra were recorded using Brucker Alpha II. 1H NMR spectra were recorded on Brucker W.M. 400 Spectrometer (Bruker AG, Fallanden, Switzerland) at 360 MHz using tetramethylsilane (TMS) as internal standard.
2.3 Synthesis and characterization
2.3.1 General procedure of synthesis of 4- Amino Benzhydrazide II
Mixture of 0.009 mol (1.5 g) of p-nitrobenzhydrazide, 0.0295 mol (3.5 g) of powdered stannous, and 7.5 ml of conc. hydrochloric acid was taken in a 1-liter round-bottomed flask with a reflux condenser. The mixture was gently refluxed for 20 min, with frequent shaking, after about 20 min till a clear solution was obtained. The mixture was cooled and decanted, wash the filtrate was using water and a filter. Mix the filtrates and add concentrated ammonia until the solution turns alkaline and concentrates on a water bath. A 1.5 g filter aid was added and filtered via pressure and rinsed with hot water. The filter cake was transferred to a beaker of 20 ml water and heated on a water bath to ensure product extraction and refiltration. The filtered solutions were concentrated, acidified using glacial acetic acid, evaporated in the water bath, and cooled.
IR [υ/cm−1 (KBr)] = 3420, 1670, 1620, 1598, 1245. 1H NMR (300 MHz, CDCl3) δ = 4.49 (d, 2H, NH2), 9.64 (t, 2H, NH), 6.54 (d, 2H, Ar—H), 5.48 (s, 2H, NH2).13C NMR (100 MHz, CDCl3) δ = 167.2, 122.0, 130.2, 114.3, 151.8, 114.3, 130.2 ppm. HRMS (ESI): m/z calcd for C7H9N3O = 151; found: 152.
2.3.2 General procedure of synthesis of 5-(4-aminophenyl)-1,3,4-oxadiazole-2-thiol III
In a beaker mixture of 10 ml of ethanol and 1.2 ml of KOH solution, 0.02 mol (3.8 g) of compound II was taken and stirred for 15 min in an ice bath, after which 1.5 ml of CS2 was added dropwise and stirred for 15 min. Then the reaction mixture was refluxed at 60 °C for 4–5 h with stirring and the solvent was evaporated. The solid residue was dissolved in 25 ml of cold water, and 10% HCl solution was added and filtered to form yellow crystals. The yellow crystals were then recrystallized with ethanol.
IR [υ/cm−1 (KBr)] = 3366, 1687, 1646, 1514, 1461, 1272. 1H NMR (300 MHz, CDCl3) δ = 6.8 (s, 1H, SH), 7.62 (d, 2H, Ar—H), 6.58 (d, 2H, Ar—H), 5.24 (s, 2H, NH2). 13C NMR (100 MHz, CDCl3) δ = 164.5, 116.1, 128.3, 115.1, 145.6 ppm. HRMS (ESI): m/z calcd for C8H7N3OS = 193; found: 194.
2.4 Procedure for synthesis of (E)-5-(4-(benzylideneamino)phenyl)-1,3,4-oxadiazole-2-thiol derivatives (Wang et al., 2021)
2.4.1 Synthesis of 5-(4-{(E)-[(2-methoxyphenyl)methylidene]amino}phenyl)-1,3,4-oxadiazole-2-thiol (R1)
In 20 ml of toluene, compound III was dissolved, 2-Methoxy benzaldehyde was added, and the mixture was refluxed for 2 h. The mixture was then filtered, and the solvent was evaporated and crystallized using ethanol.
IR [υ/cm−1 (KBr)] = 1245, 1686, 1665, 1044, 2848. 1H NMR (300 MHz, CDCl3) δ = 6.9 (s, 1H, SH), 7.97 (d, 2H, Ar—H), 7.65 (d, 2H, Ar—H), 10.4 (s, H, CN), 7.49 (d, H, Ar—H), 7.14 (d, H, Ar—H), 3.82 (s, 3H, OCH3). 13C NMR (100 MHz, CDCl3) δ = 164.5, 124.6, 131.3, 122.8, 152.0, 156.5, 124.9, 157.6, 111.2, 132.0, 121.1 ppm. HRMS (ESI): m/z calcd for C16H13N3O2S = 311; found: 312.
2.4.2 Synthesis of 5-{4-[(E)-{[4-(dimethylamino)phenyl]methylidene}amino] phenyl}-1,3,4-oxadiazole-2-thiol(R2)
In 20 ml of toluene, compound III was dissolved, 4-N,N-dimethylaminobenzaldehyde was added, and the mixture was refluxed for 2 h. The mixture was then filtered, and the solvent was evaporated and crystallized using ethanol.
IR [υ/cm−1 (KBr)] = 1269, 1516, 1447, 1075, 1188, 2866. 1H NMR (300 MHz, CDCl3) δ = 6.9 (s, 1H, SH), 7.9 (d, 2H, Ar—H), 7.5 (d, 2H, Ar—H), 9.7 (s, H, CN), 7.50 (d, H, Ar—H), 6.82 (d, H, Ar—H), 3.02 (s, 6H, CH3).13C NMR (100 MHz, CDCl3) δ = 168.5, 129.3, 133.2, 120.0, 154.7, 166.0, 125.9, 128.3, 111.9, 153.4, 41.3 ppm. HRMS (ESI): m/z calcd for C17H16N4OS = 324; found: 325.
2.4.3 Synthesis of 5-(4-{(E)-[(3-bromophenyl)methylidene]amino}phenyl)-1,3,4-oxadiazole-2-thiol(R3)
In 20 ml of toluene, compound III was dissolved, 3-Bromo benzaldehyde was added, and the mixture was refluxed for 2 h. The mixture was then filtered, and the solvent was evaporated and crystallized using ethanol.
IR [υ/cm−1 (KBr)] = 1230, 1553, 1498, 1082, 1203, 695. 1H NMR (300 MHz, CDCl3) δ = 6.5 (s, 1H, SH), 6.89 (d, 2H, Ar—H), 7.12 (d, 2H, Ar—H), 9.64 (s, H, CN), 7.70 (d, H, Ar—H), 7.83(d, H, Ar—H). 13C NMR (100 MHz, CDCl3) δ = 168.5, 122.8, 134.7, 120.1, 152.8, 160.0, 135.9, 132.7, 123.2, 139.9 ppm. HRMS (ESI): m/z calcd for C15H10BrN3OS = 358; found: 359.
2.4.4 Synthesis of 5-(4-{(E)-[(2-nitrophenyl)methylidene]amino}phenyl)-1,3,4-oxadiazole-2-thiol(R4)
In 20 ml of toluene, compound III was dissolved, 2-Nitro benzaldehyde was added, and the mixture was refluxed for 2 h. The mixture was then filtered, and the solvent was evaporated and crystallized using ethanol.
IR [υ/cm−1 (KBr)] = 1230, 1588, 1530, 1064. 1H NMR (300 MHz, CDCl3) δ = 6.7 (s, 1H, SH), 7.9 (d, 2H, Ar—H), 7.42 (d, 2H, Ar—H), 10.2 (s, H, CN), 7.98 (d, H, Ar—H), 8.08 (d, H, Ar—H). 13C NMR (100 MHz, CDCl3) δ = 168.3, 126.1, 138.5, 155.6, 147.8, 128.4, 124.0, 130.1 ppm. HRMS (ESI): m/z calcd for C15H10N4O3S = 326; found: 327.
2.4.5 Synthesis of 5-(4-{(E)-[(2-chlorophenyl)methylidene]amino}phenyl)-1,3,4-oxadiazole-2-thiol(R5)
In 20 ml of toluene, compound III was dissolved, O-Chloro benzaldehyde was added, and the mixture was refluxed for 2 h. The mixture was then filtered, and the solvent was evaporated and crystallized using ethanol.
IR [υ/cm−1 (KBr)] = 1235, 1605, 1584, 1090, 1210, 856. 1H NMR (300 MHz, CDCl3) δ = 6.54 (s, 1H, SH), 7.33 (d, 2H, Ar—H), 7.81 (d, 2H, Ar—H), 10.7 (s, H, CN), 7.75 (d, H, Ar—H) 7.51 (d, H, Ar—H). 13C NMR (100 MHz, CDCl3) δ = 161.3, 126.8, 135.9, 121.3, 157.0, 133.4, 133.9, 132.4, 126.9 ppm. HRMS (ESI): m/z calcd for C15H10ClN3OS = 315; found: 317.
2.5 Anti-tuberculosis activity
Preparation of Inoculum: 100 µL of the Middlebrook 7H9 broth.
Requirements:
Synthesized compounds were evaluated for anti-mycobacterial activity against M. tuberculosis (H37Rv). The anti-mycobacterial activity was assayed by using the Alamar Blue Assay (MABA). The microplate Assay was used to detect M. tuberculosis. Alamar Blue reagent is used in this method and indicates a high correlation with BACTEC relative and radiometric assays. The plates were incubated at 37 °C for five days with 100 to 0.8 µg/mL concentrations. A mixture of Alamar Blue reagent and 10% Tween 80 were added and incubated for 24 h. Bacterial growth was indicated by blue, and the absence of bacterial growth was judged to be told by a pink color. The MIC was the minor drug concentration that prevented the color change from blue to pink. The synthesized compounds were compared to standards such as Isoniazid, Ethambutol, and Rifampicin (Petrou et al., 2021).
Standard Strain used: MYCOBACTERIUM TUBERCULOSIS CINE strain, H37 RV strain): ATCC No-27294. Standard values for the Anti-TB test that was performed.
2.5.1 Cytotoxicity studies (MTT ASSAY)
In CDRI-CSIR Lucknow in vitro, cytotoxicity studies were performed (in accordance with their protocol.) Several concentrations of synthesized compounds were evaluated in vitro for cytotoxic activity on cell lines representing M. tuberculosis, namely Vero cells ATCC CCL-81. It is based on the principle of uptake of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) by metabolically active cells, where it is metabolized by active cells into a blue colored formazan product. In brief, tuberculous cells were seeded in 96-well culture plates and incubated in a CO2 incubator at 37 °C with various concentrations of 0.8 to 100 µg/ml, with relevant controls in triplicate wells. After 72 h, the assay was completed by adding 25 ml of MTT solution (5 mg/ml) to each well. The percentage cytotoxicity was calculated as follows.
Percentage cytotoxicity = 100 * [1 − (X/R1)].
X = absorbance of the treated sample at 540 nm.
R1 = absorbance of control sample at 540 nm.
The IC50 values were determined using Prism software v 4.01 via by nonlinear regression model (Zheng and Av-Gay, 2017).
3 Results
3.1 Chemistry
In Fig. 1 (Fig. 1), the final product i.e., (E)-5-(4-(benzylidene amino) phenyl)-1,3,4-oxadiazole-2-thiol derivatives,were synthesized in three steps; starting with the reduction of 4-Nitrobenzhydrazide to 4-Aminobenzhydrazide by the reduction process. Compound III was formed by cyclization of 1,3,4- oxadiazole ring in the presence of EtOH, KOH, and CS2. Then, the mixture was refluxed by stirring for 5 h. The final step was based on a Schiff base reaction; an equal amount of aromatic aldehyde and 5-(4-aminophenyl)-1,3,4-oxadiazole-2-thiol (III) was reacted to get a final product.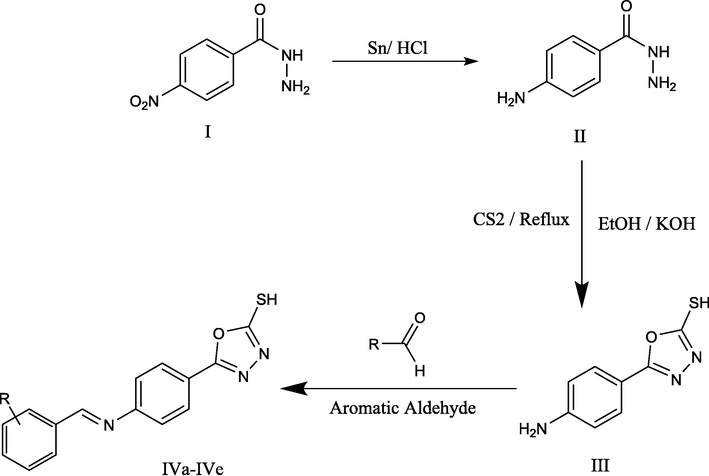
(Scheme 1) Synthesis of (E)-5-(4-(benzylideneamino) phenyl)-1,3,4-oxadiazole-2-thiol derivatives 4- Nitrobenzhydrazide(I), 4-Aminobenzhydrazide (II), 5-(4-aminophenyl)-1,3,4-oxadiazole-2-thiol (III), R1-R5: (E)-5-(4-(benzylideneamino) phenyl)-1,3,4-oxadiazole-2-thiol derivatives.
3.2 Docking of the ligands with Enoyl CoA reductase protein
The thirty compounds were computationally designed and optimized with the Enoyl-CoA reductase targeting its active binding site to observe the binding energy involved in forming complex and molecular interactions. The docking results are summarized in Table 1.
Number
Structure
Ligand
Binding Energy (Kcal/mol)
H-bonding
Interactions
1.
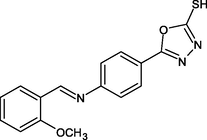
R1
−9.6
1H
LEU218, PRO193, ILE215, TYR158, PRO156
2.
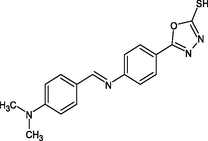
R2
−8.6
3H
LEU218, ILE215, MET155, TRP222, PRO193
3.
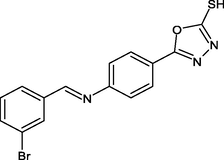
R3
−9.1
0
ALA191, MET147, PHE149, MET199
4.
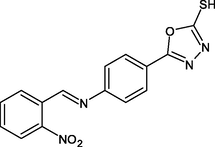
R4
−10.0
2H
TYR258, PHE149, ALA191, LYS168
5.
R5
−9.7
0
LEU218, ILE215, TRP222, MET155, PRO198
6.
R6
−8.6
0
ILE215, PHE149, MET147, LEU218, PRO193
7.
R7
−8.8
0
ILE215, LEU218, PRO193, PHE149
8.
R8
−9.3
1H
PRO193, LEU218, MET199
9.
R9
−9.1
1H
MET155, PHE149, PRO193, LEU218, MET199
10.
R10
−9.2
2H
GLU219, PRO193, LEU218, MET199
11.
R11
−9.2
0
TYR182, GLY80, GLU 178
12.
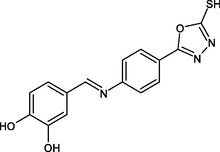
R12
−9.2
0
GLU219, MET155, MET199, LEU218, PRO193
13.
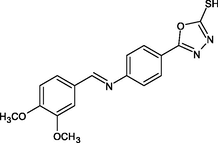
R13
−9.1
1H
SER94, ILE21, MET147, GLY14
14.
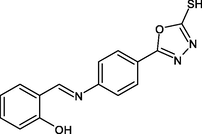
R14
−9.3
1H
PRO193, ILE215, LEU218
15.
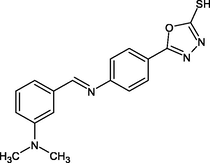
R15
−8.6
3H
SER94, GLY14, TYR196, ILE21
16.
R16
−9.3
3H
ILE215, GLU219, LEU218, PRO193
17.
R17
−9.0
1H
ILE21, ALA198
18.
R18
−8.9
1H
SER94, ILE21, GLY14
19.
R19
−9.0
3H
GLU219, ILE215, PRO193, LEU218, MET155
20.
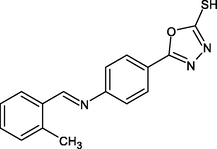
R20
−9.1
1H
TYR158, LEU218, ILE215, PRO198
21.
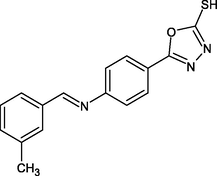
R21
−9.5
0
LEU218, ALA157, ILE215
22.
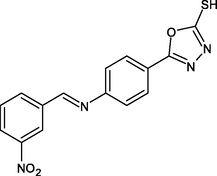
R22
−9.4
2H
ILE215, GLU219, LEU218, ALA157
23.
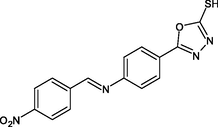
R23
−9.2
3H
ALA198, SER94, ILE21, ALA22
24.
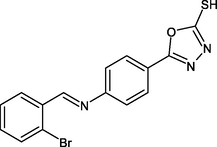
R24
−9.2
1H
PHE149, PRO193, ILE215, LEU218
25.
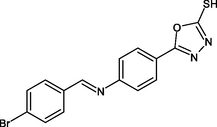
R25
−8.5
0
LEU218, ILE202, TYR158, ALA157
26.
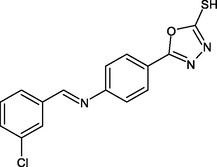
R26
−9.3
0
LEU218, ILE215, ALA157
27.
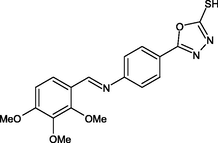
R27
−9.0
1H
ILE21, SER94, MET147
28.
R28
−9.3
1H
LEU218, PRO193, ILE215, TYR158, ILE202, VAL207
29.
R29
−9.3
1H
ILE215, ALA157, PRO193, LEU218, TYR158
30.
R30
−9.4
0
LEU215, ILE215, PRO193, ALA157, PHE149
From the thirty compounds these R5, R1 and R4 showed better binding energies at −9.7, −9.6 and −10.0 Kcal/mol, respectively.
The R4 showed the best binding energy of −10.0 Kcal/mol against the Enoyl CoA reductase domain inhibited complex formation by forming H-bond with TYR158 and π-π interaction interactions with ALA191 and PHE149 as shown in Fig. 2.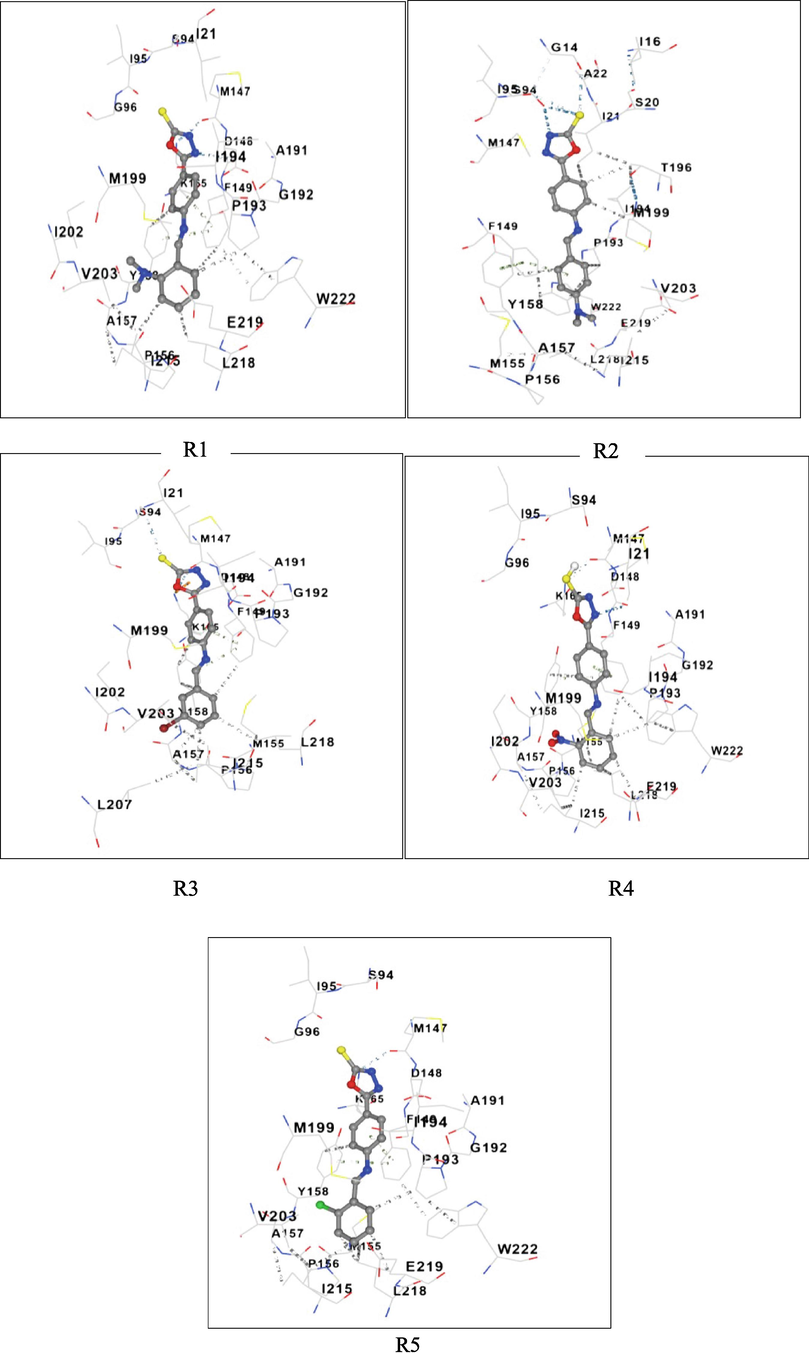
3-D Structures of the Protein-ligand complex and Molecular interactions of R1-R5.
3.3 Pharmacological and toxicological profiles of the synthesized compounds
The pharmacological attributes prediction results are given in Table 2. The TPSA (135.9) was highest with R4 ligand, which was followed by R1, R2, R5 and R3 respectively. None of the synthesized compounds showed any mutagenic, tumorigenic, and reproductive toxicities or eye irritation (Table 3). TPSA: Topological Polar Surface Area.
Ligand
Mol. Formula
Mol. wt.
TPSA
H-Donors
H-acceptors
Rot-bonds
Drug Score
R1
C16H13N3O2S
311
99.31
0
5
4
0.3
R2
C17H16N4OS
324
93.32
0
4
4
0.18
R3
C15H10BrN3OS
359
90.08
0
4
3
0.24
R4
C15H10N4O3S
326
135.9
0
6
4
0.28
R5
C15H10ClN3OS
315
90.08
0
4
3
0.26
Ligand
Mutagenic
Tumorigenic
Reproductive effects
Eye Irritant
R1
NONE
NONE
NONE
NONE
R2
NONE
NONE
NONE
NONE
R3
NONE
NONE
NONE
NONE
R4
NONE
NONE
NONE
NONE
R5
NONE
NONE
NONE
NONE
3.4 Molecular dynamic simulation
Molecular Dynamics (MD) simulations were performed using Glide XP docking to verify the binding stability. Simulation through MD has the benefit of representing the actual biological conditions with appropriate significance. With the simulation, ahigh-resolution dynamic structure of the protein and an isolated protein–ligand complex in water like a biological system and the interaction between the ligand and the protein were determined. Although Induced Fit Docking (IFD) provides complex flexibility, it cannot simulate biological conditions.
Evaluating docking scores, free binding energies, and non-bonding interactions of the significant amino acids with the pocket in MD simulations helps compare the most suitable top molecule and co-crystal ligand. Furthermore, to understand the impact of compound binding on protein conformation, Root means square deviations (RMSD), Radius of Gyrations (ROG), as well as Root mean square fluctuations (RMSF) of individual residues have been studied.
The analysis revealed that the RMSD of the protein backbone was initially observed to be fluctuating from 1.25 to 1.75 Å to 15 ns (Fig. 3A). After which it was found stabilized at around 2.0 Å, corresponding with the protein’s backbone, the ligands RMSD was also found to fluctuate around 3.5 Å up to 5 ns. In the simulated time of 5 ns, the ligand stabilized at 1.25–2.25 Å, indicating that it was quite stable inside the protein’s active site. When compared with the RMSD of the protein and the co-ligand (Fig. 3B), it was seen that after five nanoseconds, the ligand and the protein were very stable.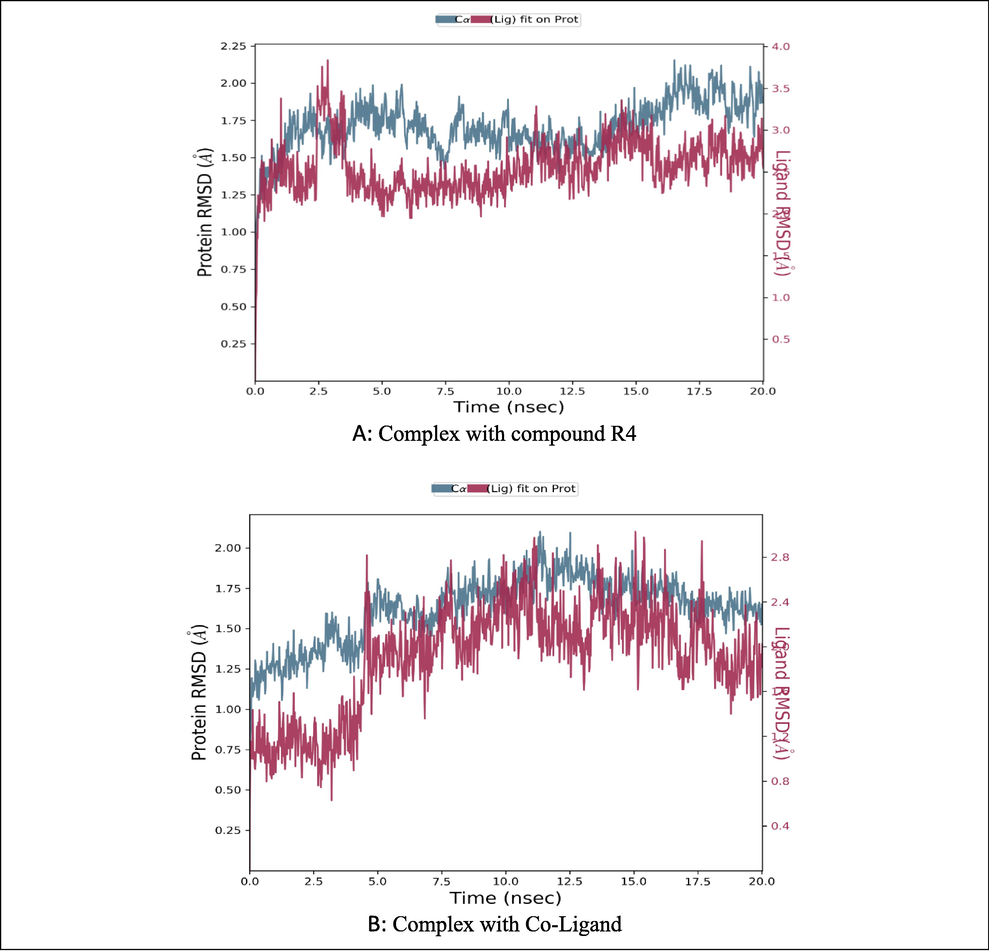
RMSD of the Enoyl CoA reductase domain backbone in complex with compound R4/Co-Ligand.
From the graph analysis of Enoyl CoA reductase in complex with the ligand (Fig. 4A), minimal fluctuations indicated that the residues were relatively stable in the presence of ligand (Fig. 4B), with few peaks up to 2 Å which are caused by the loop movements. Furthermore, the low RMSF observed is indicative of less protein deviation during simulation, signifying the system was in equilibrium.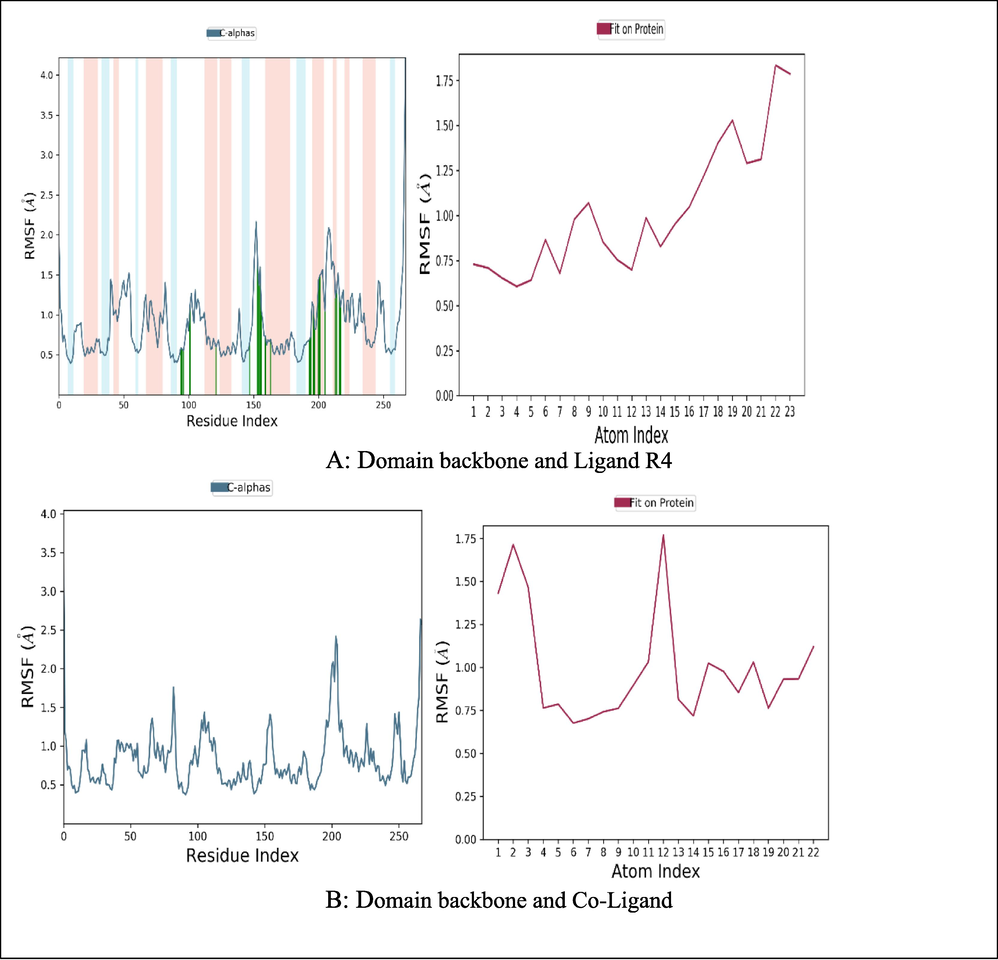
RMSF of the Enoyl CoA reductase domain backbone and Ligand R4/Co-Ligand.
3.5 Molecular interaction
The 23 contacts were found between the ligand and protein, 4 contacts were involved in hydrogen bonds, 14 in hydrophobic interactions, 9 in water bridging interactions and 2 ionic bond formations were observed, respectively (Fig. 5A). In Co-Ligand, 15 contacts were found between the ligand and protein; one contact was involved in a hydrogen bond, 14 contacts were involved in hydrophobic interactions, two contacts in water bridging interactions and were observed respectively, 13 contacts were similar with ligand R4 (Fig. 5).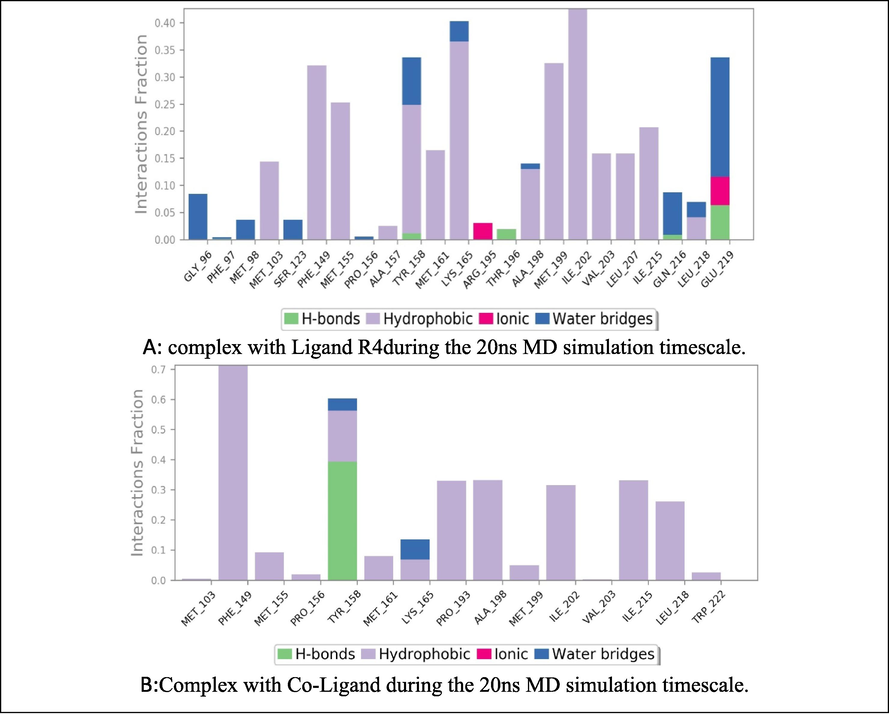
Molecular interactions observed between Enoyl CoA Reductase domain in complex with Ligand R4during the 20 ns MD simulation timescale Co-Ligand during the 20 ns MD simulation timescale.
Biological Evaluation: The synthesized compounds were evaluated for anti-mycobacterial activity against M. tuberculosis (H37Rv) using MABA assay and compared with the standards (Table 4). S – Sensitive; R – Resistant.
Cytotoxicity Studies (MTT Assay): The viability of Mycobacterium tuberculosis cell lines was tested for in vitro cytotoxicity using a range of hybrid analog concentrations. Cytotoxicity was determined via MTT assay, based on the principle that metabolically active cells consume MTT, turning it into a blue-colored formazan product because of active mitochondria. The cytotoxicity of the synthesized compounds was less than standard levofloxacin and doxorubicin (Table 5).
Sl. No.
Compound Code
MIC (µg/ml)
Cytotoxicity(% Inhibition)
IC50
Selectivity Index
Trail 1
Trail 2
Trail 1
Trail 2
Trail 1
Trail 2
1.
R1
1.6
6.6
8.07
16.14
15.03
10.08
9.393
2.
R2
1.6
7.33
6.06
20.21
17.73
12.63
11.08
3.
R3
0.8
11.74
9.52
45.03
39.05
56.28
48.81
4.
R4
0.8
12.02
10.56
49.07
41.01
61.33
51.26
5.
R5
1.6
7.91
7.73
18.11
19.02
11.31
11.88
Levofloxacin
1.5
27.64
27.24
70.01
70.01
46.67
46.67
Doxorubicin
5
35.67
30.37
7.01
7.01
1.402
1.402
4 Discussion
Based on docking results (Table 1), R4 (Fig. 2) showed the highest affinity. Hydrogen bonds between TYR158 and N⚌O group, the interaction between the phenyl ring of TYR158 and ALA191 and PHE149 were at the active sites. Further, in addition to the fragment, there was a π-alkyl interaction between ALA157, ILE 215, and ILE202, π-sigma bond interaction between VAL203 and the oxadiazole ring. Additionally, the thiol moiety attached to the oxadiazole ring showed a hydrogen bond.
Electron-withdrawing groups (EWG) and electron-donating groups (EDG) affected the binding energy score concerning ortho, meta, and para positions. While comparing both the groups i.e., EWG (NO2, Halogens) showed the least binding energy, EDG (methoxy, alkylamine, hydroxy) showed the highest binding energy. The ortho position showed the least binding energy and the para position showed the highest binding energy. With all these effects, di-substitution and trisubstituted also affected the binding energy of −9.0 to −9.7 Kcal/mol.
In Table 2 and Table 3, none of the designed molecules violated any rules. For example, compound R4 showed a molecular weight of 326 Daltons, TPSA of 135.9, no H-bond donors, 6H-bond acceptors, and four rotatable bonds.
Simulation through MD has the benefit of representing the actual biological conditions with appropriate significance. With the simulation, a high-resolution dynamic structure of the protein and an isolated protein–ligand complex in water like a biological system and the interaction between the ligand and the protein were determined. Although Induced Fit Docking (IFD) provides complex flexibility, it cannot simulate biological conditions. The MD simulation of 5-(4-{(E)-[(2-nitrophenyl) methylidene] amino} phenyl)-1,3,4-oxadiazole-2-thiol with the co-crystal ligand was carried out. The RMSD values were observed in the acceptable range of 1.25–1.75 Å units till 15 ns of the simulation timescale. The RMSF technique can detect the presence of residues that contribute to complex structural fluctuations caused by the movement of atoms in the chains and structures. The low RMSF indicates that the protein showed less variation during simulation, indicating the system is in equilibrium. The graphical analysis of the RMSF is given for the ligand (Fig. 5) and the co-crystal ligand (Fig. 4). Furthermore, it was observed that the complex showed hydrogen bond interactions with TYR158, THR196, and GLU219; hydrophobic interactions with MET103, PHE149, MET155, ALA157, TYR158, MET161, LYS165, ALA198, MET199, ILE202, VAL203, LEU207, ILE215, and LEU218; ionic bond interactions with ARG195, GLU219; and water bridges with GLY96, PHE97, MET98, SER123, PRO156, TYR158, LYS165, ALA198, GLN216, LEU218, and GLU219. The co-ligand showed few similar interactions such as hydrogen bond interaction and water bridge with TYR158, hydrophobic interactions with MET103, MET155, TYR158, MET161, LYS165, ALA198, MET199, ILE202, VAL203, ILE215, LEU218. From the RMSF and RMSD, it can be proven that the molecule shows better stability than the Co-crystal ligand. Further, the anti-tubercular activity was performed for the best-docked compounds, where the MIC was also affected by EWG and EDG. Those compounds containing EWG (NO2, Br) showed activity at 0.8 µg/ml, and EDG (methoxy, alkyl amine) showed activity at 1.6 µg/ml. It is concluded that the nitro-containing derivative, i.e., 5-(4-{(E)-[(2-nitrophenyl) methylidene] amino} phenyl)-1,3,4-oxadiazole-2-thiol showed the best activity at 0.8 µg/ml with the IC50 at 49.07 and selectivity Index at 61.33.
5 Conclusion
The tertiary protein structure of MtbEnoyl CoA reductase was prepared and analyzed. The thirty compounds were analyzed, and binding affinity was determined along with their pharmacological properties and toxicity profile. It was identified that compounds R5, R1, and R4 showed better binding energies −9.7, −9.6, and −10.0 Kcal/mol, respectively. Molecular docking analysis confirms R4 as the highest binding energy (-10.0 Kcal/mol) for Enoyl CoA reductase. R4 shows a MIC of 0.8 µg/ml, an IC50 value at 49.01, and a Selectivity Index of 61.33. Therefore, it can be concluded that among the designed compounds, 5-(4-{(E)-[(2-nitrophenyl) methylidene] amino} phenyl)-1,3,4-oxadiazole-2-thiol showed the best activity with higher IC50 values.
Funding
The authors are thankful to AlMaarefa University for their financial support.
Acknowledgment
This research was supported by the Researchers Supporting Project number (RSP2023R431) at King Saud University, Riyadh, Saudi Arabia. The authors thank the Principal and Management of Krupanidhi College of Pharmacy, Bengaluru, for their support and encouragement. We want to thank SAIF-Punjab University for the NMR spectra and VIT for the MS spectra; We also thank CSIR-CDRI Lucknow for Cytotoxicity studies.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- New oxadiazole derivatives showing partly antiplatelet, antithrombotic and serotonin antagonistic properties. Arch Pharm.. 2005;338:78-86.
- [Google Scholar]
- The promising anti-virulence activity of candesartan, domperidone, and miconazole on Staphylococcus aureus. Braz. J. Microbiol.. 2022;53(1):1-18.
- [Google Scholar]
- Synergistic effect of [1h-[1,2,4]oxadiazole[4,3-a]quinoxalin-1-one] and antidepressant drugs in the mouse forced swimming test: possible involvement of serotonergic pathway. Eur. J. Pharmacol.. 2010;630:74-78.
- [Google Scholar]
- Conjugation of n-acylhydrazone and 1,2,4-oxadiazole leads to the identification of active antimalarial agents. Bioorg. Med. Chem.. 2016;24:5693-5701.
- [Google Scholar]
- NASN Sch. Nurse. 2016;31:83.
- Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998:1639-1662.
- [Google Scholar]
- Antioxidant and anti-inflammatory properties of 1,2,4-oxadiazole analogs of resveratrol. Chem.-Biol. Interact.. 2015;240:200-207.
- [Google Scholar]
- ADMET-score – A comprehensive scoring function for evaluation of chemical drug-likeness. Med. Chem. Commun. 2018
- [Google Scholar]
- Syntheses and anti-allergic activity of 2-((bis(trimethylsilyl)methylthio/methylsulfonyl)methyl)-5-aryl-1,3,4-oxadiazoles. Eur. J. Med. Chem.. 2013;62:84-88.
- [Google Scholar]
- The synthesis of an antitussive action derivative of 1,2,4-oxadiazole, 3-(2,2-diphenylethyl)-5-(2-piperidinoethyl)-1,2,4-oxadiazole] Arzneim. Forsch.. 1966;16:615-617.
- [Google Scholar]
- Novel 1,2,4-oxadiazole derivatives in drug discovery. Pharmaceuticals. 2020;13:111.
- [Google Scholar]
- Biological aspects of 1,3,4-oxadiazole derivatives. Asian J. Pharm. Clin. Res.. 2014;7(4):11-20.
- [Google Scholar]
- Local anesthetic activity of new amidoxime derivatives. Pharm. Chem. J.. 2011;45:468-471.
- [Google Scholar]
- Synthesis of novel 1,2,4-oxadiazoles and analogues as potential anticancer agents. Eur. J. Med. Chem.. 2011;46:3085-3092.
- [Google Scholar]
- New gaba-modulating 1,2,4-oxadiazole derivatives and their anticonvulsant activity. Eur. J. Med. Chem.. 2007;42:873-879.
- [Google Scholar]
- Novel 1-methyl-1-pyrazole-5-carboxamide derivatives with potent anthelmintic activity. J. Med. Chem.. 2019;62(7):3367-3380.
- [Google Scholar]
- Synthesis and analgesic profile of novel n-containing heterocycle derivatives: arylidene 3-phenyl-1,2,4-oxadiazole-5-carbohydrazide. IlFarmaco. 1999;54:747-757.
- [Google Scholar]
- Novel 1,3,4-oxadiazoles as antitubercular agents with limited activity against drug-resistant tuberculosis. Future Med. Chem.. 2019;11:499-510.
- [Google Scholar]
- Novel 1,3,4-oxadiazoles as antitubercular agents with limited activity against drug-resistant tuberculosis. Future Med. Chem.. 2019;11(6):499-510.
- [Google Scholar]
- Synthesis, spectroscopy, single-crystal structure analysis and antibacterial activity of two novel complexes of Silver (I) with Miconazole drug. Int. J. Mol. Sci.. 2021;22(4)
- [Google Scholar]
- Design and synthesis of 3-(4-pyridyl)-5-(4-sulfamido-phenyl)-1,2,4-oxadiazole derivatives as novel gsk-3β inhibitors and evaluation of their potential as multifunctional anti-alzheimer agents. Eur. J. Med. Chem.. 2021;209:112874.
- [Google Scholar]
- System for efficacy and cytotoxicity screening of inhibitors targeting intracellular Mycobacterium tuberculosis. J Vis Exp.. 2017;122:1-8.
- [Google Scholar]
- Simultaneous improvement of ketoconazole solubility, antifungal and antibiofilm activity by multicomponent complexation. Ther. Deliv.. 2020;11(11):701-712.
- [Google Scholar]




