

Translate this page into:
Screening potential inhibitor from actinomycetes for Dihydrofolate reductase of Staphylococcus aureus – In vitro and in silico studies
⁎Corresponding author at: Biochematics Laboratory, Department of Bioinformatics, Bharathiar University, Coimbatore 641 046, Tamil Nadu, India. jeyam@buc.edu.in (Jeyam Muthusamy)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract

Abstract
Cellulitis is a skin disease which is caused by the pathogen Staphylococcus aureus. The protein Dihydrofolate reductase is an important drug based target for Trimethoprim. Dihydrofolate reductase is reversibly inhibited by the drug trimethoprim. One of the major enzymes responsible for converting dihydrofolic acid into tetrahydrofolic acid. The screening of microbial natural products, Soil isolation is a rich source of actinomycetes. The actinomycetes sp. KK21-2 strain isolated from the soil, the aqueous extract of the isolates yielded 33 active metabolites from the LC-MS analysis. Based on the ADMETox analysis, 8 compounds were excluded and finally 25 compounds were selected for the docking analysis. Protein structure of S. aureus retrieved from PDB, drug and compounds were obtained from the PubChem Database. Molecular docking (XP) and Induced Fit Docking (IFD) were completed utilizing the Glide Module of Schrodinger. Molecular dynamics simulation employing the Schrodinger Desmond module was used to examine the stability of the docked complex. From the results of docking analysis and MD Simulation result 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl) phenylhydantoin have suggest this compound have multi- targeting potency against the cellulitis skin disease from the pathogen Staphylococcus aureus, in future need to be verified through in vitro and in vivo analysis.
Keywords
Actinomycetes
Cellulitis
Staphylococcus aureus
Molecular docking
Dihydrofolate reductase
MD simulation

1 Introduction
Actinomycetes are the most important microorganism in nature that primarily survives in the soil (Pandey et al., 2011) which produced a variety of essential bioactive compounds of high value. Frequent screening of actinomycetes for new bioactive compounds resulted with around two thirds of natural antibiotics, many of which are medicinally important (Okami and Hotta, 1988). Actinomycetes are filamentous, sporulating gram-positive bacteria rich in G + C contact which accounts for 55–75 percent of DNA. Cellulitis, a bacterial skin infection, affects more than 14 million people annually in the United states (Raff and Kroshinsky, 2016). The disease cellulitis is caused mainly by methicillin-sensitive Staphylococcus aureus and Streptococcus pyogenes (Cranendonk et al., 2017).
The current antibiotic used against cellulitis diseasis is Cephalosporins, aminoglycosides, penicillin, trimethoprim, cephalexin, clindamycin (Patil et al., 2018). and the adverse effect were observed while using this drugs are Eosinophilia, Thrombocytopenia Hypersensitivity reaction, clostridioides and Leukopenia (Norrby, 2012). To overcome this issue we need to develop a new drug for reduce the side effects. At earlier phases of the drug development process, in silico approaches have the ability to anticipate negative side effects for example drug- target interactions (Hodos et al., 2016).
In drug designing, computational approach aids in identifying a ligand that can effectively bind to the target protein molecule and also to predict the properties of the docked complex (Kaapro and Ojanen, 2002). In recent years, molecular docking has been widely used in academic and professional contexts as a speedy and inexpensive method. The main purpose of ligand protein docking is to investigate the superior ligand binding modes with the target protein. Molecules orientation and conformation within a macromolecular target's binding site are examined using a technique called molecular docking. Possible postures are generated by search algorithms and ranked by scoring functions. The two primary processes of molecular docking computations are posing and scoring, which provide a prioritized list of potential complexes between target and ligands (Torres et al., 2019) The two primary causes of drug failure are safety and lack of effectiveness. ADMET stands for Absorption, Distribution, Metabolism, Excretion and Toxicity. In addition to having sufficient activity against the therapeutic target, a high-quality drug candidate should display the proper ADMET properties at a therapeutic dose. (Guan et al., 2019). This study focused on antibacterial activity by inhibiting the growth of S.aureus, identifying the compounds present in the actinomycetes extract with significant activity through LC-MS analysis and identifying the novel lead compound.
2 Material and methods
2.1 Sample collection
Fifty soil samples were collected from foot hills of Siruvani (Latitude is 10.9378° N and Longitude is 76.6870° E.), Velliangiri (Latitude is 10.9833° N and Longitude is 76.6927° E.) and surrounding areas of foot hills in Coimbatore district. The samples were collected in small clean, sterile poly vinyl pack bags properly labeled with place and date of collection, immediately transferred to the laboratory and stored in the refrigerator for further analysis.
2.2 Isolation of pure actinomycetes
Actinomycetes were isolated using the standard microbiological method. One gm of dried soil sample was suspended in 100 ml of sterile water and serial dilution from 10-2 to 10-7 were prepared. Maximum 10-4, 10-5 and 10-6 dilution are maximum number of actinomycetes colonies were produced. The Starch casein nutrient agar (SCNA) medium composition are soluble starch- 10 g, casein- 0.3 g, KNO3- 2 g, NaCl- 2 g, K2HPO4- 2 g, MgSO4·7H2O- 0.05 g, CaCO3- 0.02 g, FeSO4 7H2O- 0.01 g, Agar- 20 g and distilled water- 1000 ml, pH 7.0 ± 0.2 was used for isolation of actinomycetes. The plates were incubated at room temperature (28 °C) after inoculation for 7–14 days. After the 7 to 9 days of incubation period small colonies, white pin point, powdery colonies were observed from which pure cultures were separated and maintained in both plates and slants for further analysis. For longer period storage glycerol stock was prepared and maintained in refrigerator at 4 °C.
2.3 Protein data Bank (PDB)
A 3D structural database for large biological entities like proteins and nucleic acids is known as Protein Data Bank (PDB) (Mir et al., 2017). The data, normally collected by X-ray crystallography, NMR spectroscopy and cryo-electron microscopy and provided by biologists and biochemists from throughout the world. The target protein structures of S. aureus retrieved from PDB for this study were Dihydrofolate reductase (PDB ID: 6E4E).
2.4 Pubchem database
Pubchem is a database which provides the compounds chemical molecules and the activity observed in the biological assays. The National Centre for Biotechnology Information (NCBI) is responsible for maintaining the database. Pubchem database had a wide range of characteristics for the molecules including chemical structure fragments of name molecular weight chemical formulation donor and acceptor bond count XLogP (Kim et al., 2019). The selected 33 LC-MS compounds structures were retrieve using Pubchem Database.
2.5 Drug Bank database
The DrugBank database is an extensive, openly available online resource that was built and is continuously updated with data on medications and therapeutic targets. It includes proprietary describing clinical data on drugs like side effects and drug interactions of the drugs (Wishart et al., 2008). The Trimethoprim structure is retrieved from Drug Bank Database.
2.6 LC-MS analysis
LC-MS methods are separates chemicals in a sample by liquid chromatography and further examines and identifies them by a mass spectrometer. Polar solvents were used like methanol and water (Pitt, 2009). Liquid chromatography-mass spectrometry (LC-MS) was used to further analyze the crude water extract of isolated soil actinomycetes KK21-2 and determine the secondary metabolites. The LC-MS was performed by SAIF IIT Bombay. The Acquisition method used is 30mins_+ ESI_10032014_MSMS and the Ion source is Dual AJS ESI. Using European Mass Bank, the detected compounds were compared.
2.7 ADME properties
ADME stands for “absorption, distribution, metabolism, and excretion” in pharmacokinetics and pharmacology. The four factors together affect the drug levels and kinetics of tissue exposure, which in turn affects the effectiveness and pharmacological activity of the chemical as a medicine (Tibbitts et al., 2016). ADME properties was evaluated for LC-MS derived compounds using the Qikprop 3.5 of Schrodinger module (Ganjuly and Debneth, 2014).
2.8 Protein preparation
The protein molecule was preprocessed, missing hydrogen atoms were added, bond orders were given, disulfide connections were created, and the orientation of the amino acid's hydroxyl groups, amide groups of Asn, and Gln, was optimised in the protein production wizard. The grid was created to encompass the active site residues identified by the multiple sequence alignment. The force field OPLS3e was fixed by constructing potential ionised and tautomerized structures using the Ligprep tool within a predetermined pH range. Through XP, ligand docking was carried out utilising chosen ligands and chosen pathogen targets (Syed Abuthahir et al., 2019).
2.9 Active site prediction
Protein functions depends on active sites. This either inhibits or stimulates protein functions. The main database for information on proteins is Uniprot, particularly information on the active site's various sorts of binding capacities with substances including tiny molecules, metals, DNA, and smaller proteins, among other things (Bairoch et al., 2005). Sitemap is a tool developed by Schrodinger to predict with a high degree of accuracy the binding site of protein molecules and the druggability of the sites. (Schmidtke and Barril, 2010). protein crystal structures that have been solved and multiple sequence alignment utilising the Clustal Omega server, and Schrodinger site map software were used to identify the active site of the chosen targets of S. aureus. (Zhang et al., 2008).
2.10 Ligand preparation
The LC-MS ligands employed in this investigation were those from Actinomycetes aqueous extract that showed action against bacterial infections. According to Peach et al. (2012), the QikProp module of Schrodinger was used to analyse the 3D structures for ADMETox. Features including the Rule of Five, oral absorption, QPlog Po/w, QplogS, Qpp Caco, Qplag HERG, QPP MDCK, PSA, HB Donars, and HB Acceptor.
2.11 Molecular docking – XP docking
The four important steps were followed to use the Schrodinger Glide module for protein–ligand docking. They are 1. Protein preparation, 2. Receptor Grid generation, 3. Ligand preparation and 4. Ligand docking (Ban et al., 2018).
The preprocessing of the protein molecules includes the insertion of hydrogen atoms, the distribution of bond orders, the creation of disulfide bonds, improvement of the orientation of amide and hydroxyl groups. The energy minimization force filed used was OPLS3e.The grid was built to encompass the active site residues. The same force field was used to prepare the ligands, and the Ligprep tool was used to optimise the ligand molecules. Finally the ligand docking was carried out using the chosen ligands for specific pathogen targets through extra precision (XP) docking (Abuthakir et al., 2022).
2.12 Induced Fit docking
Using Schrodinger software, Induced fit docking was used, in which the binding site is adjusted in accordance with the ligand's binding manner and the receptors are flexible. For the docking and the structure refinement, used the Glide and Prime modules. Induced Fit Docking includes protein preparation, grid generation and selecting active site residues with length of <20 Å for binding. Based on the XP docking results, the top three ligand molecules were selected for the IFD docking. At the end of IFD, multiple poses were generated, the optimum pose was chosen after all poses were examined for their dock score interactions with active site residues (Clark et al., 2016).
2.13 Molecular dynamic simulation
The complexes that are docked and chosen based on the IFD analysiswere conducted to test the stability of the complexes employing use of Schrodinger Biosuite for molecular dynamics simulation. The docked complex of Dihydrofolate reductase protein and 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5- phenylhydantoincompound were recorded during a 50 ns time span for Molecular Dynamic Simulation. System builder was initially responsible for starting the MD simulation and further force field,solvent model and boundary conditions. The complexwas centered with a 10 Å distance as boundary conditions for the MD simulation, this was performed with the help of the OPLS 2005 force field (Abuthakir et al., 2021).The steepest descent algorithm was used to reduce the energy of complicated structureswhich Desmond uses as an equilibration technique (Fu et al., 2018).The RMSD of protein was produced using simulation event analysis and Understanding the stability of the ligand inside the protein's binding site through ligand bond proteins, protein–ligand interactions, and RMSF of the protein and ligand (Balakrishnan et al., 2022).
3 Results
3.1 Isolation of actinomycetes
A total of 50 soil samples were taken from foot hills of Siruvani, Velliangiri and surrounding area, 75 strains were isolated using SCNA medium from the isolate KK21-2 produce the good results with comparing with all other isolates (Figs. 1–4). So, this KK21-2 strain is further performed for the LC-MS analysis.
Isolates of actinomycetes from soil samples.
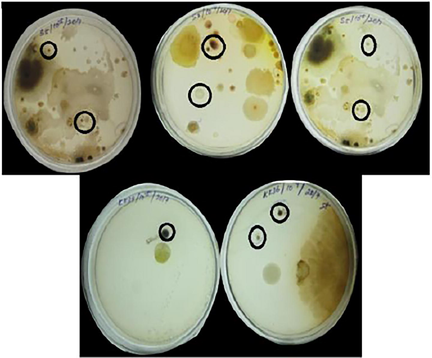
Cultures from different soil samples.
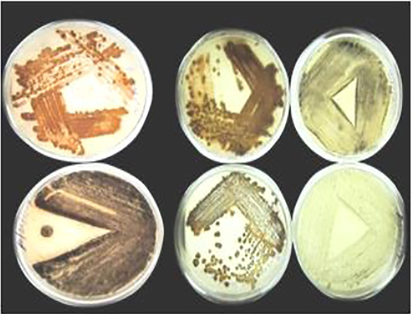
Some of the pure cultures.
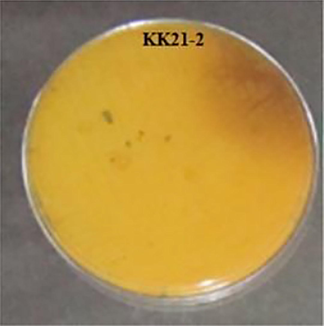
Pigment produced by KK21-2 strain.
3.2 Protein data Bank (PDB)
The 3D structures of Staphylococcus aureus target protein Dihydrofolate reductase (PDB ID: 6E4E) is retrieved from Protein Data Bank (Fig. 5).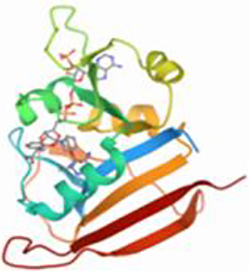
Dihydrofolate reductase (PDB ID: 6E4E).
3.3 LC-MS analysis
By using LC-MS analysis, the isolated strain KK21-2 aqueous 's extract had a total of 33 compound that could be identified. (Fig. 6). The compound name, retention time, mass of the compounds, molecular weight, molecular formula are listed in Table.1.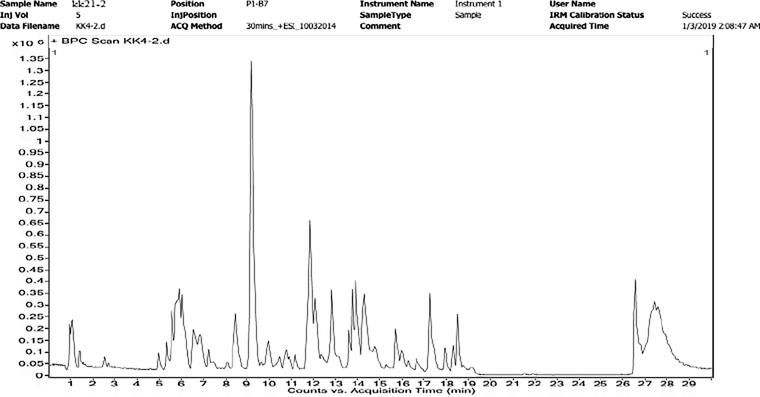
LC-MS study of KK21- 2 Aqueous extract.
S.No
Compound Name
Retention time
Mass from LC-MS
Molecular weight
Molecular formula
1
N-Ethylaniline
1.007
121.0886
121.0886
C8 H11 N
2
3-Pyridylacetic acid
1.015
137.0468
137.0468
C7 H7 N O2
3
Dextroamphetamine
1.057
135.104
135.104
C9 H13 N
4
Paramethasone
5.034
393.1982
392.1982
C22 H29 F O5
5
10-Deoxymethymycin
5.667
453.3132
453.3132
C25 H43 N O6
6
Naphthylglucuronide
6.629
320.0911
320.0911
C16 H16 O7
7
3′-Hydroxytrimethoprim
6.854
276.1184
276.1184
C13 H16 N4 O3
8
Umbelliferone
7.357
162.0305
162.0305
C9 H6 O3
9
5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5-phenylhydantoin
8.404
286.0949
286.0949
C15 H14 N2 O4
10
D-Biotin
8.442
244.0853
244.0853
C10 H16 N2 O3 S
11
Dihydrodeoxy streptomycin
9.163
567.2839
567.2839
C21 H41 N7 O11
12
PhePhe
9.437
312.1519
312.1519
C18 H20 N2 O3
13
IrigeninTrimethyl Ether
10.447
402.1273
402.1273
C21 H22 O8
14
Haematoxylone
11.144
372.1179
372.1179
C20 H20 O7
15
His Met Lys
11.768
414.2012
414.2012
C17 H30 N6 O4 S
16
Arg Val Trp
11.77
459.2581
459.2581
C22 H33 N7 O4
17
TrpGlu Pro
11.87
392.2191
430.1743
C21 H26 N4 O6
18
Hydroxycortisol
11.872
378.5000
378.5000
C21H30 O6
19
4-Biphenylamine
12.324
169.0885
169.0885
C12 H11 N
20
Fluorenylacetamide
12.407
223.0983
223.0983
C15 H13 N O
21
Dactinomycin
13.157
254.6195
1254.6195
C62 H86 N12 O16
22
N-acetyl aspartyl glutamicAcid
13.309
304.0863
304.0863
C11 H16 N2 O8
23
Phthalic acid Mono-2-ethylhexyl Ester
14.238
278.1502
278.1502
C16 H22 O4
24
Gemeprost
14.462
394.2662
394.2662
C23 H38 O5
25
Hydroxyhydroquinone
14.556
126.0333
126.0332
C6 H6 O3
26
PGF2alpha isopropylester
14.785
396.2838
396.2838
C23 H40 O5
27
GlnGln Lys
15.385
402.2214
402.2214
C16 H30 N6 O6
28
2-Amino-3-hydroxy-5-nitrobenzophenone
15.977
258.0582
258.0582
C13 H10 N2 O4
29
16b-Hydroxyestradiol
16.054
288.1721
288.1721
C18 H24 O3
30
Leupeptin
16.067
426.2921
426.2921
C20 H38 N6 O4
31
5-Amino-6-(5′-phosphoribosylamino)uracil
17.673
354.0577
354.0583
C9 H15 N4 O9 P
32
3,3′,5,5′-Tetra-tertbutyl-4,4′-dihydroxybiphenyl
17.987
410.3177
410.3177
C28 H42 O2
33
12beta-Hydroxy-3-oxo-5beta-cholan-24-oic Acid
18.535
390.2735
390.2735
C24 H38 O4
3.4 ADME properties
The 3D structures of 33 LC-MS compounds detected by of Aequous extract of actinomycetes were retrieved, more investigation on these compounds ADMET analysis using QikProp. It revealed that 25 compounds were within the range, hence proceed for further analysis. The remaining 8 compounds viz., His Met Lys, Arg Val Trp, Phe Phe, Gln Gln Lys they are amino acids which may be the broth component so excluded. 10-Deoxymethymycin, Dactinomycin, were excluded as they didn’t obey Lipinski’s rule, had three reactive groups and four violation in Rule of 5. N-acetyl aspartyl glutamic acid and Leupeptint the Human Oral Absorption < 25%. The remaining twenty five compounds were listed and further analysed in Table.2. QPlogPo/w-Octonal/water partition coefficient, QPlogS-Water solubility, QPPCaco = Predicted apparent Caco-2 cell permeability in nm/sec, QPlogHERG = Predicted IC50 value, QPPMDCK = Predicted apparent MDCK cell on 0 to 100% scale. % of HOA = <25% PSA = Van der Waals surface area of polar nitrogen and oxygen atoms, HD- Hydrogen bond Donor, HA- Hydrogen bond Acceptor.
S.No
Lead molecules
QPlog
Po/w
QplogS
Qpp
Caco
Qplag
HERG
QPP
MDCK
% of HOA
PSA
HB
Donars
HB Acceptor
1
N-Ethylaniline
1.249
−2.737
70.802
−4.776
28.274
67.371
115.488
2
3.75
2
3-Pyridylacetic acid
1.273
−1.501
105.192
−2.358
55.165
70.588
63.161
1
3.5
3
Dextroamphetamine
7.599
−8.451
5378.368
−4.692
3048.574
100
30.234
2
1.5
4
Paramethasone
2.78
−2.787
2579.965
−4.836
1378.021
100
26.305
1.5
1
5
Naphthylglucuronide
−2.877
−1.668
0.05
−0.36
0.019
77.42
241.007
8.5
15.1
6
3′- Hydroxytrimethoprim
2.788
−3.308
590.366
−4.704
309.627
92.871
86.553
1
11.1
7
Umbelliferone
3.816
−5.255
41.785
−2.063
20.336
78.30
97.061
2
5.7
8
5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)
-5-
phenylhydantoin2.448
−3.544
498.896
−3.797
233.311
89.566
65.04
3
4.15
9
D-Biotin
0.875
−0.982
66.494
5.476
68.914
15.571
363.96
2
32.5
10
Dihydrodeoxystreptomycin
1.302
−3.045
25.619
−1.911
18.484
59.77
107.152
3
4.5
11
IrigeninTrimethyl Ether
1.759
−0.523
920.538
−4.459
500.453
90.297
24.988
2
1
12
Haematoxylone
−5.787
−0.389
0.055
−5.763
0.014
94.02
314.517
16
24.2
13
Hydroxycortisol
2.254
−2.961
1544.214
−4.354
791.262
100
87.814
10
7.75
14
4-Biphenylamine
−2.608
−0.787
0.132
−2.133
0.149
67.99
193.134
6.5
9.5
15
Fluorenylacetamide
0.584
−2.934
92.18
−3.505
37.605
65.53
126.041
4
9.85
16
Phthalic acid Mono-2-ethylhexyl Ester
3.642
−3.739
5759.916
−4.799
3282.992
100
75.659
2
7
17
Gemeprost
1.28
−4.589
23.806
−4.528
16.766
59.079
172.933
1
10.5
18
Hydroxyhydroquinone
−0.818
−0.516
0.031
3.808
0.039
80.80
214.406
3.25
9.25
19
PGF2 alpha isopropyl ester
2.257
−1.651
7216.27
−4.025
4188.75
100
12.791
1
1
20
2-Amino-3-hydroxy-5-nitrobenzophenone
1.938
−3.792
191.673
−3.878
127.979
79.145
106.937
3
8.15
21
16b-Hydroxyestradiol
4.14
−5.691
293.215
−5.369
131.353
95.343
98.458
3
7.1
22
Leupeptin
6.229
−1.856
28.19
−2.24
19.996
100
101.383
3.25
4.75
23
5-Amino-6-(5′-phosphoribosylamino)uracil
3.771
−4.564
130.498
−3.505
69.64
86.888
78.334
1
4
24
3,3′,5,5′-Tetra-tertbutyl-
4,4′-dihydroxybiphenyl1.28
−4.589
23.806
−4.528
16.766
59.079
172.933
1
10.5
25
12beta-Hydroxy-3-oxo-5beta-cholan-24-oic Acid
0.709
−1.42
623.261
−3.749
296.765
81.115
63.234
1
3.25
3.5 Active site prediction
The active site of selected target S.aureus is identified from the Uniprot and Schrodinger sitemap module. From the active site residues some may identified as important binding from the Uniprot. (Table 3).
S.No
Targets
Active site residues
1
Dihydrofolate reductase
L5, V6, A7, I14, I15, G16, F17, N18, N19, Q20, P22, W23,L24,L25,D27,D28,K30,H31,M43,G44,R45, T46, S49, S50, P54,R58, V62, L63, T64, F92, F93, G94, G95, Q96, T97,L98, E101, I111, G120, T121, T122
3.6 Docking analysis
The selected target of Dihydrofolate reductase (6E4E) was docked with selected twenty five compounds and the specific drug by using XP docking and top 3 compounds were put thru induced fit docking (IFD).
3.6.1 Staphylococcus aureus: Dihydrofolate reductase
The leading three substances from XP docking with Dihydrofolate reductase were 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5-phenylhydantoin with the dock score of −10.3 kcal/mol and 3H- bonds, PGF2alpha isopropyl ester with the dock score of −10.0 kcal/mol and 4H- bonds and Naphthyl glucuronide with the dock score of −9.2 kcal/mol and 3H- bonds. The drug Trimethoprim produced the dock score as −7.4 kcal/mol with 4H- bond interactions. Of the 25 compounds, 22 had docking with Dihydrofolate reductase protein. The XP docking results of top ten compounds with Dihydrofolate reductase are presented in Table. 4.
S.No
Compounds
Dock score
(kcal/mol)
H- bonds
Bond length (Å)
Compounds
1
5-(3,4-Dihydroxy-1,5-
cyclohexadien-1-yl)-5-
phenylhydantoin−10.3
Ala 7 (N–H),
Il 14 (O-H)
Ser 49 (O-H)2.14
2.43
2.48
2
PGF2alpha isopropylester
−10.0
Ala 7 (N–H)
Leu 24 (N-H)
Asp 27 (O-H),
Thr46 (O-H)2.01
2.13
2.39
2.26
3
Naphthyl glucuronide
−9.2
Leu 5 (O–H)
Asp 27 (O-H)
Phe 92 (O-H)1.97
1.70
1.81
4
D-Biotin
−8.5
Ala 7 (N–H)
Ile 14 (O-H)
Asn 18 (O-H)2.11
2.11
2.06
5
16b-Hydroxyestradiol
−8.3
Asp 27 (O–H)
Ser 49 (O-H)1.82
2.02
6
Hydroxycortisol
−8.1
Val 6 (O–H)
Asp 27 (O-H)2.02
2.18
7
Paramethasone
−7.8
Asp 27 (O–H)
2.09
8
Gemeprost
−7.8
Ala 7 (N–H)
Leu 24 (N-H)
Thr 46 (O-H)
2.40
2.15
2.13
9
3′-Hydroxytrimethoprim
−7.4
Leu 5 (O–H)
Asp 27 (O-H)
Asp 27 (O-H)
Phe 92 (O-H)1.86
2.13
2.07
1.71
10
12beta-Hydroxy-3-
oxo-5beta-cholan-24-oic Acid−7.0
–
–
Drug
Trimethoprim
−7.4
Leu 5 (O–H)
Asp 27 (O–H)
Asp 27 (O–H)
Phe 92 (O–H)
1.91
2.15
2.09
1.69
3.6.2 Induced fit docking
The IFD results of Dihydrofolate reductase exhibited variation in positions of the top three compounds and the dock scores. In the top position was 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5-phenylhydantoin with dock score of −12.8 kcal /mol and it had 5H- bonds with Ala 7, Ile 14, Thr 46, Phe 92, Thr 121. The second compound Naphthyl glucuronide which was in the third position in XP docking had dock score of −12.0 kcal/mol and 6H- bonds with Ala 7 (2), Ile 14, Thr 46, Phe 92 (2). The third compound in IFD was PGF2alpha isopropylester with dock score of −11.6 kcal /mol and it had 4H- bonds with Ala 7 (2), Asp 27, Phe 92. The drug Trimethoprim had the dock score of −10.7 kcal/mol and had 4H- bonds with Ala 7 (2), Ile 14, and Phe 92 (Table 5 and Fig. 7).
S.No
Compounds
Dock score
(kcal/mol)
H- bonds
Active site Residues
Bond length (Å)
Compounds
1.
5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)
-5-
phenylhydantoin−12.8
Ala 7 (H–O)
Ile14(O-H)
Thr 46 (H–O)
Phe 92 (O-H)
Thr 121 (O-H)Ala 7
Ile 14
Thr 46
Phe 92
Thr 1212.45
2.31
2.05
1.83
2.02
2
Naphthyl glucuronide
−12.0
Ala 7 (H–O)
Ala 7 (O-H)
Ile 14 (O-H)
Thr 46 (H–O)
Phe 92 (O-H)
Phe 92 (O-H)Ala 7
Ile 14
Thr 46
Phe 922.09
1.96
1.67
2.03
1.66
1.57
3
PGF2alpha isopropyl
ester−11.6
Ala 7 (H–O)
Ala 7 (O-H)
Asp 27 (O-H)
Phe 92 (O-H)
Ala 7
Asp 27
Phe 922.05
1.73
2.05
2.22
Drug
Trimethoprim
−10.7
Ala 7 (O–H)
Ala 7 (H–O)
Ile 14 (O–H)
Phe 92 (O–H)
Ala 7
Ile 14
Phe 92
1.82
2.10
1.89
2.06
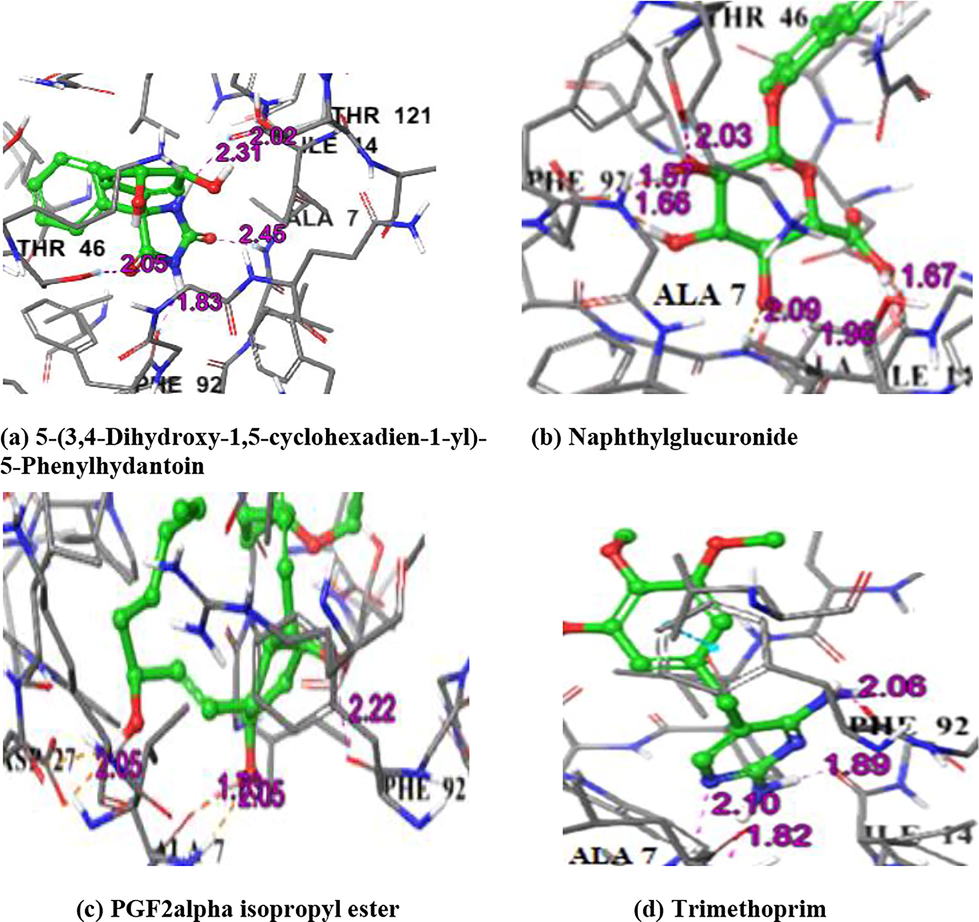
IFD complex structure of Dihydrofolate reductase with compounds and Trimethoprim drug.
3.7 Molecular dynamic simulation
3.7.1 RMSD analysis
The complex docked structure of Dihydrofolate reductase component with 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5- phenylhydantoin was done for MD simulation to assess stability. The RMSD values show the stability of complex structures via the examination of the RMSD plot. The equilibrium of protein and ligand are same and fluctuation of protein C alpha atoms and heavy ligand atoms were within the range 3 Å. Hence, the complex was more stable (Fig. 8).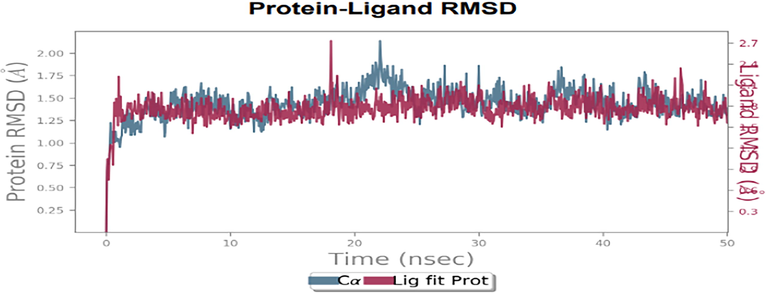
RMSD plot of the docked complex structure of Dihydrofolate reductase protein with 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5- phenylhydantoin.
3.7.2 RMSF analysis
Characterizing the changes in the protein chain during the simulation was done using RMSF analysis. The fluctuations of the aminoacids residues were less than all but N and C terminal residues. The residues from the region of 62–75 were slightly fluctuated but not the above the range of 3 Å (Fig. 9).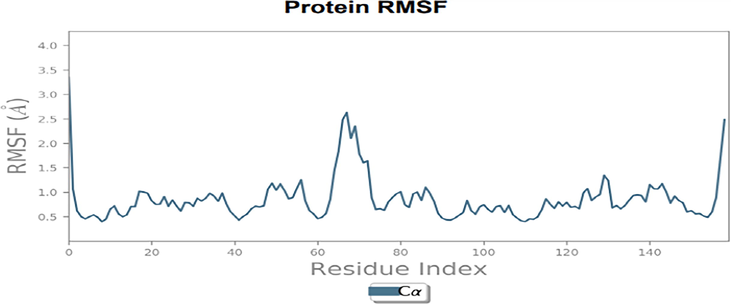
RMSD plot of Dihydrofolate reductase protein with 5-(3,4-Dihydroxy-1,5 cyclohexadien-1-yl)-5- phenylhydantoin.
3.7.3 Protein-ligand interactions
There are four different forms of protein–ligand interactions: hydrogen bonds, hydrophobic bonds, water bridges andionic bonds. Since they have an impact on drug selectivity, metabolization, and adsorption, H-bonds are crucial in the design of pharmaceuticals (Abuthakir et al., 2021). The complex structure of the histogram of Dihydrofolate reductase with 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5- phenylhydantoin demonstrates the presence of a strong hydrogen bonded between the ligand and binding site, essential residues. The residues Ala 07, Ile 14, Thr 46, Phe 92,Gln 95 and Thr 121 were involved in the hydrogen bond interactions, Leu 20 and Ile 50 were in hydrophobic contacts and water bridges with Ala 7, Asn 18 and Asp 120 (Fig. 10).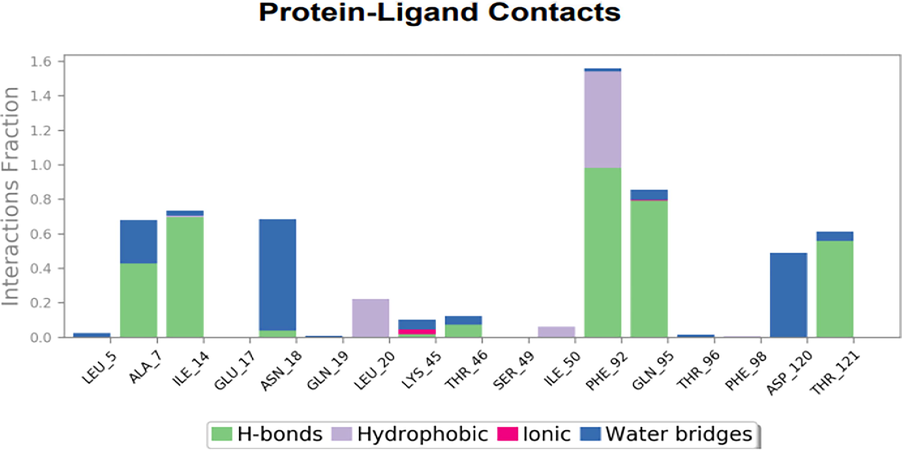
Graph illustrating the interactions between proteins and ligands in a docked complex structure of Dihydrofolate reductase protein with 5-(3,4-Dihydroxy-1,5 cyclohexadien-1-yl)-5- phenylhydantoin.
From the Timeline representation,the residues Ala 07, Ile 14, Asn 18, Phe 92, Gln 95, Asp 120 and Thr 121 maintained the interactions with maximum time during the entire simulation period.The 2D image of complexed structure was shows that the residues Ala 07, Ile 14,Asn 18, Phe 92, Gln 95 were maintained the interactions 80%, 85%, 75%, 98% and 85%, respectively of entire simulation period (Fig. 11). The 2D structure of the interaction of Dihydrofolate reductase protein with 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5 phenylhydantoin is mentioned below in (Fig. 12).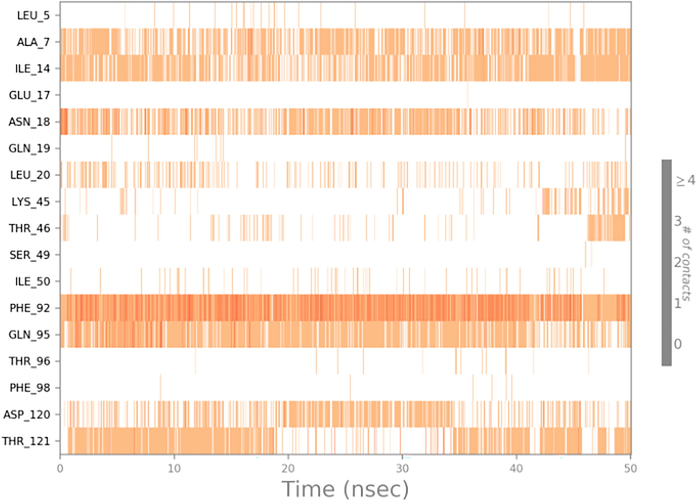
Timeline representation of complex of Dihydrofolate reductase protein with 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5- phenylhydantoin.
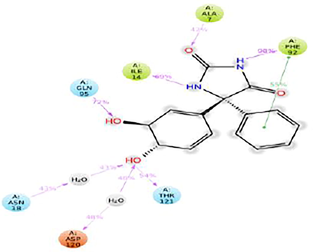
2D structure of interaction of Dihydrofolate reductase protein with 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5- phenylhydantoin.
4 Discussion
In the United States, cellulitis is a common bacterial skin illness that affects about 14 million people each year. It accounts for 3.7 billion in outpatient treatment expenses and 650,000 hospitalizations annually (Raff and Kroshinsky, 2016). Cellulitis is a deep skin infection with sore region of redness, resulting in fever for which early care with oral or intravenous antibiotics is suggested (Gabillot-Carre and Roujeau, 2007). In the present study, the targets of the pathogens causing this disease were selected and new drug / lead molecules have been identified from the compounds of soil actinomycetes by docking analysis using Glide software.
Staphylococcus aureus and Streptococcus pyogenes are the main pathogens involved in the Cellulites disease (Gunderson and Martinello, 2012). The 3D structure of the S.aureus target Dihydrofolate reductase was retrieved from PDB database. Some other targets are Dihydropteroate synthase, Penicillin binding protein 2a, Thymidylate synthase, 23 s rRNA subunit Methyltransferase these are already proven targets for the cellulites diseases. Folate metabolism inhibitors are widely acknowledged as potent treatments for rheumatoid arthritis, bacterial infections, and cancer. DHFR is a crucial enzyme for the metabolism of DNA, RNA and amino acids (Luo et al., 2006). One such protein, dihydrofolate reductase, has been a major antibacterial and anticancer therapeutic target because of its crucial function in nucleotide production.
Through LC-MS analysis of the aqueous extract, 33 compounds have been identified of which 25 compounds were subjected to ADME analysis after excluding 8 compounds that didn’t obey Lipinski’s rule and amino acids which may be from the broth component. All the 25 ligands selected from LC-MS analysis were docked with the target first in XP docking and top 3 from XP results were subjected to IFD and compared with corresponding drugs. The H-bond, ionic bond, hydrophobic bond and water bridges are the four important interactions of the protein- ligand docking of which the interaction between H-bonds is significant because of its effect on the drug specificity (Wade and Goodford, 1989).
More powerful DHFR inhibitors have recently been created through the use of structure-based design, and they are effective and selective against Staphylococcus aureus (Lam et al., 2014). dihydrofolate reductase, is the primary enzyme that produces the cofactors, essential for controlling the metabolism of folate (Schnell et al., 2004). Dihydrofolate reductase is very important in Dihydrofolate pathway as it has several roles (Fischer et al., 2010) and one of this is converting dihydrofolate to tetrahydrofolate thereby involved in purinesand thymidylate synthesis (Lin and Gerson, 2014).
The active site residues of S.aureus Dihydrofolate reductase are Val 6, Ala 7, Leu 20, Pro 25, Asp 27, Leu 28, Val 31, Ser 49, Ile 50, Arg 57, Phe 92, and Thr 111 (Kobayashi et al., 2014). The IFD results of the present study showed all the top 3 compounds, viz.5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5-phenylhydantoin (-12.8 kcal/mol), Naphthyl glucuronide (-12.0 kcal/mol) and PGF2alpha isopropyl ester (-11.6 kcal/mol) had lesser dock score than the drug Trimethoprim (-10.7 kcal/mol). The complex structure of the Dihydrofolate reductase with the 3 LC-MS compounds and the drug Trimethoprim showed the H-bond interactions with Ala 7, Tyr 121which are the active site residues reported by Kobayashi et al. (2014). With an four helices and eight-stranded sheet providing the place where substrate and cofactor are bound, The conserved fold shown in previously published crystal structures of DHFR from different species is maintained by the DHFR from the bacterium aureus. (Heaslet et al., 2009).
From the other targets the IFD results of the Dihydropteroate synthase, the selected compounds 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5-phenylhydantoin dock score was −8.1 kcal/mol and Naphthyl glucuronide dock score was −8.1 kcal/mol. This inhibitor is already known inhibitor for the cellulitis diseases and the corresponding drug sulfamethoxazole is −4.3 kcal/mol.
According to the Molecular Dynamics simulation study, the complex structure of Dihydrofolate reductase protein with 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5 phenylhydantoin was stable and active site residues are Ala 07, Ile 14, Asn 18, Phe 92, Gln 95 interacted. Based on the Molecular Dynamic Simulation study 5-(3,4-Dihydroxy-1,5cyclohexadien-1-yl) phenylhydantoin have suggest that this compound having antibacterial activity against the cellulitis skin disease for the pathogen Staphylococcus aureus. The compound 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5-phenylhydantoin is an organic compound. The present study conclude that the soil actinomycetes are rich source of antibiotics, the mentioned compound is a novel compound to the cellulitis diseases for the drug development.
5 Conclusion
The therapeutic target of DHFR inhibitors for cellulitis skin diseases was investigated using molecular docking studies. Many studies have been conducted on DHFR as a potential target for antimicrobial medicines. Of all the compounds, 5-(3,4-Dihydroxy-1,5-cyclohexadien-1-yl)-5-phenylhydantoin produced very significant docking results with all the selected target of S.aureus suggesting the possibility of broad spectrum multi-targetting efficacy, hence can be considered for further in vitro and in vivo testing for cellulitis diseases.
Acknowledgement
This work was funded by Researchers supporting Project number (RSP-2023R27), King Saud University, Riyadh, Saudi Arabia
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Screening Balanites aegyptiaca for inhibitors against putative drug targets in Microsporum gypseum – Subtractive proteome, docking and simulation approach. Infect. Genet. Evol.. 2021;90:104755
- [Google Scholar]
- Platyphylloside, a potential inhibitor from epicorp of B. aegyptiaca against CYP450 protein in T.rubrum- In vitro and in silico approaches. Saudi J. Biol. Sci.. 2022;29:3899-3910.
- [Google Scholar]
- Modifying effects of nerolidol on cell surface glycoconjugates and suppressed inflammation during DMBA-induced oral carcinogenesis: An in vivo and in silic. Biologia. 2022;78:529-541.
- [Google Scholar]
- Multiple grid arrangement improves ligand docking with unknown binding sites: Application to the inverse docking problem. Comput. Biol. Chem.. 2018;73:139-146.
- [Google Scholar]
- Prediction of protein-ligand binding poses via a combination of induced fit docking and metadynamics simulations. J. Chem. Theory Comput.. 2016;12(6):2990-2998.
- [Google Scholar]
- Cellulitis: current insights into pathophysiology and clinical management. Neth. J. Med.. 2017;75(9):366-378.
- [Google Scholar]
- The biosynthesis of folate and proteins and their enzymology. In: Lui L., Mander H.W., eds. Comprehensive natural products II chemistry and biology. Vol vol. 1. Oxford: Elsevier; 2010. p. :599-648.
- [Google Scholar]
- Insights into the molecular mechanisms of protein-ligand interactions by molecular docking and molecular dynamics simulation: A case of oligopeptide binding protein. Comput. Math. Methods Med.. 2018;2018:3502514.
- [Google Scholar]
- Acute bacterial skin infections and cellulitis. Curr. Opin. Infect. Dis.. 2007;20(2):118-123.
- [Google Scholar]
- Molecular docking studies and ADME prediction of novel isatin analogs with potent anti-ECFR activity. J. Med. Chem.. 2014;4(8):558-586.
- [Google Scholar]
- ADMET- score a comprehensive scoring function for evaluation of chemical drug likeness. Medchemcomm. 2019;10(1):148-157.
- [Google Scholar]
- A Systematic review of bacteremias in cellulitis and erysipelas. J. Infect.. 2012;64(2):148-155.
- [Google Scholar]
- Structural comparison of chromosomal and exogenous dihydrofolate reductase from Staphylococcus aureus in complex with the potent inhibitor trimethoprim. Proteins. 2009;76(3):706-717.
- [Google Scholar]
- In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med.. 2016;8(3):186-210.
- [Google Scholar]
- PUG-View: programmatic access to chemical annotations integrated in pubchem. J. Cheminf.. 2019;11(56):1-11.
- [Google Scholar]
- Identification of novel potential antibiotics against Staphylococcus using structure- based drug screening targeting dihydrofolate reductase. J. Chem. Inf. Model.. 2014;54(4):1242-1253.
- [Google Scholar]
- Structure-based design of new dihydrofolate reductase antibacterial agents: 7-(benzimidazol-1-yl)-2,4-diaminoquinazolines. J. Med. Chem.. 2014;57:651-668.
- [Google Scholar]
- Lin, Y., Gerson, S.L., 2014. Clinical Trials Using LV-P140K-MGMT for Gliomas. Gene Therapy Cancer (Third Edition) Chapter, 263, 37–391.
- Inhibition of human acetyl- and butyrylcholinesterase by novel carbamates of (−)- and (+)-tetrahydrofurobenzofuran and methanobenzodioxepine. J. Med. Chem.. 2006;49:2174-2185.
- [Google Scholar]
- PDBe: towards reusable data delivery infrastructure at protein data bank in Europe. Nucleic Acids Res.. 2017;46:D486-D492.
- [Google Scholar]
- Search and discovery of new antibiotics. In: Goodfellow M., Williams S.T., Mordarski M., eds. Actinomycetes in Biotechnology. London: Academic Press; 1988. p. :33-67.
- [CrossRef] [Google Scholar]
- Isolation and Characterization of Actinomycetes from Soil and Evaluation of Antibacterial Activities of Actinomycetes against Pathogens. Int. J. Appl. Biol. Pharm. Technol.. 2011;2:384-392.
- [Google Scholar]
- Cellulitis: A study of drug use evaluation in a tertiary care teaching hospital. Indian J. Pharmacy Practice. 2018;11(3):134-140.
- [Google Scholar]
- Computational tools and resources for metabolism-related property predictions. 1. Overview of publicly available (free and commercial) databases and software. Future Med. Chem.. 2012;4(15):1907-1932.
- [Google Scholar]
- Principles and Applications of Liquid Chromatography-Mass Spectrometry in Clinical Biochemistry. Clin. Biochem. Rev.. 2009;30(1):19-34.
- [Google Scholar]
- Understanding and predicting druggability. A Highthroughput method for detection of drug binding sites. J. Med. Chem.. 2010;53:5858-5867.
- [Google Scholar]
- Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct.. 2004;33:119-140.
- [Google Scholar]
- Syed abuthakir, Syed Abuthakir Mohamed Hussain, Sharmila Velusamy, Jeyam Muthusamy, 2019. Balanites aegyptiaca (L.) Del. for dermatophytoses: Ascertaining the efficacy and mode of action through experimental and computational approaches, Informatics in Medicine Unlocked 15, 100177.
- Key factors influencing ADME properties of therapeutic proteins: A need for ADME characterization in drug discovery and development. MAbs. 2016;8(2):229-245.
- [Google Scholar]
- Key topics in molecular docking for drug design. Int. J. Mol. Sci.. 2019;20(18):4574.
- [Google Scholar]
- The role of hydrogen-bonds in drug binding. Prog. Clin. Biol. Res.. 1989;289:433-444.
- [Google Scholar]
- DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res.. 2008;36:D901-D906.
- [Google Scholar]
- Accurate sequence- based prediction of catalytic residues. Bioinformatics. 2008;24:2329-2338.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jksus.2023.102762.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




